

Abstract
In the mouse tooth organ, shortly after birth, ameloblasts acquire their secretory phenotype, which is characterized by the prominent expression and subsequent secretion of two isoforms of amelogenin, M180 and M59 (LRAP, [A−4]). Amelogenin deposition into the ameloblast extracellular matrix promotes enamel biomineralization. A complex set of intercellular signaling events, reciprocal communications between the developing oral epithelium and its underlying dental mesenchyme, guide the expression of amelogenin mRNA, and limit it to a defined period of tooth development. In tooth germ organ culture, addition of the [A−4] isoform, lacking amelogenin exon 4 and exon 6 segments a, b, c, was shown to affect ameloblast development. To understand the basis for this regulatory activity, we have studied the effects of r[A−4] on ameloblast-like LS8 cells, and the role of the putative [A−4] cell surface receptor, LAMP1, as well as the related receptor LAMP3. In the LS8 cells, the expression of the spliced isoforms of amelogenin, LAMP1, and LAMP3 were identified by RT-PCR, and real-time PCR semi-quantitative analysis assessed the modulation of M180 message. M180 mRNA was up-regulated by exogenous [A−4], and this was further increased by blockade of LAMP1, suggesting additive effects between the intracellular signaling pathways activated by the discrete agonists. Immunofluorescence staining identified the patterns of [A−4] and LAMP1 localization in LS8 cells. Internalized r[A−4] was co-localized with LAMP1 in late endosomal/lysosomal compartments. Thus, the LAMP1 and [A−4] intracellular sorting pathways are interrelated. The nitric oxide (NO) signaling pathway was activated by exogenous [A−4]. [A−4] modulated inducible nitric oxide synthase (iNOS, NOS2) and endothelial nitric oxide synthase (eNOS, NOS3) expression, albeit, to different extents. NOS2 was significantly up-regulated after 4 h, while NOS3 increased slightly after 24 h. Co-treatment of LS8 cells with r[A−4] and anti-LAMP1 antibodies further enhanced NOS2 expression. Anti-LAMP1 antibodies did not abrogate NO production in LS8 cells treated for 4 h with r[A−4], but the iNOS inhibitor, L-Nil, down-regulated both NO production and the expression of M180 mRNA. These data suggest that [A−4] modulates M180 mRNA expression, partly, via the NO signaling pathway.
Keywords: ameloblasts, amelogenin, LAMP1, r[A−4], terminal differentiation
Introduction
Ameloblasts proceed through several distinct stages of differentiation from the beginning of tooth organ development to the terminal maturation stage, each characterized by a distinct morphology. Cuboidal preameloblasts shift to the elongated secretory stage immediately after mineralized dentin is secreted by the opposing layer of maturing odontoblasts. The secretory ameloblasts become polarized, orienting their apical pole toward the dentin (1). Generally, in polarized cells, traffic of newly synthesized proteins targeted for the cell surface follows a direct pathway from the trans-Golgi network to the plasma membrane. Some cells may, however, utilize an alternate route through the endosomes for delivery of the proteins to the plasma membrane (2). In ameloblasts, acquisition of the secretory phenotype is tightly linked to the up-regulation of amelogenin synthesis. Amelogenin, the principal structural protein of the enamel organic matrix, is synthesized, secreted, and deposited into the extracellular matrix of ameloblasts during the well-defined secretory stage of ameloblast maturation. A current hypothesis suggests that, in vivo, amelogenin molecules aggregate into nanospheres, which assemble to form the structural scaffold for the deposition of apatite crystals (3, 4). Extracellular deposition of amelogenin occurs in the space surrounding the Tome’s processes.
Several alternatively spliced isoforms of amelogenin mRNA (5, 6), and their corresponding translation products (7), have been identified in the ameloblasts of developing mouse molars. The mouse gene contains 9 exons (8), but all potential splice isoforms are not equally expressed. The major expressed amelogenin isoform, M180, is the product of exons 2,3,5,6 and 7, incorporating the peptide sequence arising from the full-length exon 6. The largest exon, exon 6, contains three cryptic internal splice sites that are variously removed. The major cryptic splice deletion removes exons 6a, b, and c yielding the isoform M59, also known as LRAP or [A−4]. Exon 4, when included and translated, adds a 14 amino acid sequence yielding the isoforms M194 and M73/[A+4]. M180 mRNA expression is initiated during the immediate postnatal developmental period (9). The expression patterns of M180 and M59 mRNA in the ameloblasts of newborn mouse molars were found to vary according to the developmental stage of the cells, and in intracellular location (10). M59/[A−4] and M73/[A+4] were also found to be present in dentin and to be expressed in odontoblasts (11). Both of these peptides demonstrated chondrogenic-inducing potential at the ectopic site, when implanted intramuscular, or in vitro, when added to embryonic muscle fibroblasts (12, 13). Further, the in vitro addition of r[A−4] and r[A+4] to the organ culture medium of developing tooth germs showed them to have distinct regulatory effects on tooth development (14). Hence, [A−4] and [A+4] might participate in epithelial-mesenchymal communication events that regulate the late stages of ameloblast and odontoblast maturation. It is possible that in developing ameloblasts, the inclusion of the polar 14 amino acid sequence encoded by exon 4, yielding M194 and M73, might confer distinct functional properties on these isoforms. Although their functions are not known, the splicing of the pre-mRNA message is obviously a major control point in amelogenin processing.
Several putative receptors for binding to discrete amelogenin isoforms have been identified. Recently, we demonstrated that both r[A−4] and r[A+4] can be taken up from the medium and internalized by fibroblasts and intact tooth germs in culture. In particular, [A−4] bound LAMP1, a member of the lysosomal-associated membrane proteins (15). Mouse LAMP3 (CD63), has been identified in the yeast 2-hybrid (Y2H) system as a binding partner for amelogenin (M180), but not for M59/[A−4] (16).
LAMPs are highly glycosylated trans-membrane proteins, associated with vesicular structures of the endocytic pathway. LAMP1 (CD107a), LAMP2 (CD107b), LAMP3 (CD63), and LimpII are prevailing isoforms, also expressed in humans. Although LAMPs are localized preferentially to the cytoplasmic compartment, alternate traffic of LAMP1 to the plasma membrane has been identified during cytokine exocytosis (17). Similarly, plasma membrane targeting has been identified for LAMP2, and both LAMP2 and LAMP1 expressed at the cell surface participate in cell-mediated adhesion to the endothelium (18). Although it is believed that plasma membrane localized LAMPs possess functional properties, delivery of newly-synthesized lysosomal membrane glycoproteins LAMP2, LEP100, and LAP to the cytoplasmic compartment involves intermediate cell-surface traffic (19).
The terminal phases of development are characterized by the expression of cell-type specific genes, and acquisition of the cellular phenotype. Cessation of cell proliferation, through cell cycle arrest, and entry into the pathway of terminal differentiation is regulated by nitric oxide (NO), which activates “coupling” between cellular proliferation and differentiation (20). NO is synthesized from L-arginine by the enzyme nitric oxide synthase (NOS). Three isoforms of NOS have been identified so far, neuronal (nNOS, NOS1), inducible (iNOS, NOS2), and endothelial (eNOS, NOS3). In chondrocytes, all NOS isozymes were identified in the hypertrophic zone of the growth plate, and the production of NO promoted cell development (21).
Regulated expression of cell-type specific genes during the process of terminal differentiation is also partly attributed to alternative splicing of the primary mRNA transcripts. The developmentally-restricted expression of alternatively spliced transcripts translates into proteins with dissimilar function, regulated by discrete signaling pathways. The M59/[A−4] peptide demonstrated signaling properties exclusive to ameloblast differentiation in embryonic mouse molars (14, 15). In contrast, in the postnatal mouse molar, [A−4] changed the expression of molecules important for matrix biomineralization (22). Selective co-expression of [A−4] and M180 in newborn mouse ameloblasts, committed toward terminal differentiation, suggests reciprocal regulation (10). Binding of [A−4] to LAMP1 has been identified in fibroblasts in vitro (15), but it is not known whether in ameloblasts, which are cells of epithelial origin, [A−4] binds to LAMP1.
