

Abstract
Human carbonyl reductase 1 (CBR1) metabolizes endogenous and xenobiotic substrates such as the fever mediator, prostaglandin E2 (PGE2), and the anticancer anthracycline drug, daunorubicin. We screened 33 CBR1 full-length cDNA samples from white and black liver donors and performed database analyses to identify genetic determinants of CBR1 activity. We pinpointed a single nucleotide polymorphism on CBR1 (CBR1 V88I) that encodes for a valine-to-isoleucine substitution for further characterization. We detected the CBR1 V88I polymorphism in DNA samples from individuals with African ancestry (p = 0.986, q = 0.014). Kinetic studies revealed that the CBR1 V88 and CBR1 I88 isoforms have different maximal velocities for daunorubicin (Vmax CBR1 V88, 181 ± 13 versus Vmax CBR1 I88, 121 ± 12 nmol/min · mg, p < 0.05) and PGE2 (Vmax CBR1 V88, 53 ± 7 versus Vmax CBR1 I88, 35 ± 4 nmol/min · mg, p < 0.01). Concomitantly, CBR1 V88 produced higher levels of the cardiotoxic metabolite daunorubicinol compared with CBR1 I88 (1.7-fold, p < 0.0001). Inhibition studies demonstrated that CBR1 V88 and CBR1 I88 are distinctively inhibited by the flavonoid, rutin (IC50 CBR1 V88, 54.0 ± 0.4 μM versus IC50 CBR1 I88, 15.0 ± 0.1 μM, p < 0.001). Furthermore, isothermal titration calorimetry analyses together with molecular modeling studies showed that CBR1 V88I results in CBR1 isoforms with different binding affinities for the cofactor NADPH (Kd CBR1 V88, 6.3 ± 0.6 μM versus Kd CBR1 I88, 3.8 ± 0.5 μM). These studies characterize the first functional genetic determinant of CBR1 activity toward relevant physiological and pharmacological substrates.
Human carbonyl reductase 1 (CBR1) is a monomeric cytosolic enzyme that catalyzes the two-electron reduction of biologically and pharmacologically active substrates by using NADPH as cofactor (Forrest and Gonzalez, 2000). For example, CBR1 converts the potent fever mediator prostaglandin E2 (PGE2) into the less active metabolite PGF2α. Recent studies have shown that the transcription of CBR1 in lung and liver is down-regulated to suppress the catabolism of PGE2 during the course of the febrile response (Ivanov et al., 2003; Ivanov and Romanovsky, 2004). CBR1 also plays a predominant role in the metabolism of the anticancer anthracyclines doxorubicin and daunorubicin by catalyzing the formation of their corresponding C-13 alcohol metabolites (doxorubicinol and daunorubicinol). Anthracycline C-13 alcohol metabolites circulate in plasma, have half-lives similar to those of the parent compounds, and are devoid of significant tumor cell-killing activity. The use of anthracyclines for cancer treatment is hampered by the development of clinical cardiotoxicity in some patients (Wouters et al., 2005). The pathogenesis of anthracycline-related cardiotoxicity is complex and appears to be mediated by a combination of oxidative stress and intracardiac metabolic perturbations induced by C-13 anthracycline alcohol metabolites (Minotti et al., 2004). In human myocardium, cytosolic carbonyl reductases (CBRs), and aldo/keto reductases catalyze the two-electron reduction of the anthracycline side chain C-13 carbonyl group to form cardiotoxic alcohol metabolites (e.g., doxorubicinol, daunorubicinol). CBR activity is the main source of quinone detoxification in humans, and biochemical studies showed that aldo/keto reductases have 7- to 18-fold lower catalytic efficiencies (Vmax/Km) for the reduction of anthracycline substrates than CBRs (Wermuth et al., 1986; Ohara et al., 1995; Rosemond and Walsh, 2004; Covarrubias et al., 2006). The key role of CBR1 during the pathogenesis of anthracycline-related cardiotoxicity has been established in biochemical studies and in experiments with murine models (Mordente et al., 2001). Mice over-expressing human CBR1 in heart showed high intracardiac levels of doxorubicinol and increased signs of myocardial damage after doxorubicin administration (Forrest et al., 2000). Conversely, mice with a null allele of Cbr1 (Cbr1+/−) treated with doxorubicin showed low plasmatic levels of doxorubicinol and significantly lower incidence of anthracycline-related cardiotoxicity compared with animals with two active Cbr1 alleles (Cbr1+/+) (Olson et al., 2003).
