

Abstract
Three new ent-trachylobane diterpenoids (1–3) were isolated and structures elucidated from Mitrephora glabra Scheff. (Annonaceae). Mitrephorone A (1) possesses a hexacyclic ring system with adjacent ketone moieties and an oxetane ring, both of which are unprecedented among trachylobanes. All compounds were evaluated for cytotoxicity against a panel of cancer cells, where 1 displayed the most potent and broadest activity, and against a battery of antimicrobial assays, where all compounds were approximately equipotent.
As part of our collaborative program to search for new compounds from the plant kingdom with promising anticancer activity,1 Mitrephora glabra Scheff. (Annonaceae) was selected for investigation. The genus Mitrephora, representing approximately 40 species distributed widely throughout tropical Asia,2 has been under-explored for biologically active secondary metabolites, and to the best our knowledge, only four species have been studied previously: M. celebica,3,4 M. maingayi,5,6 M. tomentosa,7 and M. zippeliana.8 The organic extract of M. glabra showed promising anticancer activity in cell cytotoxicity assays, and bioactivity-directed fractionation led to the isolation of a series of three new ent-trachylobane diterpenoids (1–3) and the related, known compound ent-trachyloban-18-oic-acid (4).9,10
The molecular formula of compound 1 was determined by HRMS to be C21H26O5, establishing an index of unsaturation of nine. Examination of the 1H, 13C, and DEPT-135 NMR data showed the presence of four singlet methyl groups (including a methoxy unit), six methylene groups, two methine groups, and nine quaternary carbons, including two oxygenated carbons in the aliphatic region and three carbonyl groups. These data accounted for all of the protons, indicating the absence of a free hydroxyl group. Importantly, the lack of any olefinic moieties required the presence of six rings to satisfy the degrees of unsaturation.
Analysis of the COSY data revealed two major spin systems. One of these comprised H2-1/H2-2/H2-3, which was extended to form the western part of the structure through HMBC studies (Fig. 1A). For example, H2-1 displayed HMBC correlations with C-2, C-3, and C-10, while the methyl protons of H3-20 showed correlations with C-1, C-5, and C-10; the combination of these data construed the connections of both C-1 and C-20 to C-10 and then C-10 to C-5. A methoxycarbonyl group was inferred based on the chemical shifts of C-19 and C-21 along with a relevant HMBC correlation (H3-21 to C-19). Also, the methyl protons of H3-18 displayed HMBC correlations with C-3, C-4, C-5, and C-19, and, in conjunction with an HMBC correlation from H2-3 to C-19, these data permitted the connectivities of both the C-18 methyl group and the C-19 methoxycarbonyl moiety to the quaternary C-4. The other spin-system deduced from the COSY spectrum corresponded to the H2-11/H-12/H-13/H2-14 unit, and these were confirmed in turn via HMBC correlations (Fig. 1B). Positions C-8, C-9, C-16, and C-17 were then incorporated with this four-carbon unit, based on HMBC correlations from H2-11 to C-9 and C-16, from H-12 to C-9 and cross correlations to position C-17, and from H-13 to C-8 and cross correlations to position C-17. At this point, an isolated methylene unit displaying an AB pattern (H2-15; geminal coupling of 12 Hz) was positioned between the two quaternary carbons C-8 and C-16 based upon HMBC correlations from H3-17 to C-15 and from H2-15 to C-13 and C-14. Furthermore, both H2-14 and H2-15 showed HMBC correlations with C-7 (δC 198.6), one of the two remaining carbonyl carbons, thus requiring attachment of C-7 to C-8. The two partial structures in Fig. 1 were united by connecting C-9 to C-10, and this was supported by key HMBC correlations of both H2-1 and H3-20 with C-9 and of H2-11 with C-10.
Figure 1.

Partial structures of compound 1 illustrating key COSY (—) and HMBC (→) correlations.
The remaining assignments were for the other carbonyl unit (C-6; δC 190.0) and a single oxygen atom. The downfield chemical shift of both C-5 (δC 89.0) and C-9 (δC 87.3) suggested oxygenation, and thus, an oxetane ring could be proposed to share this single oxygen atom. The relatively upfield chemical shift of both of the C-6 and C-7 carbonyl moieties suggested an adjacent diketone configuration.11,12 This assignment was supported by the lack of relevant HMBC correlations to C-6, given its position more than four bonds from any proton.
