

Abstract
Increased evidence suggests that cancer-associated inflammation supports tumor growth and progression. We have previously shown that semaphorin 4D (Sema4D), a ligand produced by different cell types, is a proangiogenic molecule that acts by binding to its receptor, plexin B1, expressed on endothelial cells (Conrotto, P., D. Valdembri, S. Corso, G. Serini, L. Tamagnone, P.M. Comoglio, F. Bussolino, and S. Giordano. 2005. Blood. 105:4321–4329). The present work highlights the role of Sema4D produced by the tumor microenvironment on neoplastic angiogenesis. We show that in an environment lacking Sema4D, the ability of cancer cells to generate tumor masses and metastases is severely impaired. This condition can be explained by a defective vascularization inside the tumor. We demonstrate that tumor-associated macrophages (TAMs) are the main cells producing Sema4D within the tumor stroma and that their ability to produce Sema4D is critical for tumor angiogenesis and vessel maturation. This study helps to explain the protumoral role of inflammatory cells of the tumor stroma and leads to the identification of an angiogenic molecule that might be a novel therapeutic target.
Semaphorins are a large family of either membrane-bound or secreted proteins that were originally described in the nervous system, where they are involved in the establishment of a correct neuronal network (1, 2). These ligands exert their activities by binding to high affinity receptors known as plexins, a family of transmembrane molecules that share structural homology with semaphorins themselves (3). An increasing amount of data show that these two protein families also exert their action outside the nervous system, as they play a critical role in cardiac and skeletal development (4, 5), immune response (6, 7), epithelial morphogenesis (8), and tumor growth and metastatization (9, 10). Moreover, studies suggest that these molecules are important players during angiogenesis, as they regulate blood vessel growth and endothelial cell (EC) homing during vessel development (11). Although secreted class III semaphorins inhibit EC migration and in vitro vessel formation (12), class IV semaphorins positively regulate angiogenesis (13, 14). Angiogenesis is a complex, multistep process in which ECs of preexisting blood vessels, challenged by appropriate biochemical and physical cues, dynamically remodel their cell-to-cell and cell-to-matrix adhesions and reorganize themselves into a mature, hierarchically organized system of hollow endothelial tubes that is eventually stabilized by interaction with pericytes.
We and others have identified semaphorin 4D (Sema4D), also known as CD100, as a potent proangiogenic molecule (13–17). Sema4D, originally discovered in the immune system, where it regulates B cell aggregation and survival and T cell activation (18, 19), can be found either as a 150-kD membrane-bound form or as a 120-kD soluble molecule; the latter is originated as a consequence of a proteolytic cleavage (17, 20, 21). Sema4D acts by binding to its high affinity receptor, plexin B1 (3), to an intermediate affinity receptor, plexin B2 (both of which are widely expressed) (22), or to a low affinity receptor, CD72, which is mainly expressed by cells of the hematopoietic lineage (23). Sema4D and plexin B1/B2 share structural homology with the tyrosine kinase receptors c-Met and Ron (24, 25). We have previously shown that activation of plexin B1 through Sema4D binding transactivates c-Met and promotes the invasive ability of tumor cells (8). Moreover, we have also demonstrated that Sema4D, upon binding plexin B1 and activating c-Met, promotes angiogenesis both in vitro and in vivo (13).
Reports have recently pointed out the close correlation between inflammatory infiltration of the tumor stroma and a high vascular grade (26, 27). Because Sema4D is produced by inflammatory cells present in the tumor microenvironment (7), we wanted to assess whether stroma-derived Sema4D could influence the tumorigenic ability of cells as well as their capacity to originate metastasis. In this paper, we show that in an environment lacking Sema4D, the ability of breast cancer cells to originate tumor masses and metastases is severely impaired. This impairment is paralleled by an altered vascularization inside the tumor. We also provide evidence that the main cells producing the semaphorin in the tumor stroma are the tumor-associated macrophages (TAMs), and that TAM-derived Sema4D is critical for tumor angiogenesis and for the maturation of blood vessels. This work thus highlights a novel function of cells involved in the inflammatory response of tumors and identifies Sema4D as a new player in the complex interaction between tumor cells and the tumor microenvironment.
RESULTS
Tumor growth and metastatic ability of breast cancer cells is impaired in Sema4D KO mice
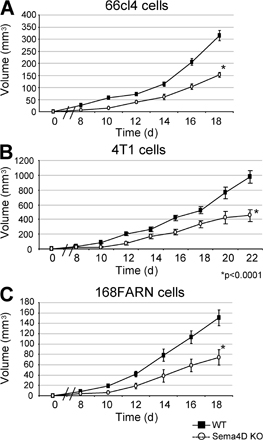
Previous works have shown that Sema4D is endowed with angiogenic properties, both in vitro and in vivo (13–15). However, Sema4D is dispensable for the formation of vessels during embryogenesis, as Sema4D KO mice are viable (19). To investigate if Sema4D might play a role in pathological vasculogenesis, such as that taking place in tumors, we grafted syngeneic tumor cells (TSA, breast cancer) both in WT and in Sema4D KO (KO) mice. As shown in Fig. 1 A, tumor growth was significantly impaired in mice lacking Sema4D in comparison to WT mice (an almost sixfold reduction at the end of the experiment; P < 0.0001). Similar experiments performed with three other breast cancer cell lines (66cl4, 4T1, and 168FARN) gave similar results, as the grafted cells originated smaller tumors in KO mice in comparison to the WT animals (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20072602/DC1).
Figure 1.

Tumor growth and metastatic ability of breast cancer cells is impaired in Sema4D KO mice. (A) TSA breast cancer cells were subcutaneously injected in WT and Sema4D KO mice. Tumor volume was evaluated at the indicated days. As shown, tumor burden was dramatically less in Sema4D KO versus WT mice. Bars indicate mean tumor volume ± SEM. *, P < 0.0001 (n = 10 mice per group). (B and C) Macro- and micrometastases present in the lungs of mice bearing tumors of comparable size were evaluated as indicated in Materials and methods and scored. Error bars indicate mean ± SD. *, P < 0.0001 (n = 10 lungs). (D) Representative pictures of lungs and lung slices for WT and Sema4D KO mice. As shown, the number of metastases was significantly lower in Sema4D KO mice. Statistical differences between groups were determined by using the two-tailed Student's t test. Bars: (whole mount) 0.5 cm; (micrograph) 0.2 mm.
We have previously demonstrated that Sema4D acts not only on ECs but also on tumor cells and that it stimulates their invasive ability (8). After proving that TSA cells do express plexin B1, the high affinity receptor for Sema4D, and display enhanced in vitro motility and invasiveness in response to the semaphorin (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20072602/DC1), we evaluated their metastatic ability in KO and WT mice. As shown in Fig. 1 (B–D), we observed a reduced incidence of metastases in KO versus WT mice (P < 0.0001). Collectively, these results indicate that the presence of Sema4D in the tumor microenvironment contributes to tumor growth and metastasis.
Maturation of tumor vessels is impaired in Sema4D KO mice
We wondered if the decreased tumorigenic ability observed in Sema4D KO mice could be caused by altered vasculogenesis. Thus, we stained slices of tumors of comparable size, grown either in WT or KO mice, with anti-CD31 antibody (a marker of ECs; Fig. 2 A). Although the total number of vessels was similar in the two types of animals (Fig. 2 B, top), the total vessel area and mean vessel size were significantly smaller in tumors grown in Sema4D KO mice (P < 0.0001; Fig. 2 B, middle and bottom).
Figure 2.