Carbonyl reductase activity varies widely among individuals and may contribute to the unpredictable pharmacodynamics of CBR1 drug substrates. We documented wide ranges of hepatic CBR activities by using the prototypical quinone substrate menadione in cytosols from white (range, <0.1–28.0 nmol/min · mg) and black donors (range, 4.1–21.5 nmol/min · mg) (Covarrubias et al., 2006). To the best of our knowledge, the potential contribution of genetic polymorphisms on CBR1 to variable CBR activity has not yet been explored. Consequently, the aim of this study was to identify and characterize the functional impact of single nucleotide polymorphisms (SNPs) on the coding region of CBR1. Therefore, we first sequenced CBR1 full-length cDNA samples from liver donors (13 blacks and 20 whites) and performed SNP database analyses to identify SNPs in the coding region of CBR1. We pinpointed a SNP on CBR1 that results in a valine-to-isoleucine substitution at position 88 (CBR1 V88I). Next, we investigated the presence of the CBR1 V88I polymorphism in human DNA variation panels, and we characterized the functional properties of the resulting CBR1 V88 and CBR1 I88 protein isoforms. Kinetic analyses with prototypical CBR1 substrates (e.g., PGE2, and daunorubicin) together with data from isothermal titration calorimetry experiments demonstrate that CBR1 V88 and CBR1 I88 have distinctive functional properties. Furthermore, we discuss the impact of CBR1 V88I in light of the recently reported X-ray crystal structure of human CBR1 (Tanaka et al., 2005). Our findings document the first genetic determinant of human CBR1 activity toward substrates of physiological and pharmacological relevance.
Materials and Methods
Human DNA and RNA Samples
Human DNA variation panels were obtained from the Coriell Institute for Medical Research (Camden, NJ). Each panel contained DNA samples from individuals representing the following groups: Mexican, South American (Andes region), Chinese, Indo-Pakistani, Japanese, Southeast Asian (excluding Japanese and Chinese), Middle Eastern, Pacific, North American (African ancestry), North American (European ancestry), African (north of the Sahara), and African (south of the Sahara). Human liver RNA samples (n = 33) were provided by the Liver Tissue Procurement and Distribution System (University of Minnesota Department of Pediatrics, Gastroenterology and Nutrition, Minneapolis, MN; National Institutes of Health Contract N01-DK-9-2310) and by the Cooperative Human Tissue Network (http://www.chtn.ims.nci.nih.gov/), respectively.
Sequencing of CBR1 Full-Length cDNA Samples
PCR amplification of CBR1 full-length cDNA followed by SNP detection by direct sequencing analysis was used to investigate genetic polymorphisms in the coding region of CBR1 (Nickerson et al., 1997). CBR1 full-length cDNA was synthesized from 1 μg of total liver RNA by using Superscript (Invitrogen, Carlsbad, CA) and oligo(dT)12–18 primers. PCR amplification primers located at the 5′- and 3′-untranslated regions were designed with PRIMER3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi), and their sequence homologies were analyzed by BLAST (http://www.ncbi.nlm.nih.gov/blast/). Primers were as follows: forward 5′-TTCTCCACGCAGGTGTTC-3′, and reverse 5′-GCATCAGAGGAAATCACAAAAG-3′. One microliter of the cDNA conversion mixture was amplified by using the Expand Long Template PCR system with buffer 1 (Roche Diagnostics, Indianapolis, IN). PCR amplification conditions were 95°C for 2 min; 45 cycles of 95°C for 35 s, 57.5°C for 40 s, and 68°C for 70 s; and final extension at 70°C for 10 min. Aliquots of PCR products were visualized by UV light after size-fractionation in 1.0% agarose gels supplemented with ethidium bromide (0.5 μg/ml). All samples presented a unique 1077-bp band.
PCR products were treated with ExoSAP-IT (U.S. Biochemical Corp., Cleveland, OH) and purified with PCR cleanup kits (QIAGEN, Valencia, CA). Purified products were sequenced with both forward and reverse PCR primers, and with the internal sequencing primer 5′-TCTGGTGCACTCCCTTCTTT-3′, by using the dye-terminator method in an ABI PRISM 3130 XL Genetic Analyzer (Applied Biosystems, Foster City, CA). The resultant trace files were assembled and analyzed with PolyPhred (http://lpgws.nci.nih.gov/perl/snp/snp_cgi.pl), and with Vector NTI (Invitrogen). PolyPhred detects the presence of single nucleotide substitutions using fluorescence sequencing of PCR products. To be considered a true variant, the SNPs were confirmed with forward and reverse primers.