The relative stereochemistry of 1 was construed from analysis of molecular models, energy minimized using the MMFF94 force field in Spartan ’04 (Wavefunction, Inc., Irvine, CA) overlaid with key correlations observed in the ROESY NMR spectrum (Fig. 2). The oxetane ring and the adjacent diketone moieties impart considerable strain to this rigid ring system. Position 10 was pivotal to the assignment of the relative stereochemistry, as strong ROESY correlations were observed to H3-20 from H3-21, H2-2, and H2-14, which were all supported by calculated interatomic distances of approximately 2.4 Å. Position C-4 was determined by strong correlations from H3-21 to H3-20, also supported by a calculated interatomic distance of 2.4 Å from the molecular model, and although not conclusive, a ROESY correlation was not observed between H3-18 and H3-20. The configurations for C-8 and C-13 were determined by ROESY correlations between H2-14 and H3-20. Moreover, the literature on ent-trachylobanes supports this configuration for the cyclopropane portion of these diterpenoids, including the configurations for C-12 and C-16.13 Finally, the formation of the oxetane ring necessitates the displayed orientation of C-5 to C-9.
Figure 2.
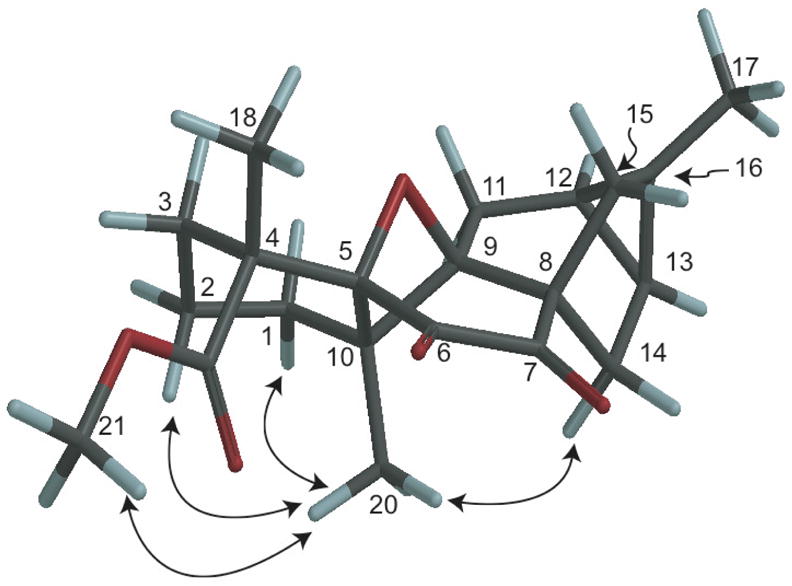
Energy minimized model of 1 illustrating the major ROESY correlations used to define the relative stereochemistry.
The trivial name mitrephorone A (1) was ascribed to this structure, which was consistent with the ent-trachylobane-type of plant metabolites,13,14 a group of diterpenoids possessing a characteristic cyclopropane ring system. This new compound, however, expands the functionalities of this core structural type by including both an unprecedented C-5, C-9 oxetane ring system and the adjacent diketone moieties.
Comparison of the NMR data for compound 2 with those of 1 revealed structural similarities, including the presence of the upfield H-12 signal as exhibited by all other ent-trachylobanes.10,13,14 The NMR and HRMS data for 2 revealed its molecular formula as C21H28O4, which has one less degree of unsaturation than 1. In this case, an oxetane ring was not present, as evident from the absence of any oxygenated aliphatic carbons, and one of the two ketone carbons of 1 (C-6) was replaced by an olefinic unit (C-5, δC 141.3; C-6, δC 142.3). Key HMBC correlations of H2-14 and H2-15 with C-7, and the C-6-OH proton with C-5, C-6, and C-7, permitted assignment of the structure of mitrephorone B as shown in 2.
The NMR data for compound 3 were comparable to those of 2, and this suggested a common core structure. In fact, the major differences between 2 and 3 were merely the presence of a new downfield ketone carbon (C-2; δC 207.1) and two pairs of isolated methylene units (H2-1 and H2-3) in 3. These data, together with the mass spectrometric information, suggested that the C-2 methylene in 2 was replaced by a carbonyl carbon in 3, and this was supported by HSQC and HMBC analyses. All protons of H2-1 and H2-3 showed strong HMBC correlations with C-2, making the unambiguous assignment of the structure of 3, as shown.
