

Abstract
The interleukin (IL)-12p40 family of cytokines plays a critical role in the development of experimental autoimmune encephalomyelitis (EAE). However, the relative contributions of IL-12 and IL-23 to the pathogenic process remain to be elucidated. Here, we show that activation of uncommitted myelin-reactive T cells in the presence of either IL-12p70 or IL-23 confers encephalogenicity. Adoptive transfer of either IL-12p70– or IL-23–polarized T cells into naive syngeneic hosts resulted in an ascending paralysis that was clinically indistinguishable between the two groups. However, histological and reverse transcription–polymerase chain reaction analysis of central nervous system (CNS) tissues revealed distinct histopathological features and immune profiles. IL-12p70–driven disease was characterized by macrophage-rich infiltrates and prominent NOS2 up-regulation, whereas neutrophils and granulocyte–colony-stimulating factor (CSF) were prominent in IL-23–driven lesions. The monocyte-attracting chemokines CXCL9, 10, and 11 were preferentially expressed in the CNS of mice injected with IL-12p70–modulated T cells, whereas the neutrophil-attracting chemokines CXCL1 and CXCL2 were up-regulated in the CNS of mice given IL-23–modulated T cells. Treatment with anti–IL-17 or anti–granulocyte/macrophage-CSF inhibited EAE induced by transfer of IL-23–polarized, but not IL-12p70–polarized, cells. These findings indicate that autoimmunity can be mediated by distinct effector populations that use disparate immunological pathways to achieve a similar clinical outcome.
Experimental autoimmune encephalomyelitis (EAE) is an inflammatory demyelinating disease of the central nervous system (CNS) induced in laboratory animals by active immunization with myelin antigens or by the adoptive transfer of myelin-specific CD4+ T cells. It is widely used as an animal model of multiple sclerosis (MS) and as a prototype of organ-specific autoimmunity. Until recently, EAE and MS were considered Th1 diseases, mediated by IL-12p70–polarized, IFN-γ–producing effector cells. This impression was based, in large part, on the association between clinical disease activity and expression of IFN-γ and IL-12p40 (a subunit of IL-12p70) in CNS tissues, cerebrospinal fluid, and circulating leukocytes (1–3). In addition, activated macrophages are the predominant leukocyte in CNS infiltrates of afflicted animals and patients, similar to the infiltrates that characterize Th1-dependent hypersensitivity and antimicrobial responses in the periphery (4, 5).
Recent findings, however, suggest that the cytokine pathways underlying encephalitogenic T cell development and function are more complex than previously appreciated. Deficiency of IL-17 or IL-23 (a heterodimeric monokine composed of IL-12p40 and p19 chains that expands and/or stabilizes Th17 cells) (6, 7) confers partial or complete resistance, respectively, against MOG35-55–induced EAE in C57BL/6 mice, whereas deficiency of IFN-γ or IL-12p70 does not (8–10). Furthermore, myelin-specific Th17 cell lines that have been expanded with IL-23 are efficient autoimmune effector cells (11). Collectively, these observations invite an alternative interpretation of the mechanism of action of IL-12p40 in neuroinflammation; namely, that its role is in the production of IL-23 and reinforcement of the Th17 effector cell population, rather than (or in addition to) the production of IL-12p70 and promotion of Th1 differentiation.
Some investigators have assumed that the newly recognized importance of IL-23/Th17-dependent events in at least some forms of EAE negates the formerly favored model of pathogenesis that highlights IL-12p70/Th1-driven pathways. However, we and others have demonstrated that IL-12p70, as well as IL-23, directly promotes encephalitogenicity because ordinarily innocuous lineage-uncommitted or tolerized myelin-specific T cells acquire the ability to transfer disease after antigenic challenge in the presence of recombinant IL-12p70 (12, 13). This suggests that myelin-specific cells, cultured under conditions that favor the development of either Th1 or Th17 cells, are capable of mediating similar clinical syndromes, most likely via engagement of distinct proinflammatory pathways.