CBR1 V88I Genotyping
The CBR1 V88I polymorphism (rs1143663) was investigated by using two genotyping techniques. The first approach utilized the TaqMan technology with specific fluorescent probes labeled with the fluorochromes 5-carboxyfluorescein (FAM) and VIC to discriminate the G and the A alleles, respectively (Applied Biosystems, Assays-by-Design). Genotyping reactions were performed according to the manufacturer’s protocol in a Bio-Rad IQ5 thermal cycler (Bio-Rad, Hercules, CA). Genotyping controls for each allele combination were prepared by diluting plasmid DNA (30 ng/μl) containing the CBR1 V88 insert (G/G genotype), the CBR1 I88 insert (A/A genotype), and a mixture (1:1) of CBR1 V88 plus CBR1 I88 inserts (G/A genotype), respectively (see further details under Cloning and Expression of CBR1 V88 and CBR1 I88). The second CBR1 V88I genotyping approach involved PCR amplification of a fragment of CBR1 (183 bp) followed by SNP detection by direct sequencing analysis. In brief, 50 ng of genomic DNA were amplified using 2.5 units of Amplitaq Gold DNA polymerase (Applied Bio-systems) with the following primers: 5′-CTTCCACCAGCTGGACATC-3′ (forward) and 5′-CCAGTGCATCGGTTCTTCTT-3′ (reverse). PCR amplification conditions were 92°C for 2 min, followed by 45 cycles of 92°C for 30 s, 57°C for 30 s, and 72°C for 70 s. PCR products were verified by agarose gel electrophoresis (2.0%). PCR products were treated with ExoSAP-IT (U.S. Biochemical Corp.), purified with QIAquick kits (QIAGEN), and sequenced with forward and reverse PCR primers by using the dye-terminator method. The resultant trace files were analyzed as described above.
Cloning and Expression of CBR1 V88 and CBR1 I88
CBR1 V88 full-length cDNA was obtained by reverse transcription-PCR using liver RNA from a donor with CBR1 V88I homozygous G/G genotype as template. Total liver RNA (1 μg) was reverse-transcribed by using Superscript (Invitrogen) and primers. The resulting cDNA was amplified with the oligo(dT)12–18 Expand Long Template PCR System (Roche Diagnostics) with the following primers: 5′-CATATGTCGTGGCATCCATGTAGC-3′ (forward), and 5′-CTCGAGTCACCACTGTTCAACTCTCTTCTC-3′ (reverse). The PCR product was purified and cloned into a pET28a expression vector (EMD, Novagen, San Diego, CA). The insert was fully sequenced with vector- and insert-specific primers to confirm its identity and the absence of mutations. The pET28a CBR1 V88 clone was used as template to generate the variant CBR1 I88 insert by site-directed mutagenesis (QuikChange; Stratagene, La Jolla, CA) with the following primers: 5′-GGCCTGGACGTGCTGATCAACAACGCGGGCATCG-3′ (forward), and 5′-CGATGCCCGCGTTGTTGATCAGCACGTCCAGGCC-3′ (reverse). The amplicon was treated with DpnI, and the resultant fragment was used to transform XL-10 Gold Ultra-competent cells (Stratagene). Cells were cultured and selected with kanamycin (30 μg/ml). After selection, plasmid DNA was extracted and purified using QIAprep Spin Miniprep kits (QIAGEN). The integrity of the CBR1 I88 construct was confirmed by sequencing. CBR1 V88 and CBR1 I88 constructs were transfected into Escherichia coli BL21 (DE3)-competent cells. Cells were plated on Luria-Bertani broth agar supplemented with kanamycin (30 μg/ml) for selection. Colonies were randomly picked and cultured overnight at 37°C in 100 ml of Luria-Bertani broth with kanamycin (15 μg/ml). Cultures were expanded to 1 liter and induced with isopropyl-β-D-thiogalactoside (1 mM). E. coli cells were pelleted and lysed in buffer containing 50 mM sodium phosphate, 10 mM imidazole, 300 mM NaCl, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 mM pepstatin, and 10 μM NADPH. Lysates were supplemented with lysozyme (1 mg/ml), DNase I (5 μg/ml), and ribonuclease (10 μg/ml). Lysates were sonicated and centrifuged for 30 min at 4°C (25,000g). The resulting supernatants were filtered through a 0.45-μm polyvinylidene fluoride membrane (Millipore, Billerica, MA) and transferred to vials equilibrated with nickel nitrilotriacetic acid His-Bind Resin (Superflow; EMD, Novagen). CBR1 V88 and CBR1 I88 proteins were eluted in buffer containing 50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 250 mM imidazole, and 1 mM dithiothreitol. Proteins were extensively dialyzed for 48 h in phosphate-buffered saline at 4°C with two buffer changes (4 liters each). After dialysis, proteins were aliquoted and stored at −80°C.