Tumor vascularization is altered in Sema4D KO mice. (A and B) Evaluation of tumor vascularization. Blood vessel staining of tumor histological sections was performed with an anti-CD31 antibody. The number and area of vessels were evaluated by fluorescence microscopy. As shown, although the number of vessels is not significantly different, vessel area and size are significantly reduced in Sema4D KO mice. Error bars indicate mean ± SD. *, P < 0.0001 (n = 8 mice per group, with 10 fields per animal). Bars, 100 μm. (C) To evaluate the presence of vessel-associated pericytes, tumor sections were costained with the pericyte-specific antibodies NG2 (red) and CD31 (green). Analysis was performed on one slice per mouse (n = 8 mice per group). As shown, vessels of tumors that originated in KO mice very rarely showed a normal interaction between ECs and pericytes, and in most cases they stained negative for the presence of pericytes (right), which were instead normally present in tumors originated in control mice (left). Statistical differences between groups were determined by using the two-tailed Student's t test. v.f., visual field. Bars, 50 μm.
Interactions between ECs and pericytes in the vessel walls have been recently identified as central processes in the regulation of vascular formation, stabilization, remodeling, and function (28). Failure of the interactions between these cell types has been described in some human pathologies, including tumor angiogenesis (29). Because pericytes are recruited by stromal-derived cytokines (30), we decided to verify if they were normally localized along the altered vessels of tumors grown in Sema4D KO mice. We thus stained tumor slices with anti-NG2, an antibody that identifies pericytes. As shown in Fig. 2 C, vessels of tumors originated in KO mice very rarely showed a normal interaction between ECs and pericytes. Most of them, in fact, were negative for the presence of pericytes, which were instead normally present in tumors that originated in control mice. Collectively, these observations suggest that Sema4D plays a role in tumor angiogenesis, where it contributes to the maturation of tumor vessels by acting on ECs and favoring the recruitment of pericytes.
Bone marrow–derived cells produce Sema4D that contributes to tumor growth and vessel organization
The results of the previous experiments suggest that cells of the tumor microenvironment produce the Sema4D that is required to promote tumor angiogenesis. To identify these cells, we performed immunofluorescent stainings on tumor slices. As shown in Fig. 3 A, most of the cells positive for Sema4D were also stained by an anti-CD45 mAb, suggesting that they belong to the leukocyte lineage (P < 0.0001).
Figure 3.

Bone marrow–derived cells produce Sema4D that contributes to tumor growth and vessel organization. (A) CD45+ cells express Sema4D. To evaluate whether CD45+ cells express Sema4D, tumor slices were costained with CD45 and Sema4D antibodies. Double-positive cells were then counted on 10 slices per group (one slice per tumor). (right) As shown, ∼85% of the leukocytes express Sema4D. Error bars indicate mean ± SD. *, P < 0.0001. Bar, 50 μm. (B) WT-derived bone marrow cells rescue tumor burden. (top) The mean volume ± SEM of grafted tumors in WT mice, Sema4D KO mice, and Sema4D KO mice transplanted with WT-derived bone marrow (BMT; n = 6 tumors per group). As shown, tumors grown in transplanted KO mice were bigger than those grown in nontransplanted KO mice. (bottom) PCR amplification of the ZFY1 gene on the Y chromosome from DNA derived from peripheral leukocytes. As shown, transplanted mice were chimeric, displaying ∼70% of leukocytes derived from the donors (for experimental details, see Materials and methods). W1 and W2 indicate representative transplanted mice, and female and male DNA are shown. (C) Sema4D KO bone marrow transplantation leads to decreased tumor burden in WT females. Lethally irradiated (900 cGy) WT females were transplanted with bone marrow cells derived from KO or WT male mice (n = 8 tumors per group). Bars indicate mean volume ± SEM. As in B, engraftment was confirmed by PCR analysis (bottom). K, Sema4D KO mice. (D and E) Tumor vessels resemble the phenotype of the original bone marrow donor when stained by CD31 antibody (compare with Fig. 2). D shows vessels, stained with CD31 antibodies, in a tumor grown in Sema4D KO mice transplanted with WT bone marrow. E shows slices derived from tumors grown in WT animals transplanted with WT (top) or KO (bottom) bone marrow. Statistical differences between groups were determined by using the two-tailed Student's t test. Bars, 100 μm.
To prove that bone marrow–derived cells are indeed the major producers of the semaphorin, we performed bone marrow transplantation experiments. We injected WT male–derived bone marrow cells into female KO animals. PCR analyses performed 2 wk later (evaluating the presence of the Y chromosome in peripheral leukocytes) showed that the mice were chimeric, with ∼70% of their leukocytes derived from WT marrow (Fig. 3 B, bottom). We thus injected these chimeric mice with tumor cells and evaluated tumor growth. As shown in Fig. 3 B, tumors grown in chimeric KO mice were bigger than those grown in KO animals. As expected, we found many CD45+ cells expressing Sema4D in tumors grown in KO mice transplanted with WT bone marrow (unpublished data).
We also performed the reverse experiment by analyzing tumor growth in lethally irradiated (900 cGy) WT female mice transplanted with bone marrow cells derived from KO male mice (Fig. 3 C, bottom). As shown in Fig. 3 C, tumors grown in WT mice transplanted with KO-derived bone marrow cells were smaller compared with those grown in WT animals transplanted with WT bone marrow. When we counted CD45+ cells inside tumors, we observed a slight decrease of bone marrow–derived cells in mice transplanted with KO bone marrow versus those transplanted with WT bone marrow (mean CD45+ cells/field = 48.5 ± 8.7 and 38.7 ± 7.8, respectively). When we analyzed tumor vascularization, we found a high vascular grade only in tumors grown in mice having WT bone marrows (either endogenous or transplanted; Fig. 3, D and E). Collectively, these experiments suggest that in the tumor microenvironment, bone marrow–derived cells produce the Sema4D that contributes to vessel maturation and, eventually, tumor growth.
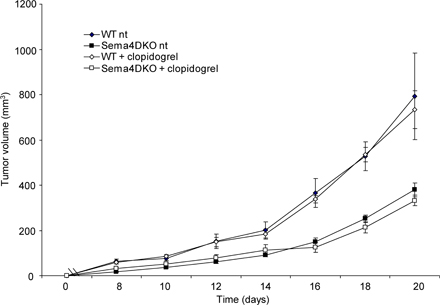
Sema4D produced by platelets does not contribute to tumor growth
It has been recently reported that activated platelets express Sema4D and that the surface expression of the semaphorin increases during platelet activation, followed by the gradual shedding of its extracellular domain (21). It is widely accepted that blood clotting can contribute to tumor growth and dissemination by creating a fibrin clot surrounding tumor cells that can protect them from apoptosis (31). We wondered if platelets could be the bone marrow–derived elements contributing the Sema4D responsible for tumor promotion. We thus grafted TSA cells in WT and in KO mice chronically treated with clopidogrel, a widely used inhibitor of platelet activation (32). Platelet inactivation was proven by analyzing the expression of CD62P on platelets obtained from treated and untreated animals. Mice treated with clopidogrel failed to expose CD62P on their membrane (unpublished data). As shown in Fig. S3 (available at http://www.jem.org/cgi/content/full/jem.20072602/DC1), we did not observe significant changes in the growth of tumor xenografts in mice in which platelet activation was inhibited. Similar results were obtained when platelet activation was inhibited by aspirin (unpublished data). These experiments indicate that platelet-derived Sema4D is not critical to elicit the tumorigenic ability of breast cancer cells.
Macrophages are the main source of Sema4D in the tumor microenvironment
As shown in Fig. 3 B, we have observed that transplantation of bone marrow from WT to KO animals could rescue the impairment of tumor growth. In recent years, compelling evidence supports the idea that TAMs, which account for the majority of the infiltrating inflammatory cells into the tumor mass and exhibit a phenotype distinct from any other activated monocytes, play an integral role during tumor progression (33). These cells, in fact, produce cytokines that aid the proliferation and survival of tumor cells, as well as their ability to invade surrounding tissues and metastasize (34–36). Immunofluorescence revealed that most of the leukocytes in the tumor stroma were indeed macrophages (Fig. 4 A), and that they expressed Sema4D (Fig. 4, B and C).