In the present work, using an in vitro model of newborn ameloblast-like LS8 cells (23), we have identified one [A−4]-related signaling pathway. We demonstrate that [A−4] stimulates M180 mRNA expression in ameloblasts, partly, through the activation of the NO signaling pathway, and LAMP1 participates in the intracellular traffic of [A−4].
Materials and methods
Purification of recombinant [A−4]
The recombinant [A−4] peptide (r[A−4]) was prepared and purified according to the published protocols (12). Briefly, PCR-amplified [A−4] was inserted into the pGEMT vector, reamplified, and purified. Thereafter, the amplified product was cloned into the GST expression vector pGEXT4 and introduced into the Escherichia coli strain BL21(DE3). One colony was incubated in LB media, and expressed by the addition of 1 µM isopropyl-β-D-thiogalactosidase. The peptide was affinity purified by passage of the media over a glutathione-Sepharose column (Amersham Pharmacia Biotech, Piscataway, NJ), and bound r[A−4] was cleaved with thrombin. The released r[A−4] peptide was dissolved in 1% trifluoroacetic acid (TFA) in water, and then purified on a C-18 reverse phase column (The Separations Group, Inc, GraceVydac, Hesperia, CA) using an increasing gradient of acetonitrile containing 0.1% TFA.
Cell culture
LS8 cells (immortalized, newborn ameloblast-like cells) (23), a kind gift from Dr. Malcolm L. Snead (USC, Los Angeles, CA), were grown in monolayer culture, in low-glucose Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 50 units penicillin, 50 units streptomycin (Invitrogen, Eugene, OR), and 10% fetal bovine serum (FBS) (Mediatech Inc, Herndon, VA). LS8 cell cultures were maintained at 37°C in a humidified 95% air/5% CO2 atmosphere, until the cells formed confluent layers. On the day prior to initiation of treatment, fresh DMEM containing antibiotics (as per above), and supplemented with 10% or 2% FBS was added to the cell cultures. Cells were treated without (control), or with 10 ng/ml r[A−4] in fresh media containing antibiotics and the selected serum concentrations. In parallel, cells were treated with 2.5 µg/ml anti-LAMP1 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), to analyze the involvement of plasma membrane LAMP1 in LS8 intracellular signaling events. In studies which determined the production of NO, the LS8 cells were treated without (control), or with 10 ng/ml r[A−4], and 0.5 mM L-N6-(1-iminoethyl)lysine (L-Nil) (Sigma-Aldrich, St. Louis, MO), to inhibit the inducible isoform of nitric oxide synthase (iNOS, NOS2). Treatments were carried out for 4 and 24 h. When required, all chemicals and reagents were cellculture grade.
RT-PCR and real-time PCR
Total RNA was isolated from LS8 cells with the TRIzol reagent (Invitrogen, Eugene, OR) following the manufacturer’s instructions. The RNA was purified from the aqueous phase of the phenol-chloroform extract, following precipitation with 75% ethanol. All oligonucleotide primers were synthesized by IDT (Coralville, IA). Primer sets were designed to discrete sequences of mouse amelogenin cDNA (Accession D83067), inducible nitric oxide synthase (iNOS, NOS2) (Accession NM012611), endothelial nitric oxide synthase (eNOS, NOS3) (Accession NM008713), LAMP1 (Accession NM010684), LAMP3 (CD63) (Accession NM007653), ARHGAP6 (Accession NM009707), and glyceraldehyde-phosphate dehydrogenase (GAPDH) (Table 1). In RT-PCR, 1–3 µg RNA was reverse transcribed with 50 units of SuperScript II reverse transcriptase, using the reagents of the SuperScript First-Strand Synthesis System (Invitrogen, Eugene, OR). First-strand cDNA was amplified in 2 mM MgCl2 with Platinum Taq DNA polymerase (Invitrogen, Eugene, OR). Optimum conditions were determined for each amelogenin and ARHGAP6 specific primer, and are given in the Legend of Figure 1. RT-PCR-amplified products were separated on 1.5 to 2% agarose gels, visualized by ethidium bromide staining, and identified with molecular weight markers obtained from the Hae III digest of the ϕX174 phage. The gels were photographed in the “Electrophoresis documentation and analysis system 120” by Kodak Digital Science.
Table 1.
Ameloblast gene expression analyzed by RT-PCR.
| Gene | Primer pair | |
|---|---|---|
| mM180 | F | 5’-CAGCAACCAATGATGCCAGTTCCT-3’ |
| R | 5’-ACTTCTTCCCGCTTGGTCTTGTCT-3’ | |
| m[A−4], [A+4] | F | 5’-GCCCTGGTTATATCAACTTAAGCTATGAG-3’ |
| R | 5’-AATGGGGGACAGGGGCGGCTGCCTTATCAT-3’ | |
| mARHGAP6 | F | 5’-TTGCGTGAAGAATTTGACCGTGGG-3’ |
| R | 5’-TGTGGAGCAGGTTGGGTCCAAATA-3’ | |
| mLAMP1 | F | 5’-TAGTGCCCACATTCAGCATCTCCA-3’ |
| R | 5’-TCCTGCCAATGAGGTAGGCAATGA-3’ | |
| mLAMP3 (CD63) | F | 5’-TGTTGCCTGTGGTCATCATTGCAG-3’ |
| R | 5’-AGCCACCAGCAGTATGTTCTTCCT-3’ | |
| rNOS2 | F | 5’-AATAACCTGAAGCCCGAAGACCCA-3’ |
| R | 5’-TTCAAAGTGGTAGCCACATCCCGA-3’ | |
| mNOS3 | F | 5’-AGTTCCCGGAAAGAGGGATTGTGT-3’ |
| R | 5’-AGCATATGAAGAGGGCAGCAGGAT-3’ | |
| GAPDH | F | 5’-CTTCACCACCATGGAGAAGG-3’ |
| R | 5’-CTTACTCCTTGGAGGCCAT-3’ | |
Figure 1. [A−4] stimulates gene expression in non-polarized ameloblasts.