Western Blotting
Recombinant CBR1 V88 and CBR1 I88 proteins were electrophoresed on 4 to 20% precast polyacrylamide gels (Pierce, Rockford, IL) and transferred to Hybond polyvinylidene difluoride membranes (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). Membranes were first incubated with a polyclonal anti-human CBR1 antibody (1:1000 dilution; Abcam Inc., Cambridge, MA), followed by incubation with a secondary rabbit anti-goat IgG antibody conjugated with horseradish peroxidase (1:10,000 dilution; Sigma-Aldrich, St. Louis, MO). Immunoreactive bands were visualized with the ECL Plus Western blotting detection system (Amersham Biosciences) by using a ChemiDoc XRS gel documentation system equipped with Quantity One software (Bio-Rad).
Kinetic Analysis
CBR1 V88 and CBR1 I88 enzymatic activities were measured by recording the rate of oxidation of the NADPH cofactor at 340 nm (NADPH molar absorption coefficient, 6220 M−1 cm−1) (Wermuth, 1981; Bohren et al., 1987; Lakhman et al., 2005). Experiments were performed in a Cary-Varian Bio 300 UV-visible spectrophotometer equipped with thermal control and proprietary software for enzyme kinetics analysis (Varian, Walnut Creek, CA). Reactions were incubated at 37°C and monitored for 3 min at an acquisition rate of 5 readings/s (900 readings). Assay mixtures (1.0 ml) contained 200 μM NADPH (Sigma-Aldrich), 100 mM potassium phosphate buffer (pH 7.4), CBR1 enzyme (1 mg/ml), and one of the following substrates: menadione (20–500 μM; Sigma-Aldrich), daunorubicin (20–650 μM; Sigma-Aldrich), or PGE2 (40–2000 μM; Cayman Chemical, Ann Arbor, MI). The kinetic constants of the cofactor were determined by recording activities with different NADPH concentrations (range, 10–200 μM) and with menadione at a fixed concentration of 250 μM. Enzymatic velocities (V0) were automatically calculated by linear regression of the Δabs/Δtime points. CBR1 protein concentrations were determined by recording the absorbance at 280 nm (molar extinction coefficient, 21,500 M−1 cm−1; http://us.expasy.org/). Enzyme kinetic parameters (Km and Vmax) were calculated by nonlinear regression analysis using a one-site binding model. Kinetic and statistical analyses were performed with GraphPad Prism (version 4.03; GraphPad Software Inc., San Diego, CA). CBR1 V88 and CBR1 I88 kinetic parameters were compared by using GraphPad’s built-in algorithm based on the F-test. Statistical results were further confirmed by performing unpaired Student’s t tests with Excel XP 2003 (Microsoft, Redmond, WA). Both statistical approaches resulted in essentially identical p values. Differences between means were considered to be significant at p < 0.05.