Figure 4.

TAMs produce Sema4D that acts on ECs. (A and B) Immunofluorescence analysis of tumors grown in WT or Sema4D KO animals. In A, staining with CD45 and F4/80 (specific for macrophages) reveals that most of the leukocytes in the tumor stroma are macrophages (∼90%; not depicted). In B, staining with F4/80 and Sema4D antibody reveals that TAMs express Sema4D. Bars, 50 μm. (C) Western blot analysis of total cell lysates of TAMs derived from WT or Sema4D KO mice. The blot was probed with anti-Sema4D antibodies and, as a loading control, with anti–β-actin. (D) PCR analysis performed on RNAs obtained from peritoneal macrophages of control mice (left) or of mice injected i.p. with CFA. As shown, only activated macrophage express Sema4D. (E) Transwell motility assays. The ability of supernatants of cultures of TAMs, purified from tumors grown either in WT or KO mice, to promote EC (HUVEC) migration was evaluated. Supernatants of WT TAMs efficiently promoted EC migration, whereas supernatants of KO TAMs were significantly less effective. Moreover, the chemoattractive activity of the WT supernatant was significantly decreased in the presence of Sema4D or plexin B1 blocking antibodies, and of the c-Met inhibitor PHA-665752. Chemoattractive positive controls included Sema4D (4D) and HGF, whereas anti-VSV was used as a control antibody. Bars represent the mean ± SD (experiments were performed twice in triplicate). Statistical differences between groups were determined by using the two-tailed Student's t test. *, P = 0.0003; **, P = 0.0002; ***, P = 0.0013; and #, P = 0.0001.
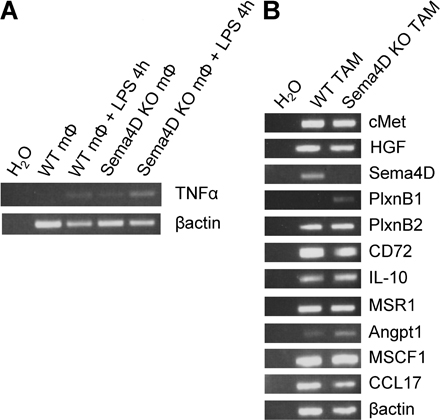
To evaluate if the expression of Sema4D is linked to monocyte/macrophage differentiation or to macrophage activation, we performed some in vivo experiments. We induced experimental peritonitis in mice by injection of CFA; 3 d later, we collected peritoneal macrophages and compared the expression of Sema4D on these cells versus resident peritoneal macrophages obtained from control mice. As shown in Fig. 4 D, although Sema4D was not expressed in resting macrophages, it was up-regulated in activated macrophages. Similar results were also obtained from the analysis of human cells; in fact, although monocytes did not express Sema4D, activated macrophages did (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20072602/DC1). Moreover, we found that, in vitro, Sema4D expression was not induced by differentiating stimuli (M-CSF, PMA, IL-10, or TGF-β), whereas it was induced by activating molecules (such as LPS; Fig. S4). All of these data suggest that the expression of Sema4D is not strictly liked to monocyte/macrophage differentiation, and that this molecule is expressed on different types of activated macrophages.
We then analyzed whether macrophages of KO mice behaved normally in term of response to activating stimuli and of cytokine production. As shown in Fig. S5 (available at http://www.jem.org/cgi/content/full/jem.20072602/DC1), macrophages from Sema4D KO mice were indeed able to respond to LPS by increasing the production of cytokines such as TNF-α. Moreover, we did not observe significant differences in cytokine production between WT- and KO-derived TAMs (Fig. S5), or in their ability to differentiate in M2 TAMs (as shown by the expression of macrophage scavenger receptor 1 [MSR1], a marker of this cell type). However, we observed that the mean number of macrophages recruited to the tumor was lower in Sema4D KO than in WT mice (19.03 ± 5.2 and 35.57 ± 4.2 F4/80+ cells/field, respectively).
To prove the role of TAM-produced Sema4D in tumor angiogenesis and growth, we performed several experiments. To begin, we purified and cultured TAMs from tumors grown both in WT and KO mice (37). Although the supernatant of cultures of WT TAMs was able to efficiently promote EC migration, the supernatant of KO TAMs was significantly less effective; moreover, the chemoattractive activity of WT supernatant was partially lost in the presence of Sema4D or plexin B1 blocking antibodies (Fig. 4 E) (16, 38).
Because we have previously shown that recruitment of the c-Met tyrosine kinase receptor by the activated Sema4D receptor (plexin B1) is required for the angiogenic ability of Sema4D, we wondered if c-Met inhibition impaired TAM-induced EC motility. In line with our previous results, inhibition of c-Met with the small molecule PHA-665752 (39) resulted in a significant decrease of EC motility in response to WT TAM supernatant (Fig. 4 E).
To evaluate the in vivo angiogenic role of TAMs, Matrigel plugs containing either WT or KO TAMs were subcutaneously implanted in KO mice. After 14 d, mice received dextran-FITC and were killed. As shown in Fig. 5 A, Matrigel plugs containing known angiogenic factors (such as hepatocyte growth factor [HGF] or basic fibroblast growth factor [b-FGF]) displayed several well-organized vessels, plugs containing WT TAMs were also well vascularized and showed well-formed and branched vessels, and plugs containing KO TAMs displayed small and poorly organized vessels. Addition of blocking plexin B1 antibodies (ec6.9), as well as of the c-Met inhibitor PHA-665752, resulted in the lack of an organized vessel tree, suggesting that activation of these two receptors is mandatory for the Sema4D-induced angiogenic response.
Figure 5.

Sema4D produced by TAMs promotes angiogenesis and increases the tumorigenic ability of cancer cells. (A) Matrigel plugs containing either WT or KO TAMs were subcutaneously implanted in KO mice. After 14 d, mice received dextran-FITC and were killed, and plugs were microscopically analyzed. As shown, plugs containing WT TAMs were well vascularized and showed well-formed and branched vessels, whereas plugs containing KO TAMs displayed small and poorly organized vessels. Addition of blocking plexin B1 antibodies, as well as of the c-Met inhibitor PHA-665752, resulted in the lack of an organized vessel tree. Plugs containing PBS were used as a negative control, whereas plugs containing known angiogenic factors (HGF and b-FGF) were used as positive controls. (n = 3 plugs per group). Bar, 2 mm. (B) TSA tumor cells were injected in KO mice alone or together with TAMs obtained either from WT or KO animals. TSA cells alone were also injected in WT mice as a control. Coinjection of TSA cells and WT TAMs resulted in the formation of significantly larger tumor masses in comparison to those originated by TSA plus KO TAMs. Bars represent the mean ± SEM.
Sema4D produced by TAMs increases the tumorigenic ability of cancer cells
To finally prove that the ability of TAMs to produce Sema4D contributes to the tumorigenic ability of cancer cells, we coinjected TSA tumor cells and TAMs obtained either from WT or KO animals in KO mice. Coinjection of TSA cells and WT TAMs resulted in the formation of significantly larger tumor masses in comparison to those originated by TSA plus KO TAMs (Fig. 5 B). We found a comparable number of TAMs (F4/80+ cells) in the tumors of both groups of animals; moreover, we could detect positivity for Sema4D only in KO mice injected with WT TAMs (unpublished data). As expected, in the presence of WT or KO TAMs, tumor growth was enhanced in comparison to tumor cells alone, in agreement with the previously described protumorigenic role of these cells (35).
DISCUSSION
Tumor development is a complex event that involves not only tumor cells but also the surrounding stroma. In recent years, in fact, data collected both from experimental systems and clinical observations have strengthened the idea that the tumor microenvironment and neoplastic cells act in concert and contribute as a functional whole to the growth and progression of the tumor mass (33). This idea was originally proposed by Paget's famous “seed and soil” hypothesis, which highlighted the possible contribution of stromal cells to the process of tumorigenesis (33).