Indeed, here we show that IL-12– and IL-23–modulated T cell lines, derived from proteolipid protein (PLP)139–151/IFA-primed SJL donors, trigger a clinically indistinguishable myelopathy upon transfer into naive syngeneic hosts. Despite their similarities, the disease induced by each of these cell lines differs in CNS chemokine expression patterns as well as in the extent of optic nerve involvement and the composition and positioning of infiltrating leukocytes within the spinal cord at peak disability. Of greater therapeutic relevance, the two forms of EAE differ in responsiveness to specific immunomodulatory interventions.
RESULTS AND DISCUSSION
IL-12p70– and IL-23–polarized T cells induce EAE after adoptive transfer
We harvested draining LN cells (LNCs) from SJL mice that had been primed with PLP139–151 in IFA and cultured them with antigen under either neutral conditions (i.e., with antigen and an anti–IL-12p40–neutralizing antibody), or conditions favorable to the generation of Th17 (IL-23, IL-1α, anti–IL-4, and anti–IFN-γ), or Th1 (IL-12p70, IFN-γ, anti–IL-4, and anti–IL-23p19) cells. Cells cultured with IL-12p70 up-regulated the Th1-associated transcription factor, T-bet, and secreted IFN-γ and no detectable IL-17 during a 4-d primary culture. In contrast, cells stimulated with IL-23 up-regulated the Th17-associated transcription factor, RORγt, and secreted large quantities of IL-17 and relatively small quantities of IFN-γ (Fig. 1, A and B). Cells from all three groups proliferated and secreted IL-2 to a comparable extent in response to antigenic stimulation (not depicted). As we have previously reported (13), PLP/IFA-primed LNCs failed to transfer EAE to naive syngeneic hosts after culture with antigen and anti–IL-12p40 or antigen alone. In contrast, cells stimulated with antigen and either IL-12 or IL-23 reproducibly induced EAE, manifested as an ascending paralysis, in 100% of hosts. Peak and cumulative disease scores and rate of disability progression were similar between the two groups (Fig. 1 C).
Figure 1.

IL-12– and IL-23–modulated myelin-specific T cells induce EAE in naive hosts. (A) PLP139–151/IFA-primed T cells were cultured with antigen under conditions favorable to the generation of Th1 cells (IL-12, IFN-γ, αIL-4, and αIL-23p19), Th17 cells (IL-23, IL-1, αIFN-γ, and αIL-4), or lineage-uncommitted cells (αIL-12p40). Supernatants were harvested every 24 h and analyzed by ELISA. (B) RNA was extracted at 96 h from LNC cultures as described in A for measurement of transcription factors by real-time RT-PCR. Levels were normalized to GAPDH. The data are shown as fold induction over the level in cells cultured under neutral conditions. (C) Purified T cells from IL-12– and IL-23–modulated cultures were transferred to naive syngeneic recipients (25 × 106 cells/mouse). Mice were killed at peak disease for histological analysis. The data shown are representative of four independent experiments. Asterisks indicate a significant difference when compared with the control group. *, P < 0.05 compared with recipients of lineage uncommitted T cells.
Patterns of CNS infiltration and proinflammatory molecule expression distinguish EAE induced by IL-23– versus IL-12p70–modulated cells
There was no significant difference in the number of CNS-infiltrating cells per mouse when comparing recipients of IL-12– and IL-23–modulated cells. CNS mononuclear cells from host mice that received IL-12p70–modulated cells consisted almost exclusively of IFN-γ producers, indicating that the donor cells had maintained a Th1 phenotype after adoptive transfer (Fig. 2, A and B). Conversely, the majority of CNS mononuclear cells from recipients of IL-23–modulated cells secreted IL-17 (at a 30-fold higher frequency over recipients of IL-12p70–modulated cells). These cells also included a significant population of IFN-γ producers.
Figure 2.