Daunorubicinol Quantification
The amount of daunorubicinol synthesized by CBR1 V88 and CBR1 I88 was quantified by HPLC analysis (Cowens et al., 1993). CBR1 V88 and CBR1 I88 incubation mixtures (1.0 ml) contained potassium phosphate buffer (pH 7.4, 100 mM), enzyme (1 mg/ml), NADPH (200 μM), and daunorubicin (125–500 μM). Mixtures were incubated for 30 min at 37°C, and reactions were stopped by placing samples on ice. Samples were stored at −80°C until HPLC analysis. Samples were diluted with 150 μl of ammonium formate buffer (16 mM, pH 4.0) and directly injected (25 μl) into a Waters HPLC system (Waters, Milford, MA), consisting of an M600 pump, an M717Plus autosampler, and an M474 scanning fluorescence detector with excitation and emission wavelengths set at 490 and 590 nm, respectively. Separation was performed on a reverse phase column (C8, 5 μm, 150 mm; Alltech Associates, Deerfield, IL), with a mobile phase gradient of ammonium formate and tetrahydrofuran. Doxorubicin was used as internal standard. Calibration curves with authentic daunorubicin and doxorubicin standards (0.5–20 μg/ml) were run for each determination. Daunorubicinol/internal standard ratios of the peak areas were used for quantification. For the lack of daunorubicinol standard, the daunorubicinol peak was quantified in daunorubicin equivalents. The capacity factors (K′) for doxorubicin, daunorubicinol, and doxorubicinol were calculated from chromatograms by using the formula: K′ = (TA − To)/To, where TA is the retention time of the analyte and To is the retention time of nonretained compounds. K′ values were 6.6 (doxorubicin), 6.9 (daunorubicinol), and 7.8 (doxorubicinol), with resolution values of ~1 (doxorubicin), 2.1 (daunorubicinol), and 2.7 (doxorubicinol), respectively.
CBR1 V88 and CBR1 I88 Inhibition Studies
CBR1 V88 and CBR1 I88 activities were inhibited with the flavonoid rutin (Sigma-Aldrich) in the presence of the substrate menadione (Wermuth et al., 1986; Covarrubias et al., 2006). Assay mixtures (1.0 ml) contained 200 μM NADPH, 100 mM potassium phosphate buffer (pH 7.4), CBR1 enzyme (1 mg/ml), 40 μM menadione, and rutin (5–300 μM). The concentration of rutin required to inhibit CBR1 activity by 50% (IC50) was calculated by nonlinear regression analysis using a one-site model with GraphPad Prism 4.
CBR1 V88 and CBR1 I88 Thermolability
The thermolability of both CBR1 isoforms was assessed by incubating protein aliquots (1 mg/ml) in phosphate buffer (pH 7.4, 100 mM), for 5 min at the following temperatures: 46°C, 50°C, and 60°C (Weisberg et al., 2001). Control reaction mixtures were incubated at 37°C for 5 min. Incubations were performed in an Isotemp 125D dry block (Fisher Scientific Co., Pittsburgh, PA) with digital temperature control. After incubation, protein aliquots were immediately transferred to ice. CBR1 activities were assayed as described above by using menadione (200 μM) and daunorubicin (500 μM) as substrates.
Determination of CBR1 V88 and CBR1 I88 NADPH Binding Affinities
The NADPH binding affinities of CBR1 V88I and CBR1 I88 were measured by isothermal titration calorimetry using a MicroCal microcalorimeter equipped with the VP Viewer 2000 Origin software (version 7SR2.V7; MicroCal, Northampton, MA). Freshly expressed and purified CBR1 isoforms were extensively dialyzed in phosphate-buffered saline solution (PBS, pH 7.2) and Tris (2-carboxyethyl)-phosphine (1 mM) up to a final concentration of 20 to 40 μM. Protein samples were degassed and added to the sample cell. PBS was added to the reference cell, and both cells were allowed to reach a stable temperature of 30.0°C. CBR1 isoforms were titrated with the NADPH cofactor (200 μM) and with PBS to subtract for heat dilution (Pey and Martinez, 2005). Titration experiments were performed at 30.0°C with a stirring rate of 300 rpm. A total of 29 NADPH aliquots were injected (10 μl/injection) to the sample cell at 300-s intervals. Changes in heat due to enzyme-cofactor binding were automatically recorded and plotted as kcal/mol of ligand versus ligand/enzyme molar ratio. The binding affinities (Ka), dissociation constants (Kd = 1/Ka), enthalpies (ΔH), entropies (ΔS), and Gibbs’ free energies (ΔG) were calculated by nonlinear regression analysis using a one-site binding model.
Docking and Modeling Analysis
The Molecular Operating Environment (MOE 2005.08; Chemical Computing Group, Montreal, QC, Canada) software package was used for substrate docking and modeling analysis. The site finder option of MOE automatically and correctly delineated the active site. Default parameters including partial charges for amino acids and ligands were turned on, and full conformational searches for the substrate PGE2 were carried out. The pose with the best score was selected as the model for the docked substrate. To account for conformational flexibility of the active site pocket, the complex was energy minimized by restrained minimization within 9 Å of the substrate molecule. In addition, energies of the ternary CBR1-PGE2 complexes with V88 and I88 side chains were also minimized by imposing default positional restraints on atoms within 9 Å of the cofactor atoms and fixing all atomic positions outside. PyMOL routines were used for analysis and illustrations (PyMOL Molecular Graphics System; DeLano Scientific LLC, Palo Alto, CA. http://www.pymol.org).