The mechanisms promoting tumor angiogenesis are not yet fully understood, but it is believed that the most important factor is tumor hypoxia, which induces the release of angiogenic molecules from tumor cells, attracting ECs and inflammatory cells. Recruited ECs migrate toward the hypoxic areas, whereas inflammatory cells, in turn, secrete molecules that intensify the angiogenic stimulus. Once ECs are engaged, perivascular support cells stabilize nascent vessels and guarantee appropriate blood flow (28). Many molecules involved in these processes have been identified, and it has recently become clear that most of them originate from stromal cells recruited to the tumor microenvironment (30). Tumor-infiltrating inflammatory cells undergo a process of maturation or “education” within the tumor microenvironment (40), and pharmacological or genetic inhibition of molecules that are up-regulated in these cells was indeed shown to be able to impair tumor progression in animal models (33).
In this scenario, a critical role has been demonstrated for TAMs that represent the major inflammatory component of the stroma of many tumors (41). Experimental models have demonstrated that the lack of recruitment of macrophages at the tumor site results in decreased tumorigenic ability (42), and clinical evidence has shown that there is a correlation between a high TAM content inside the tumor and a poor prognosis (41). These results have been recently confirmed by gene profiling data showing that genes associated to leukocyte or macrophage infiltration are part of the molecular signatures linked to a poor prognosis in breast cancer (43). In this line, the identification of macrophage-derived molecules that support the protumoral activity of these cells becomes very important.
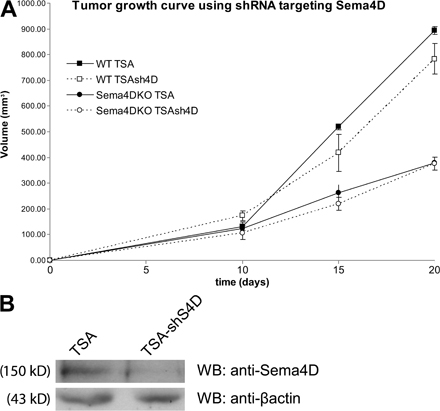
Sema4D is a proangiogenic factor (13–17), but this activity is dispensable for “normal” angiogenesis because Sema4D KO mice are viable (19). It has been previously shown that tumor angiogenesis is partially different from the norm (in fact, tumor vessels are often abnormal in structure and function) and is characterized by an altered vessel maturation, possibly because of an inappropriate balance between vasculogenic and angiogenic factors (44). We thus wondered if Sema4D could play any role in tumor angiogenesis. In this paper, we show that Sema4D is required for proper vessel maturation in tumors, as its deficiency results in the presence of vessels of smaller diameter that are poorly lined by perimural cells, such as pericytes. Interestingly, we have observed that the number of lymphatic vessels in the tumors is not altered in Sema4D KO mice (unpublished data). We also demonstrate that the angiogenic Sema4D is produced by stromal cells infiltrating the tumor. Although another paper has shown an angiogenic role of tumor-derived Sema4D (16), we could not observe a different behavior of breast cancer cells, either producing Sema4D (TSA, 4T1, and 168FARN; unpublished data) or not (66cl4; reference 45), when they where subcutaneously injected in KO mice. Moreover, when we silenced the expression of this semaphorin in the producing cells, we did not observe any significant difference compared with control cells (Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20072602/DC1). A possible reason for these divergences can be caused by the different tumor types used in the two experimental systems. In breast cancer, in fact, a very good correlation has been established between the presence of a strong inflammatory component, mainly containing TAMs, and the malignant behavior (35, 43). It is possible that the head and neck carcinoma cells used by Basile et al. (16) rely on different mechanisms to promote angiogenesis and are less dependent on the tumor microenvironment.
The tumor stroma contains several cell types, either “resident” (such as fibroblasts or adipocytes) or “infiltrated.” Because most of the Sema4D+ cells were also positive for leukocyte markers, we hypothesized that they were blood-derived cells. Bone marrow transplantation experiments formally proved that the Sema4D-producing cells are indeed of hematopoietic origin, as we could recover the WT phenotype (concerning tumor growth and angiogenesis) by transplanting a WT bone marrow into Sema4D KO mice.
A recent report has highlighted the role of Sema4D in platelet activation and thrombus formation (21). Because it is known that blood clotting contributes to tumor growth and dissemination by creating a fibrin clot surrounding tumor cells that can protect them from apoptosis (31), we investigated whether platelet-derived Sema4D could contribute to tumor growth. Chronic treatment of mice with clopidogrel and aspirin did not result in significant changes in the growth of grafted tumors either in WT or KO mice. These results suggest that platelet-derived Sema4D is not required for tumor angiogenesis.
Among the cells of the tumor stroma, we found the highest levels of Sema4D in TAMs, expressing markers of the M2 type. These cells are known to tune inflammatory responses and adaptive Th2 immunity, to scavenge debris, and to promote angiogenesis, tissue remodeling, and repair (35). We found that M2-type TAMs were also present in the microenvironment of Sema4D KO mice and that the absence of this molecule did not significantly affect their functional response to activating stimuli and their ability to produce cytokines. However, the inability of TAMs to produce Sema4D strongly decreased their capacity to attract ECs, in vitro and in vivo, and to promote vessel maturation. Moreover, WT-derived TAMs coinjected with tumor cells in KO animals were able to promote tumor growth at similar levels as in WT mice.
We show that Sema4D exerts its activity on ECs through its high affinity receptor plexin B1, as demonstrated by the lack of chemotactic and angiogenic responses in the presence of plexin B1 blocking antibodies. Moreover, as previously reported (8, 13), plexin-induced c-Met activation is a crucial event in Sema4D signal transduction, as c-Met inhibition resulted in the loss of Sema4D-induced biological responses.
On the basis of the reported results and of data derived from the literature, how can we figure the activity of Sema4D in the tumor stroma? We propose the following model. Cancer cells start to grow and reach a mass that becomes hypoxic. Hypoxia induces several changes in cancer cells and is a strong recruiting factor for TAMs. TAMs, indeed, accumulate preferentially in poorly vascularized tumor regions, where they stop and “adapt” to hypoxia by increasing the expression of hypoxia-inducible and proangiogenic genes (46, 47). Most of these activities are mediated by the hypoxia-responsive transcription of hypoxia-inducible factor 1; its inhibition, in fact, resulted in in vivo impairment of macrophage motility and activation (48). Among the genes induced by hypoxia are the c-Met tyrosine kinase (49) and Sema4D (17). TAM-derived Sema4D can thus trigger the plexin expressed by macrophages and activate c-Met. This receptor, in turn, promotes the production of several cytokines that can contribute to the protumorigenic activity of these cells (50). Among the several proteases produced by TAMs is membrane type 1 metalloproteinase (whose expression is also induced by hypoxia) (51), which can cleave the membrane-bound Sema4D and induce its release as a soluble protein (17) able to act on distant cells. Production of the soluble form has been shown to be critical for the angiogenic activity of Sema4D (17), as it can diffuse and act on ECs, stimulating angiogenesis, and on mural cells, promoting their recruitment. Secreted Sema4D can also stimulate the invasive ability of cancer cells (which very frequently express its receptor). In previous works, we have shown that this biological activity requires the recruitment of c-Met, probably facilitated by hypoxia-induced c-Met overexpression (8, 13, 49). Loss of c-Met activation can be one of the mechanisms contributing to the decreased metastatic ability of cancer cells grown in KO mice.
In conclusion, we have identified a novel molecule produced in the tumor stroma that can contribute to the interaction between the tumor microenvironment and tumor cells by promoting tumor angiogenesis and invasion. This molecule is produced by macrophages, a cell type that is strongly implicated in tumor progression and associated with a poor prognosis of several tumor types. As many attempts are now ongoing to target the tumor stroma, we propose Sema4D as a new candidate for antiangiogenic and antimetastatic cancer therapy.