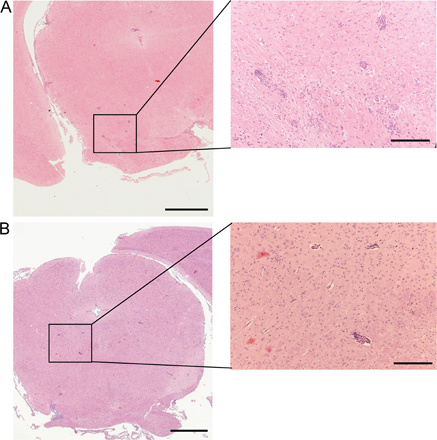
IL-12– and IL-23–modulated T cells induce forms of EAE with distinct cytokine profiles and histological characteristics. (A) RNA was isolated from spinal cords of the recipients of IL-12– and IL-23–modulated T cells at the time of peak disease. Cytokine mRNA levels were measured by real-time RT-PCR and normalized to GAPDH. The data are shown as fold increase over levels in naive cords. (B) Mononuclear cells were isolated from pooled spinal cords at peak EAE for measurement of cytokines by ELISPOT assay. (C) Representative spinal cord sections are from adoptive transfer recipients that were killed upon reaching a clinical score of 3. Bars: left, 200 μm; right, 50 μm. (D) Optic nerves from representative mice with a clinical score of 3 were fixed and stained with hematoxylin and eosin. Histological scores were calculated from a total of 25 sections (obtained from 10 optic nerves) per group using ImageJ software. Bar, 100 μm. **, P < 0.001; *, P < 0.05 compared with recipients of IL-12–modulated cells.
The spatial distribution of leukocytes within the spinal cord differed consistently between the two groups. Inflammatory infiltrates were restricted to the meninges and subpial white matter in sections analyzed from recipients of IL-12–modulated cells (Fig. 2 C, top). In contrast, infiltrating leukocytes typically extended from the subpial surface deep into the white matter parenchyma in recipients of IL-23–modulated cells (Fig. 2 C, bottom). In addition, optic nerve inflammation was more severe in the recipients of IL-23–modulated cells based on a significantly higher number of infiltrating cells per section (Fig. 2 D). Brainstem inflammation was scant in both groups and consisted of occasional perivascular infiltrates (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20080159/DC1).
Neutrophils are a conspicuous component of CNS infiltrates in mice injected with IL-23–modulated, but not IL-12–modulated, T cells
The cellular composition of CNS–infiltrating cells in each group at peak EAE was determined by flow cytometric analysis. A significant subpopulation of MHC class II− CD115− Ly6G+ 7/4+ leukocytes was present among the CNS mononuclear fraction isolated from recipients of IL-23–modulated, but not IL-12–modulated, cells (Fig. 3 A, C, and D). The identification of these cells as neutrophils was corroborated by cytospin preparation and Giemsa staining after FACS sorting (not depicted). The accumulation of neutrophils in the CNS was intriguing because granulocytes have been shown to play a critical role in EAE induced by active immunization in SJL mice and to increase blood–brain barrier permeability in other models of neuroinflammation (14, 15). Spinal cords from recipients of IL-23–modulated cells expressed elevated levels of G-CSF, a neutrophil activation and growth factor (Fig. 4 C). G-CSF was detectable in the sera of mice with IL-23–driven EAE at peak disease, but not in the sera of mice with EAE mediated by IL-12–modulated cells (unpublished data). Interestingly, G-CSF mRNA has been detected in MS lesions, and administration of recombinant G-CSF has been associated with acute MS flares in patients undergoing bone marrow transplantation (16, 17).
Figure 3.

CNS inflammatory infiltrates induced by IL-12– and IL-23–modulated T cells differ in cellular composition. (A and B) Mice were killed at peak EAE, and flow cytometry was performed on mononuclear cells isolated from pooled brains and spinal cords (five to six mice/group). Peripheral blood monocytes were analyzed in parallel. Plots in A are gated on class II− cells. (C and D) Absolute cell numbers were calculated as the product of the average number of CNS mononuclear cells harvested per mouse (pooled from brain and spinal cord) and the percentage of CD11b+ class II+ or Ly6G+7/4+ cells (determined by flow cytometry). The data shown are representative of three independent experiments.
Figure 4.

IL-12– and IL-23–modulated T cells induce distinct panels of chemokines and effector molecules in the CNS. Real-time RT-PCR was performed using RNA isolated from individual spinal cords at peak disease. Levels of ELR+ CXC chemokines (A), ELR− CXC chemokines (B), and G-CSF and NOS2 (C) were normalized to GAPDH and averaged over four to six mice per group. The data shown are representative of three independent experiments. **, P < 0.001; *, P < 0.05 by comparison to recipients of IL-12–modulated T cells.