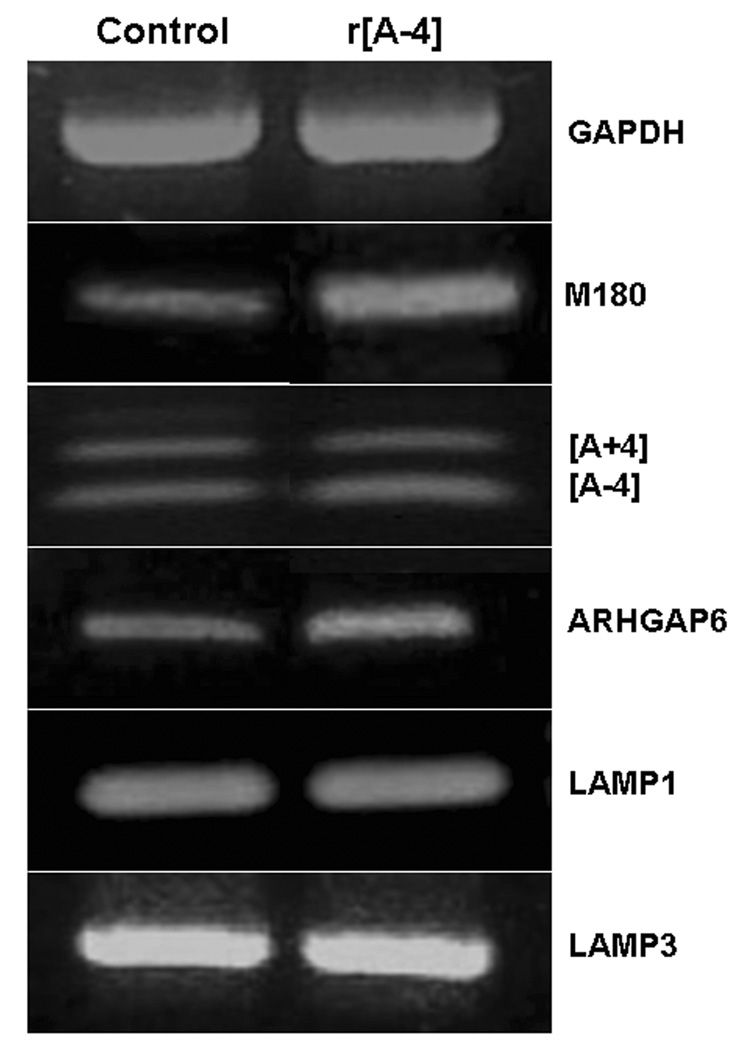
LS8 cells, newborn mouse molar ameloblast-like cells, were cultured in 10% FBS-DMEM and treated without (Control), or with 10 ng/ml r[A−4] for 24 h. Total RNA was extracted with the TRIzol reagent, and amplified by RT-PCR. 1 µg total RNA was used with the LAMP1, LAMP3 and GAPDH (loading control)-specific primers. 2 to 3 µg total RNA were used with the amelogenin isoform-specific primers (M180, [A−4], [A+4]), and the ARHGAP6-specific primers. Cycle times were: M180 – 35 ×(94°C-1 min; 60°C-1 min; 72°C-2 min); r[A+/−4]− 40 ×(94°C-0.45 min; 62°C-1 min; 72°C-2 min); ARHGAP6 – 35 ×(94°C-1 min; 62°C-1 min; 72°C-2 min). Amplified products were separated on 1.5 to 2% agarose gels stained with ethidium bromide. Left lane: control; right lane: treatment with 10 ng/ml r[A−4].
In real-time PCR, RNA was reverse transcribed with M-MLV reverse transcriptase (Promega, Madison, WI), and first-strand cDNA was amplified with the SYBR Green universal master mix, and AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA). Semi-quantitative analysis was performed with the ABI PRISM 7900HT detection system (Applied Biosystems, Foster City, CA). The amplification efficiency (E) for each primer set was calculated according to the slope value of an amplification plot obtained from a series of 10-fold dilutions of cDNA, where E = 10[−1/slope] (24). The mRNA was semi-quantitated using the ΔCt mathematical model developed by M.W. Pfaffl (25), where Ct represents the crossing point value. Relative fluorescence intensity was expressed as fold-increase in target gene expression, as defined by Pfaffl, normalized to the expression of the housekeeping gene GAPDH.
Biotinylation of the r[A−4] peptide
The r[A−4] peptide was biotinylated with the “Biotin-XX microscale protein labeling kit” (Invitrogen, Eugene, OR), following the manufacturer’s instructions. The Biotin-XX, sulfosuccinimidyl ester (biotin-XX, SSE) reacts with the primary amines of the peptide, and forms stable conjugates of biotin-r[A−4]. Briefly, 1 mg/ml r[A−4] stock solution was suspended in 1/10 volume of 1 M sodium bicarbonate, with an approximate pH 8.3, and incubated with 14.93 nmol/µl reactive biotin solution (r[A−4]/biotin ratio = 15) at room temperature for 15 min. Biotinylated r[A−4] was separated from the non-reacted biotin by filtration through a Bio-Gel P-6 fine resin column; the spin-filter was centrifuged at 16000 X g for 1 min. In the final product, conjugated r[A−4] is separated from the biotin moiety by a spacer arm of 14 atoms, thus, biotin can precisely attach to streptavidin-binding pockets (protocol MP 30010, Molecular Pobes, Eugene, OR).
Immunocytochemistry
LS8 cells were grown on cover-slips, in low-glucose DMEM supplemented with 50 units penicillin/streptomycin and 10% FBS. Prior to the initiation of treatment, LS8 cells were equilibrated overnight in DMEM with antibiotics and 2% FBS. Thereafter, the cells were incubated with approximately 10 ng/ml biotinylated r[A−4] in DMEM with antibiotics, and 2% FBS for 1 h at 4°C or 37°C. Treatment was terminated by a brief rinse with 1 X phosphate-buffered saline (PBS). Then, LS8 cells were fixed in 2% neutral buffered formalin for 10 min, followed by incubation with 100% chilled methanol for 3 min. The permeabilization step was omitted from experiments identifying cell surface proteins. The immunofluorescence cell staining protocol provided by Santa Cruz Biotechnology was applied, with minor modifications, for the study of r[A−4] binding and/or endocytosis in LS8 cells. LS8 cells on cover-slips were incubated with blocking serum (2 % BSA (Sigma, St. Louis, MO) in PBS) for 20 min at room temperature, to block out non-specific binding. LAMP1 antibody was diluted in blocking serum to a final 2.5 µg/ml concentration, and cells were incubated with the primary antibody for 60 min at room temperature. Following 3 washes of 5 min with 1 X PBS, cells were incubated with 2% goat serum (Zymed, San Francisco, CA) in blocking serum for 20 min, followed by 2 washes with 1 X PBS. Thereafter, cells were incubated with FITC-conjugated IgG (Santa Cruz Biotechnology, Santa Cruz, CA) in 2% blocking serum for 45 min in the dark, followed by Cy3-streptavidin (Zymed, San Francisco, CA) for 15 min in the dark. LS8 cells were counter-stained with DAPI, the cover-slips were washed 5 times with 1 X PBS, and mounted. The LS8 cells were observed in a Dialux 20 fluorescence microscope (Leitz, Wetzlar, Rockleigh, NJ), and images were captured and analyzed with the SPOT software (Diagnostics Instruments, Sterling Heights, MI).
Production of NO
In the aqueous cell culture system, NO is transformed into nitrate and nitrite. The NO assay kit (EMD Biosciences, San Diego, CA) quantitates the total nitrites in an aqueous solution by converting nitrates to nitrites using the NADH-dependent enzyme nitrate reductase. Thereafter, the Griess reagent enables the spectrophotometric quantitation of total nitrites produced by the LS8 cells. The cells were stimulated without (control), or with 10 ng/ml r[A−4] ± 0.5 mM L-Nil, or ± 2.5 µg/ml anti-LAMP1 antibodies. The nitrate reductase was reconstituted to a concentration of 1 U/µl in 1.5 ml glycerol buffer solution. A nitrate standard solution, provided in the kit, was used to create a series of standards ranging from 0.5 to 500 µM. Equal volumes (85 µl) of standards and samples of cell culture media were incubated with 10 units of nitrate reductase, and 10 µl of 2 mM NADH, for 20 min at room temperature. Following a brief incubation with the Griess reagent, absorbance of standards, and samples was determined at 540 nm.
Isolation and quantitation of protein
Total protein was isolated from LS8 cells with the TRIzol reagent (Invitrogen, Eugene, OR), following the instructions of the manufacturer. Protein was isolated from the phenol-chloroform phase, following precipitation with 100% isopropanol, and three 20 min washes in 0.5 M guanidine HCl. Total cellular protein was analyzed spectrophotometrically at 595 nm, using the Bradford method (Bio-Rad, Hercules, CA), and quantitated by linear regression. Thereafter, NO content was expressed as [µmoles] total nitrites, normalized to [mg] total cellular protein.
Statistical analysis
Significance was calculated with Student’s t test, where P<0.05. The production of NO was assayed in duplicate samples. Real-time PCR reactions were repeated a minimum of 3 times.