The relative stereochemistries of mitrephorone B (2) and mitrephorone C (3) were established independently based on similar ROESY data analysis as described for 1, and all three adopt analogous configurations as shown. The absolute stereochemistry for all three compounds was presumed to belong to their enantio- series, as determined by crystallographic analysis9 and chemical transformations10 of other known ent-trachylobane-type diterpenoids.
For the bioactivity-directed fractionation studies, the human KB carcinoma assay was used,15,16 and all pure compounds were evaluated also using a human cancer cell panel of MCF-7, NCI-H460, and SF-268 cell lines.17 Of the three new compounds, 1 displayed the broadest spectrum of cytotoxicity, having a modest IC50 value against all cell lines (Table 3), even SF-268, a slow-growing tumor cell line. Compound 2 had modest activity against KB cells, but was inactive otherwise, while compound 3 lacked any discernable cytotoxicity against any of the four cancer cell lines.
Table 3.
Cytotoxicity and Antimicrobial Activity of Compounds Isolated from Mitrephora glabra
| IC50 values (μg/mL)a | minimal inhibitory concentration (μg/mL)b | |||||||
|---|---|---|---|---|---|---|---|---|
| compound | KB | MCF-7 | H460 | SF-268 | M. luteus | M. smegmatis | S. cerevisiae | A. niger |
| 1 | 8.0 | 15.7 | 23.3 | 30.9 | 125 | 63 | 63 | 63 |
| 2 | 7.0 | Inactive | Inactive | Inactive | 88 | 88 | 88 | 88 |
| 3 | Inactivec | Inactive | Inactive | Inactive | 63 | 31 | 31 | 63 |
| 4 | Inactive | Inactive | Inactive | Inactive | 19 | 75 | 75 | 75 |
| positive controlsd | 0.005 (camptothecin) | 0.025 (camptothecin) | 0.002 (camptothecin) | 0.021 (camptothecin) | 0.1 (ampicillin) | 0.2 (kanamycin) | 25 (amphotericin B) | 25 (amphotericin B) |
KB: human oral epidermoid carcinoma; MCF-7: human mammary adenocarcinoma; H460: human non-small cell (large cell) lung carcinoma; SF-268: human astrocytoma. IC50 values are determined as the concentration required to reduce cellular staining with sulforhodamine B by 50% relative to untreated controls following 72 h of continuous exposure.17
Minimal inhibitory concentration is the lowest concentration of compound completely inhibiting growth (as turbidity) expressed in μg/mL.17
Inactive = IC50value > 30 μg/mL.
Average values.
Trachylobane diterpenoids are reported to have antimicrobial activity.3,13,18 As such, the minimal inhibitory concentration (MIC) of all the compounds were measured against bacteria (Micrococcus luteus and Mycobacterium smegmatis), a yeast (Saccharomyces cerevisiae), and a filamentous fungus (Aspergillus niger).17 Compounds 1–3 exhibited modest MIC values and were equipotent against all organisms. This is in sharp contrast to the anticancer data, where only compound 1 had broad-spectrum activity (Table 3). Moreover, the least active compound in the anticancer assays, compound 3, appeared to have the strongest antimicrobial activity of the three, possessing an MIC value against the yeast, S. cerevisiae, that was similar to that of the standard antifungal antibiotic, amphotericin B.
In summary, this is the first reported investigation of Mitrephora glabra for bioactive compounds, and three new ent-trachylobane-type diterpenoids, mitrephorones AC (1–3), were obtained. The oxetane ring in mitrephorone A (1) was not present in any of the known members of this class, making it the first representative of this novel hexacyclic ring system. Based on a limited set of human tumor cell cytotoxicity data, this unique structure seems to impart a greater degree of anticancer activity than in the other trachylobanes. In the antimicrobial assays, all of the compounds had similar levels of activity, regardless of the presence or absence of the oxetane ring.
Supplementary Material
Supporting Information Available: Experimental details and 1- and 2-D NMR spectra of mitrephorone A (1). This material is available free of charge via the Internet at http://pubs.acs.org.
Table 1.