In contrast, CNS-infiltrating cells from the recipients of IL-12–modulated cells were composed almost exclusively of monocytes and lymphocytes. Spinal cords from these mice contained a higher percentage and absolute number of MHC class II+ CD11b+ cells and large amounts of NOS2, a product of activated macrophages and microglia (Figs. 3, B–D, and 4 C).
Adoptive transfer of IL-12– and IL-23–modulated T cells induces distinct chemokine profiles in the inflamed CNS
The above findings suggest that IL-23–modulated, as opposed to IL-12p70–modulated, cells induce a CNS microenvironment that is favorable for the accumulation of neutrophils. Because the cellular composition of inflammatory infiltrates is influenced by local chemokine production, we compared CNS expression of a panel of chemokines in IL-12– and IL-23–driven EAE by real-time RT-PCR. Adoptive transfer recipients of IL-23–modulated cells preferentially up-regulated the neutrophil-attracting ELR+ CXC chemokines, CXCL1 and CXCL2, in the brain and spinal cord (Fig. 4 A and not depicted). Conversely, recipients of IL-12–modulated cells preferentially up-regulated the monocyte- and lymphocyte-attracting ELR− CXC chemokines, CXCL9, CXCL10, and CXCL11 (Fig. 4 B).
Mice with EAE induced by IL-12– or IL-23–modulated T cells respond differently to specific immunomodulatory interventions
We next questioned whether IL-12– and IL-23–driven forms of EAE would respond differently to specific immunomodulatory interventions. Administration of a neutralizing antibody against IL-17 blocked disease mediated by IL-23–modulated, but not IL-12–modulated, T cell lines (Fig. 5 A). This result supports our contention that Th17 cells and/or IFN-γ/IL-17 double producers within the IL-23–modulated cell lines are critical for disease transfer. Furthermore, it indicates that EAE induced by IL-12–modulated cell lines is not mediated by a contaminating subset of Th17 cells.
Figure 5.

EAE mediated by IL-12– or IL-23–polarized T cells responds differently to specific immunomodulatory interventions. Adoptive transfer recipients were injected with neutralizing antibodies specific for IL-17 (A), GM-CSF (B), TNF-α (C), IFN-γ (D), or isotype control. Antibody injections were given either every 4 d (A, C, and D) or every 2 d (B) from day 4 after transfer (0.4 mg/dose). Each treatment group consisted of five to seven mice. Data shown are representative of two or more experiments with similar results. **, P < 0.005; *, P < 0.05 compared with the group treated with isotype control.
GM-CSF is induced by Th17 cells and drives myeloid cell mobilization and differentiation during inflammation (18). GM-CSF plays a critical role in the development of EAE in MOG35–55–immunized C57BL/6 mice because its deficiency or neutralization results in resistance (19). Within our experimental paradigm, we found that GM-CSF neutralization is selectively effective for the suppression of EAE induced by IL-23–modulated cells (Fig. 5 B).
Th1 cells frequently secrete TNF-α. In addition, TNF-α acts synergistically with IL-17 to trigger cytokine expression and to promote autoimmune responses in animal models of arthritis (18). Neutralization of TNF-α has been shown to have beneficial as well as disease-exacerbating effects in EAE depending on the particular model used and the timing of antibody administration (20, 21). As illustrated in Fig. 5 C, administration of a monoclonal antibody to TNF-α ameliorated disease in recipients of either IL-12– or IL-23–modulated, PLP-reactive cells. In contrast, neutralization of IFN-γ did not suppress adoptively transferred EAE, regardless of the polarizing conditions used to generate encephalitogenic cell lines (Fig. 5 D). In fact, animals treated with anti–IFN-γ tended to experience a more severe course. The latter result is consistent with previously published studies demonstrating that IFN-γ deficiency paradoxically increases susceptibility to autoimmune CNS demyelination, even in models that are IL-12p40 or IL-12p70 dependent (10). We have found that IL-12 directly stimulates myelin-reactive T cells to up-regulate P-selectin ligand and CCR5 by IFN-γ–independent pathways, thereby facilitating CNS homing (22, 23). On the other hand, a Th1-driven model of uveitis, elicited with retinal antigen-pulsed dendritic cells, is dependent on recipient-produced IFN-γ (24). Hence, even among Th1-mediated, organ-specific autoimmune diseases, cytokine requirements are variable.