Results
[A−4] modulates the expression of discrete genes
The newborn mouse ameloblast-like cell line LS8 was used because it expressed M180, the major amelogenin isoform (23). As shown in Figure 1 (Control, left panel), LS8 cells, grown in the DMEM medium supplemented with 10% FBS, showed the constitutive expression of low levels of message for M180 and both short isoforms of amelogenin, M59/[A−4] and M73/[A+4], as well as ARHGAP6, as compared with GAPDH mRNA. Also, messages for LAMP1 and LAMP3, which are constitutively expressed endosomal/lysosomal membrane proteins, were identified in the LS8 cells. The addition of 10 ng/ml r[A−4] to the LS8 culture for 24 h, led to the distinct upregulation of M180 and ARHGAP6 mRNAs, while there was no increase in LAMP1, LAMP3 or GADPH message levels (Figure 1, right panel). Consequently, r[A−4] specifically upregulates the expression of the M180 amelogenin isoform. These data show that, as postulated previously, ameloblasts do respond to exogenous M59/r[A−4] (15).
Quantitation of the activating potential of [A−4] by real-time PCR
LS8 cells were grown in the 10% FBS-DMEM culture medium for 4 h (Figure 2A, small check bars) and 24 h (Figure 2A, large check bars), without (control) or with the addition of the 10 ng/ml r[A−4]. Treatment with r[A−4] for 24 h led to a significant increase of 13.6 fold in M180 mRNA, as compared to the control sample. It might be possible to attribute part of this increase in M180 mRNA to cellular proliferation. Our earlier studies on the action of r[A−4] in fibroblasts demonstrated that specific effects could be seen most prominently in cultures under conditions (2% FBS culture media) less favorable for cell proliferation (14, 15). To confirm specificity of the modulatory effect of r[A−4] on M180 gene expression, LS8 cells were cultured in DMEM supplemented with 2% FBS, and treated without (control), or with 10 ng/ml r[A−4], for 4 and 24 h. Under these culture conditions, the expression of M180 mRNA following treatment with r[A−4] for 4 h was enhanced 1.7-fold when compared to the corresponding control mRNA (Figure 2B, small check bars). After 24 h treatment with r[A−4], expression of M180 mRNA was increased 1.2-fold, as compared to the corresponding control (Figure 2B, large check bars). These data suggested that indeed, part of the increase in M180 mRNA in LS8 cells maintained in 10% FBS-DMEM culture was due to the presence of r[A−4], but that it is likely that other stimulating serum factors might affect r[A−4] activity.
Figure 2. [A−4] and LAMP1 modulate the expression of M180 mRNA.
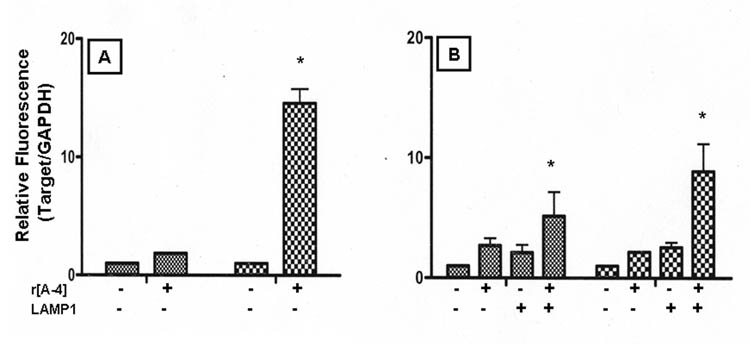
LS8 cells were cultured in 10% FBS-DMEM. 24 h prior to treatment, cultures were equilibrated in 10% FBS-DMEM (Panel A) or 2% FBS-DMEM (Panel B). Cells were treated without (control), or with 10 ng/ml r[A−4] for 4 h (small check bars) and 24 h (large check bars). Additionally, LS8 cells cultured in 2% FBS-DMEM were treated with anti-LAMP1 antibodies ± 10 ng/ml r[A−4]. M180 gene expression was analyzed by semi-quantitative real-time PCR, using the ΔCt method, where E=10−1/slope. Relative fluorescence intensity was expressed as fold-increase of target copies in the treated samples over target copies in the control samples, as normalized to GAPDH. Statistical analysis was performed with the Student’s t test. Significance (*) - P<0.05.
LAMP1 modulates [A−4] function
The effects of r[A−4] on fibroblasts had indicated that its activity was mediated by a cell surface receptor, possibly LAMP1 (15), as evidenced by the isolation of a biotinylated r[A−4]-LAMP1 conjugate. Interestingly, LAMP3/CD63, also present at the endosomal membrane, was shown to bind to M180 protein (26). Since the LS8 cells have an abundant endogenous expression of LAMP1 (Figure 1, left lane), we examined the effect of blockade of cell surface LAMP1 on the regulation of M180 mRNA by r[A−4]. As a control, LS8 cells in 2% FBS-DMEM cultures were treated with anti-LAMP1 antibodies, without exogenous r[A−4]. Although blockade of cell surface LAMP1 did not have a significant effect on M180 mRNA expression after 4 h treatment, this addition yielded the surprising result of a normalized 1.6-fold increase in M180 message as compared to LS8 cells in the 2% FBS-DMEM culture without any treatment for 24 h. Addition of r[A−4] to the anti-LAMP1 pre-treated cells led to a significant 7.9-fold increase in M180 mRNA (Figure 2B, large check bars). Thus, if the endogenously produced and secreted M59/[A−4] enhances M180 mRNA message level via an autocrine interaction with cell surface LAMP1, then the M180 message increase upon addition of anti-LAMP1 suggests that the blockade of cell surface-expressed LAMP1 changes the sorting of the exocytic vesicles, and increases the level of intracellular M180.
Localization patterns of [A−4] and LAMP1 in ameloblasts
As a putative acceptor for r[A−4], it was necessary to first establish the distribution of LAMP1 in the LS8 ameloblast-like cells, since they do not become polarized, as do true, in situ, secretory ameloblasts. Control cultures of LS8 cells in 2% FBS-DMEM were maintained at 37°C for 1 h, then fixed but not permeabilized, and then stained with FITC-labeled anti-LAMP1 antibody. As shown in Figure 3A, the LS8 cells were rounded in shape and the abundant LAMP1 was distributed along the cell surface in a punctuate fashion. The addition of r[A−4] to the culture medium for 1 h at 37°C, induced a significant change in cell shape, as they appeared more elongated, and in these non-permeabilized cells, a substantial portion of LAMP1 appeared to have moved along the plasma membrane (Figure 3B, white arrows).
Figure 3. Distribution of cell surface LAMP1 receptors in ameloblasts.
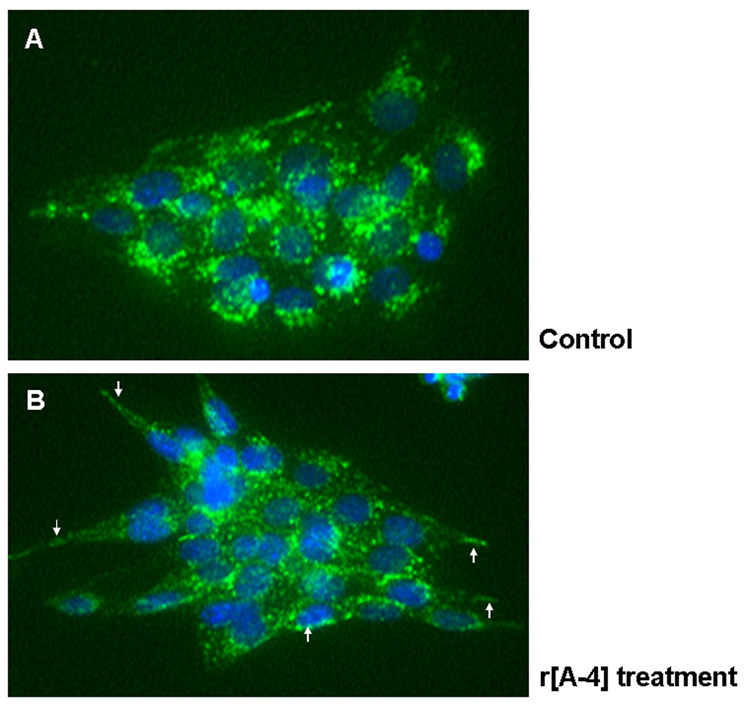
LS8 cells, cultured on coverslips, were equilibrated in 2% FBS-DMEM, and treated without (A), or with 10 ng/ml r[A−4] for 1 h at 37°C (B). Cells were fixed, but not permeabilized, and immunostained with anti-LAMP1 antibodies, followed by FITC-conjugated IgG. Nuclei were counterstained with DAPI. Images were analyzed in fluorescent microscopy using the SPOT software. Panel B: white arrows point to the translocation of endogenous LAMP1 to the cellular plasma membrane, following addition of exogenous r[A−4]. Original magnification – 400X.