MATERIALS AND METHODS
Cell culture
TSA, 66cl4, 168FARN, MLP29, and 4T1 cells were cultured in DMEM and were supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Human umbilical vein endothelial cells (HUVECs) were cultured on 1% gelatin–coated dishes in endothelial cell basal medium-2 (Lonza) and were used at early passages (II–V).
In vivo tumorigenesis assay
All animal procedures were approved by the Ethical Commission of the University of Turin and by the Italian Ministry of Health. 5–8-wk-old Sema4D-deficient (19) and WT BALB/c mice (Charles River Laboratories) were injected with 5 × 105 TSA, 66cl4, or 168FARN cells and 10 × 104 4T1 cells subcutaneously in both posterior flanks. Tumor appearance was evaluated every 2 d using a caliper. Tumor volume was calculated using the formula V = 4/3π × (d/2)2 × (D/2), where d is the minor tumor axis and D is the major tumor axis.
Platelet inactivation.
Clopidogrel treatment was administrated in the drinking water at a concentration of 0.26 mg/ml (equivalent to an oral dosage of ∼30 mg/kg/day) (32). Aspirin was administered in the drinking water at a concentration of 0.3 mg/ml (equivalent to an oral dosage of ∼30 mg/kg/day). Evaluation of platelet inactivation was performed by checking negativity of CD62p (BD Biosciences) by FACS.
Coinjection of TAMs.
2 × 105 TSA cells and 105 TAMs, WT or KO derived, were coinjected subcutaneously in the posterior flanks of Sema4D KO mice.
Lung metastases analyses.
When tumors reached a volume of ∼800 mm3, animals were killed. We did not kill all of the mice at the same time; we allowed some tumors of KO mice to grow longer, to reach a size comparable with that observed in WT mice when they were killed. Lungs were contrasted with black India ink to facilitate scoring of superficial macrometastases. The analysis of micrometastases was performed on paraffin-embedded sections stained with hematoxylin and eosin.
Vessel counting.
10 random fields of eight representative tumor sections stained with anti-CD31 antibodies were analyzed as follows: vessels were counted manually using digital images taken with a microscope (DM IRB; Leica) connected to a camera (DC350FX; Leica) and analyzed with Metamorph 6.3 software (MDS Analytical Technologies). Vessel area is the total area occupied by vessels, whereas mean vessel size is vessel area/vessel number (as determined by averaging pixels contained inside of each vessel divided by the mean number of vessels). Structures without lumen at the magnification show in the figures were examined at higher magnification, and only structures with a lumen at higher enlargement were counted.
Analysis of in vivo–activated macrophages
On day 0, WT mice received a single i.p. injection of 0.1 ml CFA (Sigma-Aldrich) or 0.1 ml PBS. 3 d later, mice were killed. Peritoneal cells from control or treated mice were harvested by washing the peritoneal cavity with RPMI 1640 medium with 10% FBS. Cells were centrifuged at 1,200 rpm and washed twice with PBS. Cells were resuspended in RPMI 1640 serum-free medium and left to adhere for 1 h; nonadherent cells were removed by washing with PBS. Total RNA was extracted using a TRIzol extraction kit (Invitrogen). 1 μg RNA was retrotranscribed using M-MLV reverse transcriptase minus H (Promega) in a final volume of 40 μl, according to the manufacturer's protocol. cDNA was later amplified by PCR using primers specific for each mouse cytokine.
Immunofluorescence
Immunofluorescent analyses were performed according to standard techniques. In brief, tumors were embedded on optimum cutting temperature–cryostat embedding compound. 4-μm sections were air dried and fixed in cold acetone. Sections were blocked for 2 h with blocking solution (2% goat serum, 1% BSA, 0.1% cold fish skin gelatin, 0.1% Triton X-100, 0.05% Tween 20, and 0.1 M PBS, pH 7.2). Primary antibodies were diluted 1:200 in antibody dilution solution containing 1% BSA, 0.1% cold fish skin gelatin, and 0.1 M PBS, pH 7.2, for 1 h. Anti-CD31, anti-CD45, and anti-Sema4D were purchased from BD Biosciences. Anti-F4/80 was purchased from AbD Serotec. Anti-NG2 was purchased from Chemicon. Sections were washed with TBS–0.1% Tween 20 and incubated for 30 min with secondary conjugated antibodies diluted 1:300 in TBS–0.1% Tween 20. Secondary antibodies were purchased from Invitrogen. Pictures were taken with a DM IRB microscope connected to a DC350FX camera and analyzed with Metamorph 6.3 software.
Transwell assay
5 × 104 TSA, 66cl4, and 4T1 cells or 3 × 104 HUVECs were seeded on the upper surface of 8-μm porous Transwell filters (Corning), coated on both sides with 3,000 ng/ml fibronectin. Upper chambers were filled with RPMI 1640 with 2% FBS, whereas lower chambers were supplied with RPMI 1640 with 2% FBS plus 6 nM Sema4D or 4 nM HGF (R&D Systems). For EC migration, RPMI 1640 with 2% FBS was placed in upper chambers, whereas TAM supernatants (WT or Sema4D KO) were placed in the lower chamber. Inhibitory molecules were added into the lower chamber compartment as follows: 100 ng/ml anti–plexin B1 (EC6.9; reference 52), 100 ng/ml anti-Sema4D (clone BMA-12; reference 19), 100 ng/ml anti–vesicular stomatitis virus (VSV; Sigma-Aldrich), and 100 nM PHA-665752 (Tocris Bioscience). As positive controls 6 nM Sema4D or 4 nM HGF (R&D Systems) were used. After 12 h of incubation, cells on the upper surface of the membrane were mechanically removed; cells that migrated to the lower face were fixed in 11% glutaraldehyde for 10 min and then stained with crystal violet for 5 min. Stained cells were quantified by counting.
Bone marrow transplantation
In one set of experiments, two tail-vein injections (with an interval of 1 h) of 8 × 107 male WT bone marrow cells were delivered into female Sema4D KO mice. In another set, lethally irradiated (900 cGy) female WT mice were injected with 5 × 106 male Sema4D KO or WT bone marrow cells. 2 wk after transplantation, genomic DNA was extracted (QIAGEN) from peripheral blood monocytes obtained from tail vein puncture. Genomic DNA was quantified and PCR amplified with ZFY1- and β-actin–specific primers: ZFY1 forward, 5′-CAATCCCAAACCTGCTTGT-3′; ZFY1 reverse, 5′-CTCCCCTTCAGCTCTTCCTT-3′; b-act forward, 5′-GCTTACACTGCGCTTCTTGC-3′; and b-act reverse, 5′-AGAAAGGGCCACAGGAAGTC-3′. As a positive control, we used genomic DNA obtained from males; as a negative control, we used genomic DNA obtained from females. To quantify the engraftment, we compared the PCR products obtained from the genomic DNA of peripheral leukocytes of transplanted mice with the products of PCR reactions performed on a standard titration curve of a DNA mix containing different ratios of male/female genomic DNA.
TAM purification
TAM purification was performed as previously described (37). In brief, tumors grown in either WT or Sema4D KO mice were surgically removed, minced, and left to disaggregate in 0.125% trypsin in PBS with constant stirring at 37°C for 45 min. Trypsin was inactivated with FBS. Cells were later centrifuged and washed at 1,500 and 1,000 rpm with PBS. Cells were later left to adhere in serum-free RMPI 1640 for 1 h. Nonadherent cells were washed away. The remaining adherent cells were 90–95% F4/80+. For supernatant collection, cells remained in serum-free conditions for 16 h and media were later collected.