Results
SNPs in the Coding Region of Human CBR1
To identify common polymorphisms in the coding region of CBR1, we first screened 33 full-length cDNA samples isolated from liver donors (13 black and 20 white). The statistical power resulting from both sample sizes was adequate to detect polymorphic alleles with frequencies ≥5% (powerwhites ≈ 0.96, and powerblacks ≈ 0.85) (Kruglyak and Nickerson, 2001). We found no common CBR1 SNPs in cDNA samples from black donors, and we detected three synonymous CBR1 SNPs in samples from white donors (Table 1). In whites, CBR1 synonymous SNPs were present in 5% (V231V) and in 20% (L73L, A209A) of the samples, respectively. Genotyping data from the public dbSNP database (built 126) showed a total of nine SNPs in the coding region of CBR1 (exon 1, four SNPs; and exon 3, five SNPs), including the synonymous SNPs V231V, L73L, and A209A. Interestingly, the dbSNP database showed only one nonsynonymous SNP on CBR1. The SNP is located in exon 1 (contig position, 23104545) and encodes for a valine-to-isoleucine substitution at position 88 of the amino acidic chain (CBR1 V88I, rs:1143663). Therefore, we sought to confirm the existence of the CBR1 V88I polymorphism by analyzing 12 human DNA variation panels representing population groups with distinctive geographical ancestries. We analyzed 211 DNA samples (422 alleles) by using a validated real-time PCR assay for CBR1 V88I genotyping, and we confirmed the presence of the variant allele (A) by direct sequencing (Fig. 1, A and B). We detected the CBR1 V88I polymorphism in the DNA panel representing individuals from the United States with African ancestry (Table 2). CBR1 V88I genotype distribution in the U.S.-African ancestry panel was as follows: 97.1% were homozygous for the G allele (G/G) and 2.9% were heterozygous (G/A). Allele distributions were in Hardy-Weinberg equilibrium (chi square test, p = 0.93; Table 2). Our observations were in agreement with the CBR1 V88I allele frequencies reported in dbSNP for a panel of 24 DNA samples representing individuals from the United States with African ancestry (p = 0.979, q = 0.021).
TABLE 1.
Genotype distributions of common polymorphism in the coding region of CBR1
| N | L73L (G312C, rs25678)
|
A209A (C720T, rs20572)
|
V231V (G786A, rs2230192)
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G/G | G/C | C/C | p | q | C/C | C/T | T/T | p | q | G/G | G/A | A/A | p | q | ||
| United States (European) | 20 | 14 | 6 | 0 | 0.850 | 0.15 | 14 | 6 | 0 | 0.85 | 0.15 | 19 | 1 | 0 | 0.98 | 0.02 |
| United States (African) | 13 | 13 | 0 | 0 | N.A. | N.A. | 13 | 0 | 0 | N.A. | N.A. | 13 | 0 | 0 | N.A. | N.A. |
N.A., not applicable.
Fig. 1.
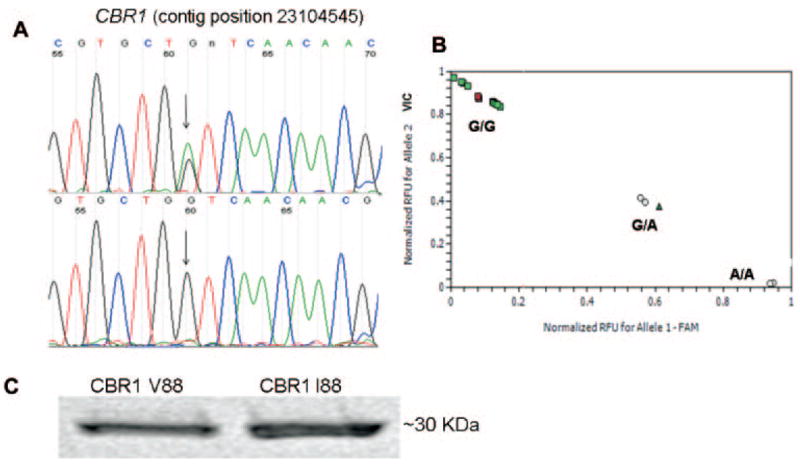
A, detection of CBR1 V88I by DNA sequence analysis. The chromatogram at the top shows the sequence trace from an individual with heterozygous (G/A) genotype. The chromatogram at the bottom shows the trace from a subject with homozygous (G/G) genotype. B, CBR1 V88I genotyping by real-time PCR. C, immunodetection of purified CBR1 V88 and CBR1 I88 with a specific anti-human CBR1 antibody.