Permeabilized preparations of LS8 cells in 2% FBS-DMEM cultures were incubated at 4°C, where uptake of potential exogenous ligands was minimized. Anti-LAMP1 staining (Figure 4G) showed a more prominent and diffuse cytosolic distribution of LAMP1 than in the non-permeabilized cells shown in Fig. 3A. The same cells (Figure 4H) showed background staining with Cy3-strepavidin, but there was no co-localization of LAMP1 and the background streptavidin staining (Figure 4I). When the permeabilized cells were incubated at 37 °C for 1 h, a condition allowing for uptake, anti-LAMP1 staining showed a more prominent and diffuse cytosolic distribution of LAMP1, and the plasma membrane was not accentuated (Figure 4A). Again, the same cells showed a diffuse background staining of Cy3-streptavidin (Figure 4B), but the LAMP1 and background streptavidin staining were not co-localized (Figure 4C). However, the addition of biotinylated r[A−4] to the culture, and incubation for 1 h at 37 °C, showed LAMP1 to have moved at the plasma membrane boundary (Figure 4D), and the Cy3-streptavidin staining indicated that the r[A−4] was prominent at the cell boundary (Figure 4E, white arrows), and was co-localized with the LAMP1 (Figure 4F) there and in the late endosomal/lysosomal compartment (Figure 4F, white arrowhead). LS8 cells treated with biotinylated r[A−4] at 4 °C showed cell membrane binding (Figure 4K, white arrow), and weak co-localization with LAMP1 in the late endosomal region (Figure 4L, white arrow), confirming that r[A−4] is indeed endocytosed by the LS8 cells treated with exogenous r[A−4] at 37°C.
Figure 4. Binding and endocytosis of [A−4] in ameloblasts.
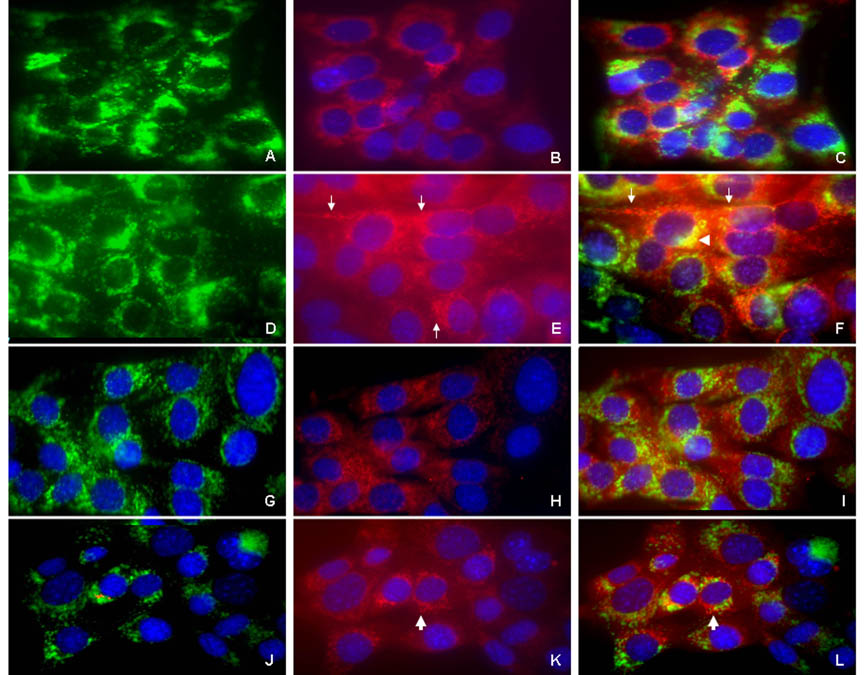
LS8 cells, cultured on coverslips, were equilibrated in 2% FBS-DMEM, and treated without (panels A–C), or with biotin-conjugated r[A−4] (panels D–F) for 1 h at 37°C. The cells were fixed, permeabilized, and immunostained with anti- LAMP1 antibodies, followed by FITC-conjugated IgG. Cell-surface bound and/or endocytosed biotinylated r[A−4] was identified with Cy3- streptavidin. Nuclei were counterstained with DAPI. (A) LAMP1, intrinsic distribution; (B) background staining with Cy3-streptavidin in the absence of exogenous biotin-conjugated r[A−4]; (C) merged A and B image. (D) LAMP1 distribution after addition of r[A−4]; (E) distribution and uptake of biotinylated r[A−4], visualized by Cy3-streptavidin; (F) D and E merged image. White arrows: localization of r[A−4] to the LS8 cell membrane (E, F). White arrowhead: the intracellular co-localization of LAMP1 and r[A−4] in the late endosomal/lysosomal compartment (F).
Cell surface binding of [A−4] in ameloblasts. Following equilibration in 2% FBS-DMEM, LS8 cells were treated without (panels G–I), or with biotin-conjugated r[A−4] for 1 h at 4°C (panels J–L). r[A−4], LAMP1 and nuclei were stained as per above. (G) LAMP1, cell surface distribution; (H) background staining of Cy3-streptavidin; (I) merged G and H image. (J) Cell surface LAMP1 distribution after addition of r[A−4]; (K) binding to the cell surface of biotinylated r[A−4], visualized by Cy3-streptavidin; (L) merged J and K image. White arrows: binding of r[A−4] to the LS8 plasma membrane (K) and co-localization of r[A−4] with LAMP1 at the LS8 plasma membrane (L). Original magnification – 400X.
[A−4] activates NOS isozymes and the production of NO
Previous work suggested that the addition of r[A−4] to neonatal fibroblasts altered the course of cell differentiation, and when added to tooth germs in culture, appeared to affect the maturation of the ameloblasts. Since NO has been shown to regulate differentiation in some systems (20, 21), we evaluated the effect of r[A−4] on the activation of nitric oxide by the inducible (iNOS, NOS2) and endothelial (eNOS, NOS3) nitric oxide synthase in LS8 cells.
Semi-quantitative PCR analysis was used to determine the specific effects of r[A−4] and LAMP1 on the expression of NOS2 and NOS3 mRNA following treatment for 4 and 24 h (Figure 5). The relative fluorescence intensity for eNOS (NOS3) was not significantly increased above control levels following addition of 10 ng/ml r[A−4], anti-LAMP1 antibodies, or r[A−4] in the presence of anti-LAMP1 (Figure 5B). However, iNOS (NOS2) mRNA expression was modulated by r[A−4] and anti-LAMP1 antibody treatments. Exogenous r[A−4] significantly up-regulated iNOS expression after 4 h of treatment. Co-treatment of r[A−4] with anti-LAMP1 for 4 h further enhanced NOS2 expression. Following 24 h of co-treatment, NOS2 expression was strongly elevated (Figure 5A).
Figure 5. [A−4] activates NOS isozymes.