It has long been proposed that autoimmune demyelinating disease and even “garden-variety” relapsing-remitting MS represent a spectrum of conditions with distinct pathophysiological etiologies but a common clinical outcome (26). Our experiments provide “proof of principle” in demonstrating that IL-12– and IL-23–modulated cells transfer a clinically indistinguishable ascending myelitis by using distinct proinflammatory pathways based on CNS chemokine, cytokine, and effector molecule profiles and on the cellular composition and spatial distribution of inflammatory infiltrates. Most strikingly, the two forms of EAE respond differently to specific immunomodulatory interventions; neutralization of either GM-CSF or IL-17 suppressed EAE induced by IL-23–modulated, but not IL-12–modulated, myelin-specific T cells. By extension, the variable responsiveness of patients with MS to therapy with IFN-β or glatiramer acetate might reflect differences in underlying immunopathogenic mechanisms.
There is growing evidence that myelin-specific Th17 cells and/or IL-17/IFN-γ double-producing cells are the critical effectors in EAE induced by the active immunization of C57BL/6 mice with MOG35–55 (8, 11, 25). Interestingly, expression of GM-CSF and IL-23p19, but not IL-12p35, is required for the manifestation of disease in this model (8, 9, 19). IL-17 is a potent inducer of GM-CSF in a wide spectrum of cell types (18). During EAE, GM-CSF could stimulate the mobilization and activation of monocytes that, in turn, present myelin antigens to autoreactive T cells and provide a source of CNS-infiltrating macrophages. Our finding that anti–GM-CSF treatment was selectively effective in suppressing IL-23–driven disease suggests that IL-12–driven disease invokes alternative, as of yet undiscovered pathways to recruit myeloid cells into the pathogenic process.
It could be argued that the histopathological and immunological features of IL-23–driven EAE are more reminiscent of rare variants of autoimmune demyelination, such as Marburg's disease, acute disseminated encephalomyelitis, and neuromyelitis optica, as well as the opticospinal form of MS that has been described in Asia. Each of those conditions is characterized by the presence of neutrophils in white matter lesions (27, 28). Elevated levels of IL-8 (in the same family as CXCL1 and CXCL2) have been detected in the cerebrospinal fluid of patients with the opticospinal variant (28). Interestingly, both the opticospinal variant and Devic's disease specifically target the spinal cord and optic nerves, and IL-23–driven EAE is manifested by myelitis with severe optic neuritis.
On the other hand, IL-12–driven EAE more closely resembles conventional relapsing-remitting MS in the prominence of activated macrophages and dearth of neutrophils in CNS infiltrates and preferential up-regulation of the chemokines CXCL9, CXCL10, and CXCL11. Our study has important implications with regard to the future use of agents that target specific cytokines, chemokines, or effector molecules in MS and related inflammatory demyelinating disease. It is likely that immunomodulatory regimens will have to be customized based on the dominant Th effector pathways that underlie each individual patient's condition.
MATERIALS AND METHODS
Mice.
6–8-wk-old female SJL/J mice were obtained from NCI Frederick and housed in microisolator cages. All animal protocols were approved by the University of Michigan Committee on Use and Care of Animals.
Immunization and T cell culture.
Mice were injected s.c. with 100 μg PLP139–151 HSLGKWLGHPDKF (Quality Controlled Biochemicals) in IFA (Difco). Draining LNs were harvested 10–14 d later and passed through a 70-μm cell strainer (BD Falcon). LNCs were cultured in vitro for 4 d with PLP139–151 under conditions favorable to the generation of either Th1 cells (5 ng/ml rmIL-12, 2 ng/ml rmIFN-γ, 10 μg/ml αIL-4 [clone 11B11], and 10 μg/ml αIL-23 p19 [CNTO209]) or Th17 cells (5 ng/ml rmIL-23, 10 ng/ml IL-1α, 25 μg/ml αIFN-γ [AN18], and 10 μg/ml αIL-4). Other cells were cultured with antigen under neutral conditions (10 μg/ml αIL-12 p40 [C17.8]). Mouse αIL-23p19 and αIL-17 were provided by J. Benson (Centocor).