NMR Data for Mitrephorone A (1)a
| position | δC | DEPT | δH (mult.; J in Hz) | HMBC (H → C) |
|---|---|---|---|---|
| 1 | 32.4 | CH2 | 2.20 (m); 1.63 (m) | 2, 3, 9, 10 |
| 2 | 17.5 | CH2 | 1.58 (m) | 1, 3, 4, 10 |
| 3 | 35.0 | CH2 | 1.78 (br d; 10); 1.64 (br d; 10) | 1, 4, 5, 19 |
| 4 | 43.0 | C | ---- | ---- |
| 5 | 89.0 | C | ---- | ---- |
| 6 | 190.0 | C | ---- | ---- |
| 7 | 198.6 | C | ---- | ---- |
| 8 | 59.0 | C | ---- | ---- |
| 9 | 87.3 | C | ---- | ---- |
| 10 | 48.5 | C | ---- | ---- |
| 11 | 26.4 | CH2 | 2.29 (dd; 17, 2); 2.11 (dd; 17, 2) | 8, 9, 10, 12, 13, 16 |
| 12 | 17.8 | CH | 0.74 (br dd; 7, 2) | 9, 17 |
| 13 | 20.9 | CH | 1.07 (dd; 7, 3) | 8, 17 |
| 14 | 30.1 | CH2 | 2.49 (dd; 13, 3); 1.74 (d; 13) | 7, 8, 9, 12, 13 |
| 15 | 40.0 | CH2 | 2.31 (d; 12); 1.63 (d; 12) | 7, 8, 9, 13, 14 |
| 16 | 21.6 | C | ---- | ---- |
| 17 | 19.1 | CH3 | 1.21 (s) | 12, 13, 15, 16 |
| 18 | 18.4 | CH3 | 1.12 (s) | 3, 4, 5, 19 |
| 19 | 174.9 | C | ---- | ---- |
| 20 | 18.3 | CH3 | 1.09 (s) | 1, 5, 9, 10 |
| OCH3-21 | 51.7 | CH3 | 3.64 (s) | 19 |
Table 2.
NMR Data for Mitrephorones B (2) and C (3)
| mitrephorone B (2) | mitrephorone C (3) | |||
|---|---|---|---|---|
| position | δC | δH (mult.; J in Hz) | δC | δH (mult.; J in Hz) |
| 1 | 30.5 | 1.19 (m); 1.69 (br d; 9) | 48.4 | 2.08 (d; 18); 2.53 (d; 18) |
| 2 | 14.5 | 1.74 (br d; 11) | 207.1 | ---- |
| 3 | 30.7 | 1.54 (br d; 11); 2.18 (m) | 44.5 | 2.44 (d; 18); 2.96 (d; 18) |
| 4 | 45.5 | ---- | 46.2 | ---- |
| 5 | 141.3 | ---- | 137.4 | ---- |
| 6 | 142.3 | ---- | 142.5 | ---- |
| 7 | 197.9 | ---- | 197.4 | ---- |
| 8 | 48.3 | ---- | 48.1 | ---- |
| 9 | 48.0 | 1.78 (br d; 11) | 47.4 | 1.95 (br d; 11) |
| 10 | 37.3 | ---- | 38.5 | ---- |
| 11 | 19.9 | 1.86 (br dd; 9, 14); 2.02 (ddd; 4,11, 14) | 19.7 | 1.85 (br dd; 9, 12); 2.05 (br dd; 4, 12) |
| 12 | 19.1 | 0.71 (br d; 6) | 18.9 | 0.76 (br dd; 3, 7) |
| 13 | 22.4 | 1.02 (dd; 3, 6) | 22.3 | 1.07 (dd; 3, 8) |
| 14 | 33.7 | 1.91 (br d; 12); 2.18 (m) | 33.7 | 1.99 (m); 2.19 (br d; 12) |
| 15 | 47.4 | 1.47 (d; 12); 2.21 (d; 12) | 47.4 | 1.50 (d; 12); 2.25 (d; 12) |
| 16 | 24.1 | ---- | 24.3 | ---- |
| 17 | 19.9 | 1.22 (s) | 19.8 | 1.24 (s) |
| 18 | 22.6 | 1.45 (s) | 23.4 | 1.46 (s) |
| 19 | 177.2 | ---- | 174.9 | ---- |
| 20 | 21.5 | 1.33 (s) | 22.9 | 1.44 (s) |
| OCH3-21 | 52.2 | 3.65 (s) | 52.8 | 3.70 (s) |
| OH-6 | ---- | 6.24 (s) | ---- | 6.32 (s) |
Acknowledgments
Supported by grant U19-CA52956 from the National Cancer Institute, National Institutes of Health. N.H.O. acknowledges partial support via a Research Scholar Grant from the American Cancer Society (RSG-02-024-01-CDD). HR-MALDI-TOF mass spectrometry data were acquired on an Applied Biosystems 4700 tandem time-of-flight instrument by the Mass Spectrometry Research Group at Research Triangle Institute. HREIMS data were acquired on a Thermo-Finnigan MAT95 BEqq instrument by the mass spectrometry core facility at the University of Florida.