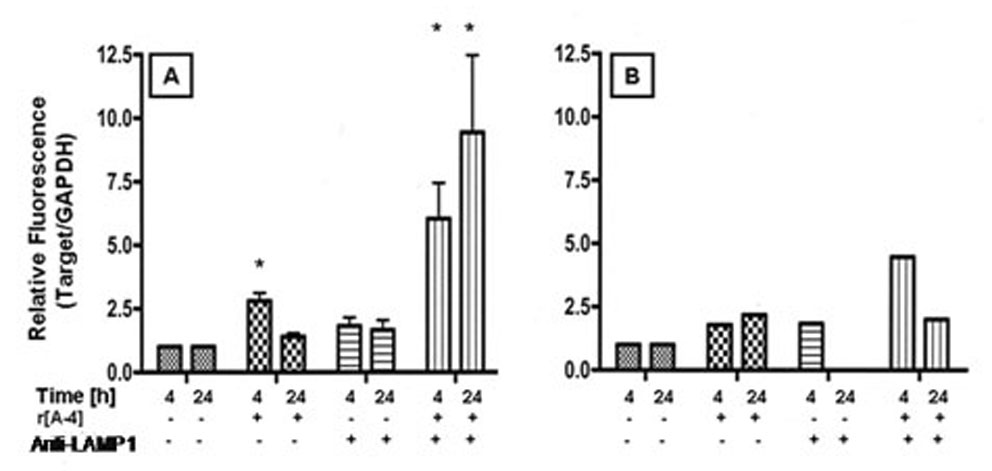
LS8 cells, cultured in 2% FBS-DMEM, were treated without (control), or with 10 ng/ml r[A−4] for 4 and 24 h. Cells were also pre-treated with 2.5 µg/ml anti-LAMP1 antibody ± 10 ng/ml r[A−4]. The expression of NOS2 mRNA (panel A) and NOS3 mRNA (panel B) was analyzed by real-time PCR, using the SYBR Green master mix. Target mRNA expression was analyzed semi-quantitatively by the ΔCt method, and normalized to GAPDH mRNA expression. Values represent fold-increase in target copy number in the treated sample over basal copy level in the control. Statistical analysis was performed with the Student’s t test. Significance (*) - P<0.05.
The addition of exogenous r[A−4] also affected NO production levels. The basal NO level of LS8 cells in 2% FBS-DMEM culture was 54.2 µmoles of total nitrites/mg cellular protein over a 4 h period. Treatment with 10 ng/ml r[A−4] approximately doubled NO production to 120.9 µmoles total nitrites/mg cellular protein within 4 h, rising to 390 µmoles total nitrites/mg cellular protein after 24 h culture (Figure 6). Co-treatment of LS8 cells with r[A−4] and 2.5 µg/ml anti-LAMP1 antibody reduced the NO content to 100 µmoles total nitrites/mg cellular protein within 4 h, and the NO was further reduced to the basal control level within 24 h.
Figure 6. [A−4] activates the production of NO in ameloblasts.
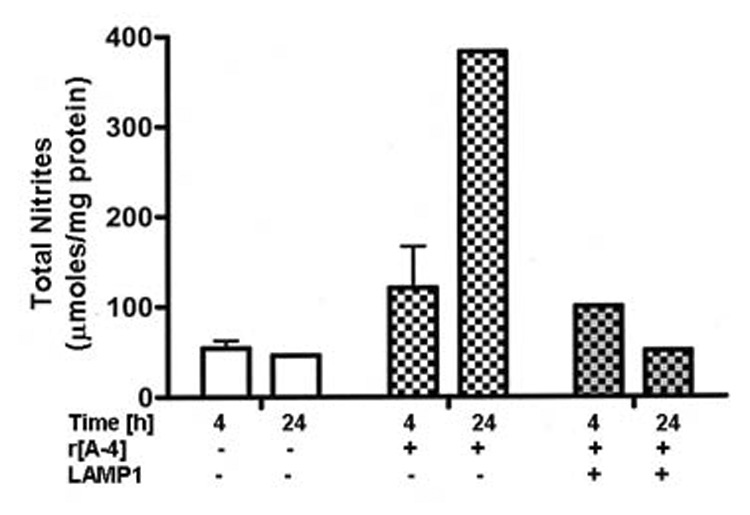
LS8 cells, cultured in 2% FBS-DMEM, were treated without (control), or with 10 ng/ml r[A−4] for 4 and 24 h at 37°C. To confirm LAMP1-mediated modulation of NO production, LS8 cells were pre-treated with 2.5 µg/ml anti-LAMP1 antibodies, prior to induction with r[A−4]. Total nitrites were quantitated spectrophotometrically with the Griess reagent, analyzed by linear regression, and expressed as µmoles/mg cellular protein.
The specific effect of r[A−4] on NOS2 expression was confirmed by the addition of the specific NOS2 inhibitor, L-Nil, to the LS8 cell cultures (Figure 7A). After 4 h of treatment in the presence of L-Nil and r[A−4], the NO level dropped to 22.2 µmoles total nitrites/mg cellular protein, well below the 120.9 µmoles total nitrites/mg cellular protein observed in the cultures treated with r[A−4], and well below the 54.2 µmoles total nitrites/mg cellular protein observed in the control cultures (Figure 6). NO levels of 33.3 µmoles total nitrites/mg cellular protein induced by L-Nil alone did not differ from the basal NO level in the corresponding control (data not shown). The addition of L-Nil also decreased by approximately 50% the expression of M180 mRNA induced by exogenous r[A−4] (Figure 7B).
Figure 7. M180 mRNA expression is partly mediated by NO signaling activated by the NOS2 isozyme.
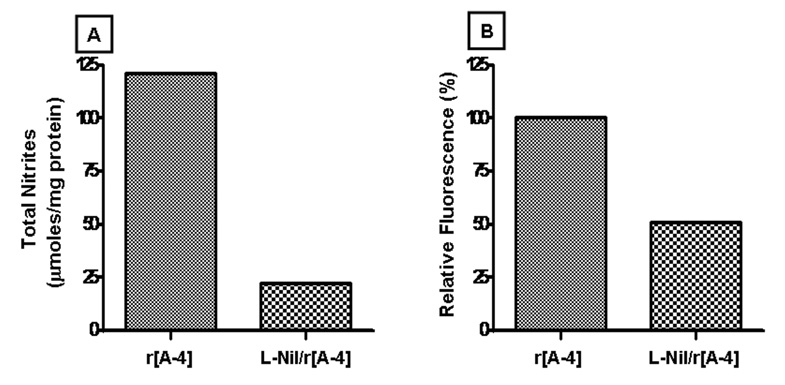
LS8 cells, cultured in 2% FBS-DMEM, were treated with 10 ng/ml r[A−4] for 4 h. Panel A: total nitrites were quantitated spectrophotometrically with the Griess reagent, analyzed by linear regression, and expressed as µmoles/mg cellular protein. To confirm NOS2-mediated production of NO, LS8 cells were pre-treated with 0.5 mM L-Nil, followed by 10 ng/ml r[A−4]. Panel B: the expression of M180 mRNA was analyzed by semi-quantitative real-time PCR, and target mRNA expression was normalized to GAPDH mRNA expression, according to the ΔCt method. The histogram depicts one representative change in M180 expression (expressed as %) in the sample treated with r[A−4] vs. the sample co-treated with L-Nil and r[A−4].