Functional screening of peritoneal macrophages and TAMs
Peritoneal macrophages from WT or Sema4D KO mice were isolated and plated in DMEM containing 10% FBS. After 1 h of adherence, cells were washed twice in PBS. 100 ng/ml LPS was administered to macrophages. Total mRNA was extracted using the TRIzol extraction kit. 1 μg RNA was retrotranscribed using M-MLV reverse transcriptase minus H in a final volume of 40 μl using the manufacturer's protocol. Amplification of 1 μl cDNA was performed using the following primers: mTNFa forward, 5′-TAGCCAGGAGGGAGAACAGA-3′; mTNFa reverse, 5′-TTTTCTGGAGGGAGATGTGG-3′; bactin forward, 5′-TGTTACCAACTGGGACGACA-3′; and bactin reverse, 5′-GGGGTGTTGAAGGTCTCAAA-3′.
TAMs were purified from tumors as described, and mRNA, obtained as described, was retrotranscribed and amplified by PCR with the following primers: mPlxnB1 forward, 5′-CAGATGTCTGGTGCCACATC-3′; mPlxnB1 reverse, 5′-CCCTCACACCACACACAGTC-3′; PlxnB2 forward, 5′-AGGGGAGCCTCTCTACAAGC-3′; PlxnB2 reverse, 5′-TCGATCCCTTCATCCTGAAC-3′; mMet forward, 5′-GTTGGTCCCGCGATGAGT-3′; mMet reverse, 5′-GAGTGAGTAAGGTGCCTCCAG-3′; mCCL17 forward, 5′-AGTGGAGTGTTCCAGGGATG-3′; mCCL17 reverse, 5′-CTGGTCACAGGCCGTTTTAT-3′; mMSR1 forward, 5′-CTGGACAAACTGGTCCACCT-3′; mMSR1 reverse, 5′-CCCCTTCTCTCCCTTTTGTC-3′; mHGF forward, 5′-AGGAACAGGGGCTTTACGTT-3′; mHGF reverse, 5′-GTCAAATTCATGGCCAAACC-3′; mCD72 forward, 5′-CTGCACATCTCTGTCCTCCA-3′; mCD72 reverse, 5′-TCAGAGTCCTGCCTCCACTT-3′; mIL-10 forward, 5′-CCAAGCCTTATCGGAAATGA-3′; mIL-10 reverse, 5′-TTTTCACAGGGGAGAAATCG-3′; mMSCF1 forward, 5′-GACCCTCGAGTCAACAGAGC-3′; mMSCF1 reverse, 5′-TGCTTCCTGGGTCAAAAATC-3′; bactin forward, 5′-TGTTACCAACTGGGACGACA-3′; and bactin reverse, 5′-GGGGTGTTGAAGGTCTCAAA-3′.
Analysis of human monocytes
PMBCs were obtained and stained as previously described (50, 53). In brief, PMBCs were obtained from healthy blood donors by Ficoll density gradient centrifugation. Mononuclear cells were washed three times in PBS, and 25 × 106 cells were plated on 10-cm diameter Petri dishes. Nonadherent cells were removed after a 1-h incubation and examined (sample labeled as day 0). A portion of the PMBCs was kept in culture for 48 h in the absence of differentiating factors (sample labeled as day 2). Surface staining of cells was performed using the following antibodies: anti-Sema4D (Beckman Coulter) and anti-CD14 (monocyte-specific marker; Invitrogen). Isotype controls were performed in each experiment.
To evaluate the response of monocytes to different stimuli, PBMCs were purified by magnetic beads and stimulated for 24 h in the presence of one of the following molecules: 100 ng/ml IL-10, 100 ng/ml TGF-β, 100 ng/ml M-CSF (PrepoTech), 100 mg/ml LPS, or 100 nM PMA (Sigma-Aldrich). Hypoxia was induced by incubating cells in a 3% O2 atmosphere. After 24 h, cells were washed with PBS and total mRNA was extracted using the TRIzol extraction kit. 1 μg RNA was retrotranscribed using M-MLV reverse transcriptase minus H in a final volume of 40 μl, according to manufacturer's protocol. cDNA was amplified using the following primers: hSema4D forward, 5′-CCAGCTCTTCCAGGACTTTG-3′; hSema4D reverse, 5′-ACTTTTATCCGGGCACACAG-3′; hTNFa forward, 5′-CCAAGCCCTGGTATGAGC-3′; hTNFa reverse, 5′-GGGCAATGATCCCAAAGTAG-3′; hIL-10 forward, 5′-GTTGCCAAGCCTTGTCTGA-3′; hIL-10 reverse, 5′-GGCCTTGCTCTTGTTTTCAC-3′; hGAPDH forward, 5′-ACAGTCAGCCGCATCTTCTT-3′; and hGAPDH reverse, 5′-GACAAGCTTCCCGTTCTCAG-3′.
Immunoblot analysis
WT or Sema4D TAMs were lysed with Laemmli buffer (67.5 mM Tris, 2% SDS), and 100 μg of proteins was run on 8% SDS-PAGE. TSA and MLP29 cells were lysed in EB buffer, as previously described (8). 500 μg of protein was immunoprecipitated with 2 μg anti–plexin B1 and 2 μg of rabbit anti–mouse Ig for 3 h in a rotating wheel at 4°C. 100 μg of protein and immunoprecipitations were subjected to 8% SDS-PAGE. Anti-Sema4D antibody was purchased from BD Biosciences; anti–β-actin (I-19) and anti–plexin B1 (A-8) were purchased from Santa Cruz Biotechnology, Inc.
Matrigel plugs
500 μl of Matrigel (Becton Dickinson) was injected subcutaneously in the ventral area of Sema4D KO mice. Matrigel plugs contained 2 × 105 TAMs (WT or Sema4D derived) in the presence or absence of 100 ng/ml anti–plexin B1 (ec6.9), anti-VSV antibodies, or 100 nM PHA-665752. Control plugs contained 36 ng/ml HGF (R&D Systems), 200 ng/ml b-FGF (R&D Systems), or an equivalent amount of PBS (negative control). Each experimental condition was analyzed in triplicate. 14 d after plug implantation, 5 min before death, mice received a 200-μl tail-vein injection containing 20 mg/ml dextran-FITC (Sigma-Aldrich). Plugs were collected, immediately fixed in formalin, and analyzed using a fluorescent microscope.
Sema4D silencing
TSA cells were transduced with MISSION shRNA lentiviral plasmid pLKO.1-puro (clone no. TRCN0000067495; Sigma-Aldrich) targeting the following sequence: 5′-CCACAGCTACACATCAGTCAT-3′. Cells were selected using 2 μg/ml puromycin.
Statistical analyses
For analyses of experimental data, comparisons of data were performed using the Student's t test. All p-values are two tailed. P < 0.001 was considered significant.
Online supplemental material
Fig. S1 shows that different syngeneic breast cancer cell lines grafted into Sema4D KO mice give rise to smaller tumors when compared with those observed in WT mice. Fig. S2 provides information regarding the expression of plexin B1 (high affinity receptor for Sema4D) in TSA cells; it also shows that Sema4D is able to promote the motogenic/invasive ability of the examined breast cancer cells. Fig. S3 illustrates that Sema4D produced by platelets does not contribute to tumor growth. Fig. S4 shows that human macrophages, but not monocytes, express Sema4D; it also shows that Sema4D expression is probably linked to macrophage activation better than differentiation. Fig. S5 confirms that macrophages obtained from Sema4D KO mice are functionally similar to those taken from WT mice. Fig. S6 shows that Sema4D silencing in mammary tumor cells does not affect tumor growth. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20072602/DC1.