ELISPOT assays.
CNS cells were cultured for 24 h in 96-well filtration plates (MAIP N4550; Millipore) at 7.5 × 103 cells/well in triplicate, with or without 50 μg/ml PLP139–151. Spots were counted using the CTL ImmunoSpot Analyzer (Cellular Technology) with ImmunoSpot software.
Induction of EAE and immunomodulatory treatment.
LNCs were harvested after 4 d of in vitro culture. CD4+ T cells were isolated via complement-mediated depletion of class II+ (clone BP107), CD24+ (clone J11D), and CD8+ cells (clone 3.155). Fico/Lite (Atlanta Biologicals) was then used to isolate live cells. CD4+ T cells (25 × 106 in 0.2 ml sterile PBS; 85–95% pure) were injected i.p. Mice were observed on a daily basis for signs of EAE and scored as described previously (10). Mean clinical scores were compared using Student's t test. The following antibodies were used for in vivo neutralization: anti–IL-17 (clone CNTO5824), anti–IFN-γ (XMG1.2), anti–GM-CSF (22E9.11), and anti–TNF-α (TN3-19). Rat IgG (Sigma-Aldrich) was used as a control for anti–IL-17, anti–IFN-γ, and anti–GM-CSF. Armenian hamster IgG (eBioscience) was used as a control for anti–TNF-α. The neutralizing efficacy of anti–IFN-γ monoclonal antibodies in vivo was demonstrated by suppression of CXCL9 mRNA in the spleen as measured by real-time RT-PCR (P < 0.01) (unpublished data).
RT-PCR.
CNS tissues were homogenized in Trizol reagent (Invitrogen). RNA was isolated and cDNA was synthesized using a Quantitect reverse transcription kit (QIAGEN). Primers and probes were designed using Beacon Designer and synthesized by Integrated DNA Technologies. For RORγt, a previously published primer/probe set was used (29). Samples were analyzed on an iCycler PCR machine (Bio-Rad Laboratories). All data were normalized to GAPDH and expressed as fold induction over naive unless otherwise stated. REST 2005 software was used to analyze data and determine statistical significance.
Flow cytometry.
Brain and spinal cord were harvested at peak disease, homogenized in 1 mg/ml DNase and 2 mg/ml collagenase, and incubated for 1 h at 37°C. Mononuclear cells were then isolated over a 30/70% Percoll gradient (GE Healthcare). Flow cytometry was performed using a BD FACSCalibur.
Histological staining and analysis.
Paraformaldehyde-fixed CNS and optic nerve sections were stained with hematoxylin and eosin using standard protocols (10). Optic nerve sections were imaged and analyzed using ImageJ software. Histological sections were scored by an investigator blinded to experimental group as follows: 0, no infiltration (<50 cells); 1, mild infiltration of nerve or nerve sheath (50–100 cells); 2, moderate infiltration (100–150 cells); 3, severe infiltration (150–200 cells); 4, massive infiltration (>200 cells).
Online supplemental material.
Fig. S1 shows perivascular infiltrates in the mesencephalon of representative mice with EAE mediated by IL-12– or IL-23–modulated myelin-specific T cells.
Supplementary Material
Acknowledgments
The authors would like to thank J. Benson of Centocor for generously providing monoclonal antibodies to mouse IL-23p19 and mouse IL-17, and Irah King for assistance in preparing monoclonal antibody to mouse GM-CSF.
This work was supported by grants from the National Multiple Sclerosis Society (RG 3866-A-3) and the National Institutes of Health (NS047687-01A1).
The authors have no conflicting financial interests.