Discussion
Regulation of tooth organ development depends upon a constant signaling communication between the neural crest-derived mesenchyme and dental lamina epithelium (27). Although the earliest signals driving tooth formation and determining tooth placement and shape appear to originate in the epithelium, reciprocal passage of information between the cells of the inner enamel epithelium and underlying condensing mesenchyme control both preameloblast and preodontoblast development and their terminal differentiation. The early signaling growth and transcription factors are common to most tissues. However, as maturation proceeds, cell specific factors, that is factors uniquely produced by the cells themselves, come into play. We demonstrated that particular amelogenin isoforms, which are translated from distinct amelogenin gene splice products, do have signaling properties in fibroblasts and tooth germs in culture (12, 13, 14, 15). Furthermore, we demonstrated that in tooth organ cultures, the M59/[A−4] isoform, secreted by maturing mouse odontoblasts before the onset of dentin mineralization, had a direct affect on ameloblast terminal differentiation (14). The present study was undertaken to examine whether r[A−4] binding and uptake in newborn ameloblasts involves LAMP1, as previously observed by us in an in vitro model of fibroblasts (15), and whether r[A−4] activates intracellular signaling events.
Figure 1 shows that the addition of r[A−4]/M59 to ameloblast-like LS8 cells in culture, up-regulated M180 mRNA expression, as well as the expression of the ARHGAP6 gene, in which the amelogenin gene is embedded. This was not a general stimulatory effect, as the expression of the housekeeping gene GAPDH, and the constitutively expressed genes LAMP1 and LAMP3 were not modulated. When LS8 cells were cultured under conditions which stimulate cell proliferation (10% FBS-DMEM), semi-quantitative PCR analysis showed a significant increase in M180 mRNA expression. The effect of r[A−4] on the expression of M180 mRNA was specific, as demonstrated by the significant up-regulation of M180 expression following addition of r[A−4] to cultures of LS8 cells grown in 2% FBS-DMEM media.
In fibroblasts, r[A−4] was shown to bind to cell surface LAMP1 (15). The conventional RT-PCR data demonstrated that untreated LS8 cells constitutively express abundant LAMP1 message (Figure 1), and subsequent examination by immunocytochemistry demonstratetd that the LS8 cells express abundant cell surface (Figure 3A) and vesicular endosomal/lysosomal LAMP1 (Figure 4A, D). In non-permeablized cells, where anti- LAMP1 antibodies do not detect intra-cytoplasmic LAMP1, exogenous r[A−4] induced a redistribution of plasma membrane-associated LAMP1, succeeded by a change in the shape of the LS8 cells, from a rounded to a more elongated form (Figure 3B). Thus, in LS8 cells, the r[A−4]-induced translocation of LAMP1 at the cell surface mimicked traffic of LAMP1 in the human peripheral blood mononuclear cells (18).
The internalization of r[A−4] into the ameloblast-like LS8 cells was detected in cultures grown at 37°C, similar to the previously observed internalization of r[A−4] into fibroblasts (15). Upon addition of r[A−4], LAMP1 accumulated in a more punctuate fashion in the late endosomal/lysosomal compartments, but was also present at the cell surface (Figure 4D), as shown previously, where LAMP1 redistributed at the plasma membrane of non-permeabilized LS8 cells (Figure 3B). The biotinylated r[A−4] co-localized with LAMP1 at the plasma membrane (Figure 4F, white arrow), as well as intra-cellular, in particular, in the late endosomal/lysosomal compartments (Figure 4F, arrowhead). Interestingly, not all LAMP1 expressed at the plasma membrane was occupied by r[A−4], suggesting that activation by r[A−4] might induce a redistribution of LAMP1 from the cytoplasmic compartment to the LS8 cell surface.
Surprisingly, blockade of LAMP1 by anti-LAMP1 antibodies moderately enhanced M180 mRNA expression to a level comparable to the effect induced by r[A−4] ( Figure 2B). Co-treatment with anti-LAMP1 and r[A−4] strongly enhanced the production of M180 mRNA (Figure 2B). More so, NOS2 activity, which is implicated in the modulation of M180 mRNA in the LS8 cells (Figure 7), was significantly up-regulated by co-treatment with r[A−4] and anti-LAMP1 (Figure 5A). These data raise a very interesting question. If indeed, LAMP1 is involved in the internalization of r[A−4] at the LS8 cell surface, and subsequently, r[A−4] activates intracellular signaling molecules, why should blockade of LAMP1 at the cell surface by the addition of anti-LAMP1 antibodies enhance the internalization and the activity of r[A−4]? Figure 4F shows the co-localization of r[A−4] with LAMP1, both at the cell surface and in cytoplasmic vesicles of the late endosomal/lysosomal compartments, in accord with our previous data from in vitro studies of the binding and internalization of 3H- r[A−4] in mouse fibroblasts (15). LAMP1 is synthesized in the rough endoplasmic reticulum and transported to the trans-Golgi network, where, after glycosylation, it is targeted to either the early/sorting endosomes, or directly to the plasma membrane. LAMP traffic is complex, since the membrane constituents in the early endosomes can move to the late endosomes and ultimately, to the lysosomes, or to a recycling endosome which can deliver some of the cargo to the plasma membrane. The plasma membrane and the early endosome compartment are both primary steps in LAMP traffic, which terminates in its delivery to the lysosomes (19). Many factors regulate these sorting steps, but LAMP traffic is rapid. Endocytosis of ligand-receptor complexes is one mechanism for attenuating the activity of the activating molecule. However, blockade of LAMP1 by anti-LAMP1 antibodies might also induce LAMP1 internalization. Hence, internalized cell-surface LAMP1 might activate amelogenin message expression, similar to the effect induced by internalized r[A−4]. At 37°C, r[A−4] binds to the LS8 cells, and induces a redistribution of LAMP1 at the plasma membrane. Concomitantly, r[A−4] is rapidly internalized into the late endosomes and then, the lysosomes (Figure 4E, F), and it is possible that here, r[A−4] activates signaling pathways that regulate M180 mRNA. One supporting finding is the modulation in M180 mRNA expression following co-treatment of LS8 cells with r[A−4] and anti-LAMP1, as well as the activation of NOS2. The co-operative effect suggests that both molecules activate M180 expression, albeit through distinct signaling pathways. We demonstrated that [A−4] activates the NOS2 pathway, while the signaling pathways activated by LAMP1 still need to be elucidated (Figure 5A, B). Specific NOS2 blockade abrogated NO production and M180 mRNA induction in parallel, although not to the same extent (Figure 7). In the bone growth plate, NOS2 and NOS3 have been detected in chondrocytes during the process of terminal differentiation (21). Hence, internalized r[A−4] might activate NOS2 at the level of the late endosomal compartment, thus, inducing a change in shape of the LS8 cells toward a more elongated form, characteristic of the mature, secretory ameloblast type. Another very interesting possibility arises from studies on Ca2+-mediated exocytosis via LAMP1, which implicate it in the process of plasma membrane resealing of skin fibroblasts wounded by contracting collagen fibers (28).
Whether LAMP1 is involved in exocytosis of M180 protein remains to be determined. Furthermore, this study suggests that M180 mRNA expression in LS8 cells might be regulated by r[A−4] either at the transcriptional level, or by modulation of message stability. In support of this hypothesis is the observation that expression of M180 in developing ameloblasts first occurs in the newborn mouse molar, and continues throughout ameloblast maturation (9), paralleled by the expression of M59/[A−4] (10). Cumulatively, our data suggest that in the newborn ameloblasts, M59/[A−4] participates in the regulation of M180 expression through a NOS2-mediated pathway of activation.
The evidence that modulation of M180 gene expression is regulated by both [A−4] functional activity and the compartmentalized distribution of LAMP1 in developing ameloblasts suggests that [A−4] may operate as an autocrine factor: when secreted into the extracellular matrix, [A−4] will bind cell surface LAMP1, subsequently internalize via LAMP1, and then will be delivered to its appropriate signaling pathway.