Supplementary Material
Acknowledgments
We are grateful to Dr. P. Gabriele and his staff for the irradiation of mice, to Dr. A. Sottile for the evaluation of platelet inhibition, to Drs. A. Elia and M. Mazzone for help in analyzing the human macrophages, and to all our colleagues that shared interesting and fruitful discussions with us. The excellent technical assistance of R. Albano, L. Palmas, M. Crudelini, and R. Lo Noce is gratefully acknowledged. J.R. Sierra was mainly responsible for the experimental work; S. Corso, V. Cepero, and P. Conrotto also helped perform experiments; L. Caione and W. Piacibello performed bone marrow transplants; A. Cignetti performed the Sema4D expression profile in human monocytes; and A. Kumanogoh and H. Kikutani provided Sema4D KO mice. All of the authors analyzed and discussed the data. S. Giordano and L. Tamagnone guided the project and contributed to the experimental design and to data interpretation.
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) grants to S. Giordano and L. Tamagnone, and Regione Piemonte grants to S. Giordano, L. Tamagnone, and S. Corso. J.R. Sierra is a Consejo Nacional de Ciencia y Tecnología–Mexico fellow, and S. Corso is an AIRC fellow.
The authors have no conflicting financial interests.
Abbreviations used: b-FGF, basic fibroblast growth factor; EC, endothelial cell; HGF, hepatocyte growth factor; HUVEC, human umbilical vein endothelial cell; MSR1, macrophage scavenger receptor 1; Sema4D, semaphorin 4D; TAM, tumor-associated macrophage; VSV, vesicular stomatitis virus.
P. Conrotto's present address is Dept. of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland.
References
- 1.Pasterkamp, R.J., and A.L. Kolodkin. 2003. Semaphorin junction: making tracks toward neural connectivity. Curr. Opin. Neurobiol. 13:79–89. [DOI] [PubMed] [Google Scholar]
- 2.Kolodkin, A.L., D.J. Matthes, and C.S. Goodman. 1993. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell. 75:1389–1399. [DOI] [PubMed] [Google Scholar]
- 3.Tamagnone, L., S. Artigiani, H. Chen, Z. He, G.I. Ming, H. Song, A. Chedotal, M.L. Winberg, C.S. Goodman, M. Poo, et al. 1999. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 99:71–80. [DOI] [PubMed] [Google Scholar]
- 4.Takegahara, N., H. Takamatsu, T. Toyofuku, T. Tsujimura, T. Okuno, K. Yukawa, M. Mizui, M. Yamamoto, D.V. Prasad, K. Suzuki, et al. 2006. Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat. Cell Biol. 8:615–622. [DOI] [PubMed] [Google Scholar]
- 5.Behar, O., J.A. Golden, H. Mashimo, F.J. Schoen, and M.C. Fishman. 1996. Semaphorin III is needed for normal patterning and growth of nerves, bones and heart. Nature. 383:525–528. [DOI] [PubMed] [Google Scholar]
- 6.Kumanogoh, A., and H. Kikutani. 2001. The CD100-CD72 interaction: a novel mechanism of immune regulation. Trends Immunol. 22:670–676. [DOI] [PubMed] [Google Scholar]
- 7.Bismuth, G., and L. Boumsell. 2002. Controlling the immune system through semaphorins. Sci. STKE. 2002:RE4. [DOI] [PubMed] [Google Scholar]
- 8.Giordano, S., S. Corso, P. Conrotto, S. Artigiani, G. Gilestro, D. Barberis, L. Tamagnone, and P.M. Comoglio. 2002. The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat. Cell Biol. 4:720–724. [DOI] [PubMed] [Google Scholar]
- 9.Tse, C., R.H. Xiang, T. Bracht, and S.L. Naylor. 2002. Human Semaphorin 3B (SEMA3B) located at chromosome 3p21.3 suppresses tumor formation in an adenocarcinoma cell line. Cancer Res. 62:542–546. [PubMed] [Google Scholar]
- 10.Christensen, C.R., J. Klingelhofer, S. Tarabykina, E.F. Hulgaard, D. Kramerov, and E. Lukanidin. 1998. Transcription of a novel mouse semaphorin gene, M-semaH, correlates with the metastatic ability of mouse tumor cell lines. Cancer Res. 58:1238–1244. [PubMed] [Google Scholar]
- 11.Carmeliet, P., and M. Tessier-Lavigne. 2005. Common mechanisms of nerve and blood vessel wiring. Nature. 436:193–200. [DOI] [PubMed] [Google Scholar]
- 12.Miao, H.Q., S. Soker, L. Feiner, J.L. Alonso, J.A. Raper, and M. Klagsbrun. 1999. Neuropilin-1 mediates collapsin-1/semaphorin III inhibition of endothelial cell motility: functional competition of collapsin-1 and vascular endothelial growth factor-165. J. Cell Biol. 146:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conrotto, P., D. Valdembri, S. Corso, G. Serini, L. Tamagnone, P.M. Comoglio, F. Bussolino, and S. Giordano. 2005. Sema4D induces angiogenesis through Met recruitment by Plexin B1. Blood. 105:4321–4329. [DOI] [PubMed] [Google Scholar]
- 14.Basile, J.R., T. Afkhami, and J.S. Gutkind. 2005. Semaphorin 4D/plexin-B1 induces endothelial cell migration through the activation of PYK2, Src, and the phosphatidylinositol 3-kinase-Akt pathway. Mol. Cell. Biol. 25:6889–6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basile, J.R., A. Barac, T. Zhu, K.L. Guan, and J.S. Gutkind. 2004. Class IV semaphorins promote angiogenesis by stimulating Rho-initiated pathways through plexin-B. Cancer Res. 64:5212–5224. [DOI] [PubMed] [Google Scholar]
- 16.Basile, J.R., R.M. Castilho, V.P. Williams, and J.S. Gutkind. 2006. Semaphorin 4D provides a link between axon guidance processes and tumor-induced angiogenesis. Proc. Natl. Acad. Sci. USA. 103:9017–9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basile, J.R., K. Holmbeck, T.H. Bugge, and J.S. Gutkind. 2007. MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J. Biol. Chem. 282:6899–6905. [DOI] [PubMed] [Google Scholar]
- 18.Hall, K.T., L. Boumsell, J.L. Schultze, V.A. Boussiotis, D.M. Dorfman, A.A. Cardoso, A. Bensussan, L.M. Nadler, and G.J. Freeman. 1996. Human CD100, a novel leukocyte semaphorin that promotes B-cell aggregation and differentiation. Proc. Natl. Acad. Sci. USA. 93:11780–11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi, W., A. Kumanogoh, C. Watanabe, J. Uchida, X. Wang, T. Yasui, K. Yukawa, M. Ikawa, M. Okabe, J.R. Parnes, et al. 2000. The class IV semaphorin CD100 plays nonredundant roles in the immune system: defective B and T cell activation in CD100-deficient mice. Immunity. 13:633–642. [DOI] [PubMed] [Google Scholar]
- 20.Elhabazi, A., S. Delaire, A. Bensussan, L. Boumsell, and G. Bismuth. 2001. Biological activity of soluble CD100. I. The extracellular region of CD100 is released from the surface of T lymphocytes by regulated proteolysis. J. Immunol. 166:4341–4347. [DOI] [PubMed] [Google Scholar]
- 21.Zhu, L., W. Bergmeier, J. Wu, H. Jiang, T.J. Stalker, M. Cieslak, R. Fan, L. Boumsell, A. Kumanogoh, H. Kikutani, et al. 2007. Regulated surface expression and shedding support a dual role for semaphorin 4D in platelet responses to vascular injury. Proc. Natl. Acad. Sci. USA. 104:1621–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masuda, K., T. Furuyama, M. Takahara, S. Fujioka, H. Kurinami, and S. Inagaki. 2004. Sema4D stimulates axonal outgrowth of embryonic DRG sensory neurones. Genes Cells. 9:821–829. [DOI] [PubMed] [Google Scholar]