TABLE 2.
CBR1 V88I genotype distribution in DNA human diversity panels
| Population | N | G/G | G/A | A/A | p | q |
|---|---|---|---|---|---|---|
| United States (African) | 70 | 68 | 2 | 0 | 0.986 | 0.014 |
| United States (European) | 50 | 50 | 0 | 0 | N.A. | N.A. |
| Indo-Pakistani | 9 | 9 | 0 | 0 | N.A. | N.A. |
| Middle Eastern | 10 | 10 | 0 | 0 | N.A. | N.A. |
| Africans north of the Sahara | 7 | 7 | 0 | 0 | N.A. | N.A. |
| Africans south of the Sahara | 9 | 9 | 0 | 0 | N.A. | N.A. |
| Chinese | 10 | 10 | 0 | 0 | N.A. | N.A. |
| Japanese | 10 | 10 | 0 | 0 | N.A. | N.A. |
| Mexican | 10 | 10 | 0 | 0 | N.A. | N.A. |
| Asia Pacific | 7 | 7 | 0 | 0 | N.A. | N.A. |
| South American Andes | 10 | 10 | 0 | 0 | N.A. | N.A. |
| Southeast Asia | 9 | 9 | 0 | 0 | N.A. | N.A. |
N, number of DNA samples from nonrelated individuals. p denotes the G allele, and q denotes the A allele, N.A. not applicable.
Kinetic Characterization of CBR1 V88 and CBR1 I88
To evaluate the functional impact of the CBR1 V88I polymorphism, we purified the recombinant CBR1 V88 and CBR1 I88 protein isoforms. Both isoforms appeared as discrete ~32-kDa bands after immunoblotting analysis with anti-human CBR1 antibody (Fig. 1C). We analyzed CBR1 V88 and CBR1 I88 kinetic activities by using the prototypical substrates menadione, PGE2, and daunorubicin. CBR1 V88 and CBR1 I88 displayed typical one-site Michaelis-Menten kinetics for the three substrates. Kinetic comparisons with the quinone menadione demonstrated a slightly higher Km value for CBR1 I88 compared with CBR1 V88 (p = 0.370). In contrast, CBR1 V88 presented a 1.3-fold higher Vmax for menadione than did CBR1 I88 (p = 0.023; Table 3). Consequently, CBR1 V88 showed a 1.3-fold higher turnover number (Kcat) for menadione. On the other hand, CBR1 V88 and CBR1 I88 Km values for the substrates PGE2 and daunorubicin did not differ significantly (PGE2, p = 0.234; and daunorubicin, p = 0.287; Table 3). However, the maximal velocities of CBR1 V88 and CBR1 I88 for PGE2 and daunorubicin differed significantly (PGE2, p <0.01; and daunorubicin, p <0.05; Table 3; Fig. 2). On average, the Vmax of CBR1 V88 was 51% (PGE2) and 50% (daunorubicin) higher than the Vmax of CBR1 I88. Consequently, CBR1 V88 showed increased turnover values for both PGE2 (1.4- fold) and daunorubicin (1.5-fold) (Table 3). To further confirm the kinetic differences, we measured the amount of the C-13 alcohol metabolite daunorubicinol synthesized by CBR1 V88 and CBR1 I88. Under Vmax conditions, CBR1 V88 synthesized 47% more daunorubicinol than did CBR1 I88 per unit of time (p < 0.0001; Fig. 3).
TABLE 3.