Acknowledgments
The authors thank Dr. Malcolm L. Snead, USC, Los Angeles, CA, for his generous gift of the LS8 cells. We are grateful to Dr. Keith Alvares, Northwestern University, Chicago, IL, for the GST-[A−4] fusion constructs used for recombinant peptide synthesis. This work was supported by the NIDCR grant DE-014758 to AV.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith CE, Nanci A. Overview of morphological changes in enamel organ cells associated with major events in amelogenesis. Int J Dev Biol. 1995;39:153–161. [PubMed] [Google Scholar]
- 2.Orzech E, Cohen S, Weiss A, Aroeti B. Interactions between the exocytic and endocytic pathways in polarized Madin-Darby canine kidney cells. J Biol Chem. 2000;275:15207–15219. doi: 10.1074/jbc.275.20.15207. [DOI] [PubMed] [Google Scholar]
- 3.Du C, Falini G, Fermani S, Abbott C, Moradian-Oldak J. Supramolecular assembly of amelogenin nanospheres into birefringent microribbons. Science. 2005;307:1450–1454. doi: 10.1126/science.1105675. [DOI] [PubMed] [Google Scholar]
- 4.Veis A. Material science. A window on biomineralization. Science. 2005;307:1419–1420. doi: 10.1126/science.1109440. [DOI] [PubMed] [Google Scholar]
- 5.Lau EC, Simmer JP, Bringas P, Jr, Hsu DD, Hu CC, Zeichner-David M, Thiemann F, Snead ML, Slavkin HC, Fincham AG. Alternative splicing of the mouse amelogenin primary RNA transcript contributes to amelogenin heterogeneity. Biochem Biophys Res Commun. 1992;188:1253–1260. doi: 10.1016/0006-291x(92)91366-x. [DOI] [PubMed] [Google Scholar]
- 6.Hu CC, Ryu QQ, Quan Q, Zhang CH, Simmer JP. Cloning, characterization, and heterologous expression of exon-4-containing amelogenin mRNAs. J Dent Res. 1997;76:641–647. doi: 10.1177/00220345970760020401. [DOI] [PubMed] [Google Scholar]
- 7.Fincham AG, Hu Y, Lau EC, Slavkin HC, Snead ML. Amelogenin post-secretory processing during biomineralization in the postnatal mouse molar tooth. Arch Oral Biol. 1991;36:305–317. doi: 10.1016/0003-9969(91)90101-y. [DOI] [PubMed] [Google Scholar]
- 8.Papagerakis P, Ibarra JM, Inozentseva N, DenBesten P, MacDougall M. Mouse Amelogenin Exons 8 and 9: Sequence Analysis and Protein Distribution. J Dent Res. 2005;84:613–617. doi: 10.1177/154405910508400706. [DOI] [PubMed] [Google Scholar]
- 9.Hu JC-C, Sun X, Zhang C, Simmer JP. A comparison of enamelin and amelogenin expression in developing mouse molars. Eur J Oral Sci. 2001;109:125–132. doi: 10.1034/j.1600-0722.2001.00998.x. [DOI] [PubMed] [Google Scholar]
- 10.Iacob S, Veis A. Identification of temporal and spatial expression patterns of amelogenin isoforms during mouse molar development. Eur J Oral Sci. 2006;114S:194–200. doi: 10.1111/j.1600-0722.2006.00287.x. [DOI] [PubMed] [Google Scholar]
- 11.Nebgen DR, Inoue H, Sabsay B, Wei K, Ho CS, Veis A. Identification of the chondrogenic-inducing activity from bovine dentin (bCIA) as a low-molecular-mass amelogenin polypeptide. J Dent Res. 1999;78:1484–1494. doi: 10.1177/00220345990780090201. [DOI] [PubMed] [Google Scholar]
- 12.Veis A, Tompkins K, Alvares K, Wei K, Wang L, Wang XS, Brownell AG, Jengh S-M, Healy KE. Specific amelogenin gene splice products have signaling effects on cells in culture and in implants in vivo. J Biol Chem. 2000;275:41263–41272. doi: 10.1074/jbc.M002308200. [DOI] [PubMed] [Google Scholar]
- 13.Veis A. Amelogenin gene splice products: potential signaling molecules. Cell Mol Life Sci. 2003;60:38–55. doi: 10.1007/s000180300003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tompkins K, Alvares K, George A, Veis A. Two related low molecular mass polypeptide isoforms of amelogenin have distinct activities in mouse tooth germ differentiation in vitro. J Bone Min Res. 2005;20:341–349. doi: 10.1359/JBMR.041107. [DOI] [PubMed] [Google Scholar]
- 15.Tompkins K, George A, Veis A. Characterization of a mouse amelogenin [A−4]/M59 cell surfaced receptor. Bone. 2006;38:172–180. doi: 10.1016/j.bone.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 16.Wang HJ, Tannukt S, Zhu DH, Snead ML, Paine ML. Enamel matrix protein interactions. J Bone Min Res. 2005;20:1032–1040. doi: 10.1359/JBMR.050111. [DOI] [PubMed] [Google Scholar]
- 17.Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A. The secretory route of the leaderless protein interleukin 1β involves exocytosis of endolysome-related vesicles. Mol Biol Cell. 1999;10:1463–1475. doi: 10.1091/mbc.10.5.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kannan K, Stewart RM, Bounds W, Carlsson SR, Fukuda M, Betzing KW, Holcombe RF. Lysosome-associated membrane proteins h-LAMP1 (CD107a) and h-LAMP2 (CD107b) are activation-dependent cell surface glycoproteins in human peripheral blood mononuclear cells which mediate cell adhesion to vascular endothelium. Cell Immunol. 1996;171:10–19. doi: 10.1006/cimm.1996.0167. [DOI] [PubMed] [Google Scholar]
- 19.Mathews PM, Martinie JB, Fambrough DM. The pathway and targeting signal for delivery of the integral membrane glycoprotein LEP100 to lysosomes. J Cell Biol. 1992;118:1027–1040. doi: 10.1083/jcb.118.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villalobo A. Nitric oxide and cell proliferation. FEBS J. 2006;273:2329–2344. doi: 10.1111/j.1742-4658.2006.05250.x. [DOI] [PubMed] [Google Scholar]
- 21.Teixeira CC, Ischiropoulos H, Leboy PS, Adams SL, Shapiro IM. Nitric oxide-nitric oxide synthase regulates key maturational events during chondrocyte terminal differentiation. Bone. 2005;37:37–45. doi: 10.1016/j.bone.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 22.Tompkins K, Veis A. Polypeptides translated from alternatively spliced transcripts of the amelogenin gene, devoid of exon 6a, b, c region, have specific effects on tooth germ development in culture. Connect Tissue Res. 2002;43:224–231. doi: 10.1080/03008200290001096. [DOI] [PubMed] [Google Scholar]
- 23.Chen LS, Couwenhoven RI, Hsu D, Luo W, Snead ML. Maintenance of amelogenin gene expression by transformed epithelial cells of mouse enamel organ. Arch Oral Biol. 1992;37:771–778. doi: 10.1016/0003-9969(92)90110-t. [DOI] [PubMed] [Google Scholar]
- 24.Rasmussen TB, Uttenthal A, de Stricker K, Belak S, Storgaard T. Development of a novel quantitative real-time RT-PCR assay for the simultaneous detection of all serotypes of Foot-and-mouth disease virus. Arch Virol. 2003;148:2005–2021. doi: 10.1007/s00705-003-0145-2. [DOI] [PubMed] [Google Scholar]
- 25.Pfaffl MW. A new mathematical model for relative quantification RT-PCR. Nucl Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shapiro JL, Wen X, Okamoto CT, Wang HJ, Lyngstadaas SP, Goldberg M, Snead ML, Paine ML. Cellular uptake of amelogenin, and its localization to CD63, and Lamp1-positive vesicles. Cell Molec Life Sciences. 2007;64(2):244–256. doi: 10.1007/s00018-006-6429-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jernvall J, Thesleff I. Reiterative signaling and patterning during mammalian tooth morphogenesis. Mechan Develop. 2000;92:19–29. doi: 10.1016/s0925-4773(99)00322-6. [DOI] [PubMed] [Google Scholar]
- 28.Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106:157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]