References
- 1.Kinghorn AD, Farnsworth NR, Soejarto DD, Cordell GA, Swanson SM, Pezzuto JM, Wani MC, Wall ME, Oberlies NH, Kroll DJ, Kramer RA, Rose WC, Vite GD, Fairchild CR, Peterson RW, Wild R. Pharm Biol. 2003;41(supplement):53–67. [Google Scholar]
- 2.Weerasooriya AD, Saunders RMK. Bot J Linn Soc. 2001;135:305–313. [Google Scholar]
- 3.Zgoda-Pols JR, Freyer AJ, Killmer LB, Porter JR. Fitoterapia. 2002;73:434–438. doi: 10.1016/s0367-326x(02)00124-7. [DOI] [PubMed] [Google Scholar]
- 4.Zgoda JR, Freyer AJ, Killmer LB, Porter JR. J Nat Prod. 2001;64:1348–1349. doi: 10.1021/np0102509. [DOI] [PubMed] [Google Scholar]
- 5.Lee NH, Xu YJ, Goh SH. J Nat Prod. 1999;62:1158–1159. doi: 10.1021/np980565x. [DOI] [PubMed] [Google Scholar]
- 6.Yu R, Li BG, Ye Q, Zhang GL. Nat Prod Res. 2005;19:359–362. doi: 10.1080/14786410412331280104. [DOI] [PubMed] [Google Scholar]
- 7.Supudompol B, Chaowasku T, Kingfang K, Burud K, Wongseripipatana S, Likhitwitayawuid K. Nat Prod Res. 2004;18:387–390. doi: 10.1080/14786410310001643902. [DOI] [PubMed] [Google Scholar]
- 8.Brophy J, Goldsack R, Forster P. J Essent Oil Res. 2004;16:95–100. [Google Scholar]
- 9.Hasan CM, Healey TM, Waterman PG. Phytochemistry. 1982;21:177–179. [Google Scholar]
- 10.Leong YW, Harrison LJ. Phytochemistry. 1997;45:1457–1459. [Google Scholar]
- 11.Sotanaphun U, Suttisri R, Lipipun V, Bavovada R. Phytochemistry. 1998;49:1749–1755. doi: 10.1016/s0031-9422(98)00290-8. [DOI] [PubMed] [Google Scholar]
- 12.Ulubelen A, Evren N, Tuzlaci E, Johansson C. J Nat Prod. 1988;51:1178–1183. doi: 10.1021/np50060a021. [DOI] [PubMed] [Google Scholar]
- 13.Fraga BM. Phytochem Anal. 1994;5:49–56. [Google Scholar]
- 14.Graikou K, Aligiannis N, Skaltsounis AL, Chinou I, Michel S, Tillequin F, Litaudon M. J Nat Prod. 2004;67:685–688. doi: 10.1021/np030423p. [DOI] [PubMed] [Google Scholar]
- 15.Gu J-Q, Graf TN, Lee D, Chai H-B, Mi Q, Kardono LBS, Setyowati FM, Ismail R, Riswan S, Farnsworth NR, Cordell GA, Pezzuto JM, Swanson SM, Kroll DJ, Falkinham JO, 3rd, Wall ME, Wani MC, Kinghorn AD, Oberlies NH. J Nat Prod. 2004;67:1156–1161. doi: 10.1021/np040027m. [DOI] [PubMed] [Google Scholar]
- 16.Oberlies NH, Burgess JP, Navarro HA, Pinos RE, Soejarto DD, Farnsworth NR, Kinghorn AD, Wani MC, Wall ME. J Nat Prod. 2001;64:497–501. doi: 10.1021/np0005006. [DOI] [PubMed] [Google Scholar]
- 17.Alali FQ, El-Elimat T, Li C, Qandil A, Alkofahi A, Tawaha K, Burgess JP, Nakanishi Y, Kroll DJ, Navarro HA, Falkinham JO, 3rd, Wani MC, Oberlies NH. J Nat Prod. 2005;68:173–178. doi: 10.1021/np0496587. [DOI] [PubMed] [Google Scholar]
- 18.Gibbons S. Nat Prod Rep. 2004;21:263–277. doi: 10.1039/b212695h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Experimental details and 1- and 2-D NMR spectra of mitrephorone A (1). This material is available free of charge via the Internet at http://pubs.acs.org.