References
- 1.Fassbender, K., A. Ragoschke, S. Rossol, A. Schwartz, O. Mielke, A. Paulig, and M. Hennerici. 1998. Increased release of interleukin-12p40 in MS: association with intracerebral inflammation. Neurology. 51:753–758. [DOI] [PubMed] [Google Scholar]
- 2.Moldovan, I.R., R.A. Rudick, A.C. Cotleur, S.E. Born, J.C. Lee, M.T. Karafa, and C.M. Pelfrey. 2003. Interferon gamma responses to myelin peptides in multiple sclerosis correlate with a new clinical measure of disease progression. J. Neuroimmunol. 141:132–140. [DOI] [PubMed] [Google Scholar]
- 3.van Boxel-Dezaire, A.H., S.C. Hoff, B.W. van Oosten, C.L. Verweij, A.M. Drager, H.J. Ader, J.C. van Houwelingen, F. Barkhof, C.H. Polman, and L. Nagelkerken. 1999. Decreased interleukin-10 and increased interleukin-12p40 mRNA are associated with disease activity and characterize different disease stages in multiple sclerosis. Ann. Neurol. 45:695–703. [DOI] [PubMed] [Google Scholar]
- 4.Merrill, J.E., D.H. Kono, J. Clayton, D.G. Ando, D.R. Hinton, and F.M. Hofman. 1992. Inflammatory leukocytes and cytokines in the peptide-induced disease of experimental allergic encephalomyelitis in SJL and B10.PL mice. Proc. Natl. Acad. Sci. USA. 89:574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Platt, J.L., B.W. Grant, A.A. Eddy, and A.F. Michael. 1983. Immune cell populations in cutaneous delayed-type hypersensitivity. J. Exp. Med. 158:1227–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oppmann, B., R. Lesley, B. Blom, J.C. Timans, Y. Xu, B. Hunte, F. Vega, N. Yu, J. Wang, K. Singh, et al. 2000. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 13:715–725. [DOI] [PubMed] [Google Scholar]
- 7.Zhou, L., I.I. Ivanov, R. Spolski, R. Min, K. Shenderov, T. Egawa, D.E. Levy, W.J. Leonard, and D.R. Littman. 2007. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 8:967–974. [DOI] [PubMed] [Google Scholar]
- 8.Cua, D.J., J. Sherlock, Y. Chen, C.A. Murphy, B. Joyce, B. Seymour, L. Lucian, W. To, S. Kwan, T. Churakova, et al. 2003. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 421:744–748. [DOI] [PubMed] [Google Scholar]
- 9.Gran, B., G.X. Zhang, S. Yu, J. Li, X.H. Chen, E.S. Ventura, M. Kamoun, and A. Rostami. 2002. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J. Immunol. 169:7104–7110. [DOI] [PubMed] [Google Scholar]
- 10.Segal, B.M., B.K. Dwyer, and E.M. Shevach. 1998. An interleukin (IL)-10/IL-12 immunoregulatory circuit controls susceptibility to autoimmune disease. J. Exp. Med. 187:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langrish, C.L., Y. Chen, W.M. Blumenschein, J. Mattson, B. Basham, J.D. Sedgwick, T. McClanahan, R.A. Kastelein, and D.J. Cua. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ichikawa, H.T., L.P. Williams, and B.M. Segal. 2002. Activation of APCs through CD40 or Toll-like receptor 9 overcomes tolerance and precipitates autoimmune disease. J. Immunol. 169:2781–2787. [DOI] [PubMed] [Google Scholar]
- 13.Segal, B.M., J.T. Chang, and E.M. Shevach. 2000. CpG oligonucleotides are potent adjuvants for the activation of autoreactive encephalitogenic T cells in vivo. J. Immunol. 164:5683–5688. [DOI] [PubMed] [Google Scholar]
- 14.McColl, S.R., M.A. Staykova, A. Wozniak, S. Fordham, J. Bruce, and D.O. Willenborg. 1998. Treatment with anti-granulocyte antibodies inhibits the effector phase of experimental autoimmune encephalomyelitis. J. Immunol. 161:6421–6426. [PubMed] [Google Scholar]