- 23.Kumanogoh, A., C. Watanabe, I. Lee, X. Wang, W. Shi, H. Araki, H. Hirata, K. Iwahori, J. Uchida, T. Yasui, et al. 2000. Identification of CD72 as a lymphocyte receptor for the class IV semaphorin CD100: a novel mechanism for regulating B cell signaling. Immunity. 13:621–631. [DOI] [PubMed] [Google Scholar]
- 24.Winberg, M.L., J.N. Noordermeer, L. Tamagnone, P.M. Comoglio, M.K. Spriggs, M. Tessier-Lavigne, and C.S. Goodman. 1998. Plexin A is a neuronal semaphorin receptor that controls axon guidance. Cell. 95:903–916. [DOI] [PubMed] [Google Scholar]
- 25.Trusolino, L., and P.M. Comoglio. 2002. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat. Rev. Cancer. 2:289–300. [DOI] [PubMed] [Google Scholar]
- 26.Torisu, H., M. Ono, H. Kiryu, M. Furue, Y. Ohmoto, J. Nakayama, Y. Nishioka, S. Sone, and M. Kuwano. 2000. Macrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: possible involvement of TNFalpha and IL-1alpha. Int. J. Cancer. 85:182–188. [PubMed] [Google Scholar]
- 27.Welm, A.L., J.B. Sneddon, C. Taylor, D.S. Nuyten, M.J. van de Vijver, B.H. Hasegawa, and J.M. Bishop. 2007. The macrophage-stimulating protein pathway promotes metastasis in a mouse model for breast cancer and predicts poor prognosis in humans. Proc. Natl. Acad. Sci. USA. 104:7570–7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.von Tell, D., A. Armulik, and C. Betsholtz. 2006. Pericytes and vascular stability. Exp. Cell Res. 312:623–629. [DOI] [PubMed] [Google Scholar]
- 29.Yamagishi, S., and T. Imaizumi. 2005. Pericyte biology and diseases. Int. J. Tissue React. 27:125–135. [PubMed] [Google Scholar]
- 30.Ganss, R. 2006. Tumor stroma fosters neovascularization by recruitment of progenitor cells into the tumor bed. J. Cell. Mol. Med. 10:857–865. [DOI] [PubMed] [Google Scholar]
- 31.Boccaccio, C., G. Sabatino, E. Medico, F. Girolami, A. Follenzi, G. Reato, A. Sottile, L. Naldini, and P.M. Comoglio. 2005. The MET oncogene drives a genetic programme linking cancer to haemostasis. Nature. 434:396–400. [DOI] [PubMed] [Google Scholar]
- 32.Chiodoni, C., M. Iezzi, C. Guiducci, S. Sangaletti, I. Alessandrini, C. Ratti, F. Tiboni, P. Musiani, D.N. Granger, and M.P. Colombo. 2006. Triggering CD40 on endothelial cells contributes to tumor growth. J. Exp. Med. 203:2441–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Albini, A., and M.B. Sporn. 2007. The tumour microenvironment as a target for chemoprevention. Nat. Rev. Cancer. 7:139–147. [DOI] [PubMed] [Google Scholar]
- 34.Zalatnai, A. 2006. Molecular aspects of stromal-parenchymal interactions in malignant neoplasms. Curr. Mol. Med. 6:685–693. [DOI] [PubMed] [Google Scholar]
- 35.Mantovani, A., T. Schioppa, C. Porta, P. Allavena, and A. Sica. 2006. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 25:315–322. [DOI] [PubMed] [Google Scholar]
- 36.Lewis, C.E., and J.W. Pollard. 2006. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 66:605–612. [DOI] [PubMed] [Google Scholar]
- 37.Sica, A., A. Saccani, B. Bottazzi, N. Polentarutti, A. Vecchi, D.J. Van, and A. Mantovani. 2000. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J. Immunol. 164:762–767. [DOI] [PubMed] [Google Scholar]
- 38.Barberis, D., S. Artigiani, A. Casazza, S. Corso, S. Giordano, C.A. Love, E.Y. Jones, P.M. Comoglio, and L. Tamagnone. 2004. Plexin signaling hampers integrin-based adhesion, leading to Rho-kinase independent cell rounding, and inhibiting lamellipodia extension and cell motility. FASEB J. 18:592–594. [DOI] [PubMed] [Google Scholar]
- 39.Christensen, J.G., R. Schreck, J. Burrows, P. Kuruganti, E. Chan, P. Le, J. Chen, X. Wang, L. Ruslim, R. Blake, et al. 2003. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 63:7345–7355. [PubMed] [Google Scholar]
- 40.Pollard, J.W. 2001. Tumour-stromal interactions. Transforming growth factor-beta isoforms and hepatocyte growth factor/scatter factor in mammary gland ductal morphogenesis. Breast Cancer Res. 3:230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balkwill, F., and A. Mantovani. 2001. Inflammation and cancer: back to Virchow? Lancet. 357:539–545. [DOI] [PubMed] [Google Scholar]
- 42.Lin, E.Y., J.F. Li, L. Gnatovskiy, Y. Deng, L. Zhu, D.A. Grzesik, H. Qian, X.N. Xue, and J.W. Pollard. 2006. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 66:11238–11246. [DOI] [PubMed] [Google Scholar]
- 43.Paik, S., S. Shak, G. Tang, C. Kim, J. Baker, M. Cronin, F.L. Baehner, M.G. Walker, D. Watson, T. Park, et al. 2004. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 351:2817–2826. [DOI] [PubMed] [Google Scholar]
- 44.Carmeliet, P. 2003. Angiogenesis in health and disease. Nat. Med. 9:653–660. [DOI] [PubMed] [Google Scholar]
- 45.Fazzari, P., J. Penachioni, S. Gianola, F. Rossi, B.J. Eickholt, F. Maina, L. Alexopoulou, A. Sottile, P.M. Comoglio, R.A. Flavell, and L. Tamagnone. 2007. Plexin-B1 plays a redundant role during mouse development and in tumour angiogenesis. BMC Dev. Biol. 7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knowles, H., R. Leek, and A.L. Harris. 2004. Macrophage infiltration and angiogenesis in human malignancy. Novartis Found. Symp. 256:189–200. [PubMed] [Google Scholar]
- 47.Talks, K.L., H. Turley, K.C. Gatter, P.H. Maxwell, C.W. Pugh, P.J. Ratcliffe, and A.L. Harris. 2000. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 157:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cramer, T., Y. Yamanishi, B.E. Clausen, I. Forster, R. Pawlinski, N. Mackman, V.H. Haase, R. Jaenisch, M. Corr, V. Nizet, et al. 2003. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 112:645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pennacchietti, S., P. Michieli, M. Galluzzo, M. Mazzone, S. Giordano, and P.M. Comoglio. 2003. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 3:347–361. [DOI] [PubMed] [Google Scholar]
- 50.Galimi, F., E. Cottone, E. Vigna, N. Arena, C. Boccaccio, S. Giordano, L. Naldini, and P.M. Comoglio. 2001. Hepatocyte growth factor is a regulator of monocyte-macrophage function. J. Immunol. 166:1241–1247. [DOI] [PubMed] [Google Scholar]
- 51.Ray, B.K., A. Shakya, J.R. Turk, S.S. Apte, and A. Ray. 2004. Induction of the MMP-14 gene in macrophages of the atherosclerotic plaque: role of SAF-1 in the induction process. Circ. Res. 95:1082–1090. [DOI] [PubMed] [Google Scholar]
- 52.Barberis, D., A. Casazza, R. Sordella, S. Corso, S. Artigiani, J. Settleman, P.M. Comoglio, and L. Tamagnone. 2005. p190 Rho-GTPase activating protein associates with plexins and it is required for semaphorin signalling. J. Cell Sci. 118:4689–4700. [DOI] [PubMed] [Google Scholar]
- 53.Granziero, L., P. Circosta, C. Scielzo, E. Frisaldi, S. Stella, M. Geuna, S. Giordano, P. Ghia, and F. Caligaris-Cappio. 2003. CD100/Plexin-B1 interactions sustain proliferation and survival of normal and leukemic CD5+ B lymphocytes. Blood. 101:1962–1969. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}