- 15.Carlson, T., M. Kroenke, P. Rao, T.E. Lane, and B. Segal. 2008. The Th17–ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 205:811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lock, C., G. Hermans, R. Pedotti, A. Brendolan, E. Schadt, H. Garren, A. Langer-Gould, S. Strober, B. Cannella, J. Allard, et al. 2002. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 8:500–508. [DOI] [PubMed] [Google Scholar]
- 17.Openshaw, H., O. Stuve, J.P. Antel, R. Nash, B.T. Lund, L.P. Weiner, A. Kashyap, P. McSweeney, and S. Forman. 2000. Multiple sclerosis flares associated with recombinant granulocyte colony-stimulating factor. Neurology. 54:2147–2150. [DOI] [PubMed] [Google Scholar]
- 18.Fossiez, F., J. Banchereau, R. Murray, C. Van Kooten, P. Garrone, and S. Lebecque. 1998. Interleukin-17. Int. Rev. Immunol. 16:541–551. [DOI] [PubMed] [Google Scholar]
- 19.McQualter, J.L., R. Darwiche, C. Ewing, M. Onuki, T.W. Kay, J.A. Hamilton, H.H. Reid, and C.C. Bernard. 2001. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J. Exp. Med. 194:873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu, J., M.W. Marino, G. Wong, D. Grail, A. Dunn, J. Bettadapura, A.J. Slavin, L. Old, and C.C. Bernard. 1998. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat. Med. 4:78–83. [DOI] [PubMed] [Google Scholar]
- 21.Probert, L., H.P. Eugster, K. Akassoglou, J. Bauer, K. Frei, H. Lassmann, and A. Fontana. 2000. TNFR1 signalling is critical for the development of demyelination and the limitation of T-cell responses during immune-mediated CNS disease. Brain. 123:2005–2019. [DOI] [PubMed] [Google Scholar]
- 22.Bagaeva, L.V., L.P. Williams, and B.M. Segal. 2003. IL-12 dependent/IFN gamma independent expression of CCR5 by myelin-reactive T cells correlates with encephalitogenicity. J. Neuroimmunol. 137:109–116. [DOI] [PubMed] [Google Scholar]
- 23.Deshpande, P., I.L. King, and B.M. Segal. 2006. IL-12 driven upregulation of P-selectin ligand on myelin-specific T cells is a critical step in an animal model of autoimmune demyelination. J. Neuroimmunol. 173:35–44. [DOI] [PubMed] [Google Scholar]
- 24.Tang, J., W. Zhu, P.B. Silver, S.B. Su, C.C. Chan, and R.R. Caspi. 2007. Autoimmune uveitis elicited with antigen-pulsed dendritic cells has a distinct clinical signature and is driven by unique effector mechanisms: initial encounter with autoantigen defines disease phenotype. J. Immunol. 178:5578–5587. [DOI] [PubMed] [Google Scholar]
- 25.Lucchinetti, C., W. Bruck, J. Parisi, B. Scheithauer, M. Rodriguez, and H. Lassmann. 2000. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47:707–717. [DOI] [PubMed] [Google Scholar]
- 26.Abromson-Leeman, S., R. Bronson, Y. Luo, M. Berman, R. Leeman, J. Leeman, and M. Dorf. 2004. T-cell properties determine disease site, clinical presentation, and cellular pathology of experimental autoimmune encephalomyelitis. Am. J. Pathol. 165:1519–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wingerchuk, D.M., V.A. Lennon, C.F. Lucchinetti, S.J. Pittock, and B.G. Weinshenker. 2007. The spectrum of neuromyelitis optica. Lancet Neurol. 6:805–815. [DOI] [PubMed] [Google Scholar]
- 28.Ishizu, T., M. Osoegawa, F.J. Mei, H. Kikuchi, M. Tanaka, Y. Takakura, M. Minohara, H. Murai, F. Mihara, T. Taniwaki, and J. Kira. 2005. Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain. 128:988–1002. [DOI] [PubMed] [Google Scholar]
- 29.Ivanov, I.I., B.S. McKenzie, L. Zhou, C.E. Tadokoro, A. Lepelley, J.J. Lafaille, D.J. Cua, and D.R. Littman. 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 126:1121–1133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}