CBR1 V88 and CBR1 I88 kinetic constants for prototypical substrates
| Substrate | CBR1 V88
|
CBR1 I88
|
||||||
|---|---|---|---|---|---|---|---|---|
| Km | Vmax | Kcat | Vmax/Km | Km | Vmax | Kcat | Vmax/Km | |
| μM | nmol/min · mg | S−1 | mg/min × 10−3 | μM | nmol/min · mg | s−1 | mg/min × 10−3 | |
| Menadionea | 42 ± 23 | 220 ± 27 | 110 | 5.20 | 55 ± 21 | 168 ± 16 | 85 | 3.06 |
| Daunorubicina | 173 ± 26 | 181 ± 13 | 91 | 1.04 | 140 ± 23 | 121 ± 12 | 61 | 0.90 |
| PGE2a | 309 ± 94 | 53 ± 7 | 25 | 0.17 | 320 ± 63 | 35 ± 4 | 18 | 0.11 |
| NADPHb | 247 ± 29 | 356 ± 30 | 179 | 1.44 | 343 ± 63 | 295 ± 31 | 149 | 0.86 |
Values represent the mean ± S.D. from four experiments performed in duplicates with two independent protein preparations.
Menadione was held constant at 250 μM. Values represent the mean ± S.D. from two experiments performed in duplicates with two independent protein preparations.
Fig. 2.
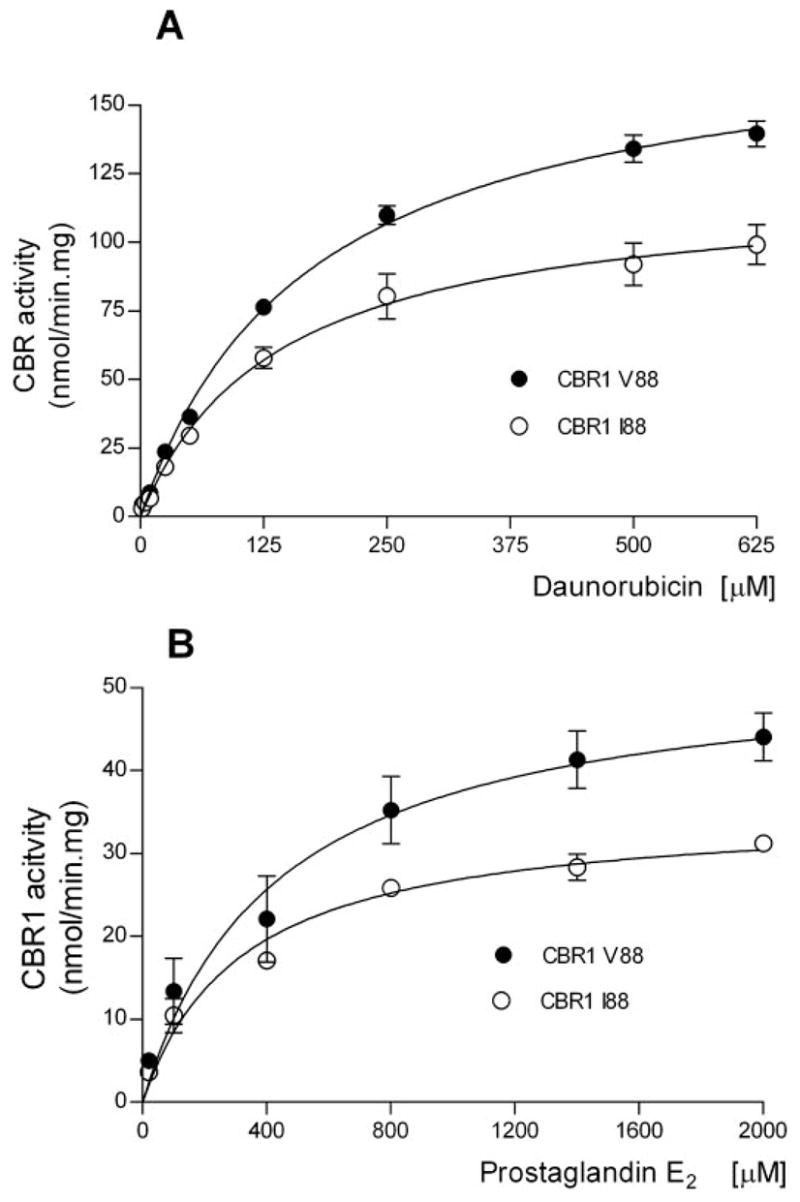
Kinetic characterization of CBR1 V88 and CBR1 I88 with the substrates daunorubicin (A) and prostaglandin E2 (B). Each point represents the mean ± S.D. of four experiments performed in duplicates with two independent protein preparations. Differences between curves were significant at p < 0.05 (A) and p < 0.01 (B).
Fig. 3.
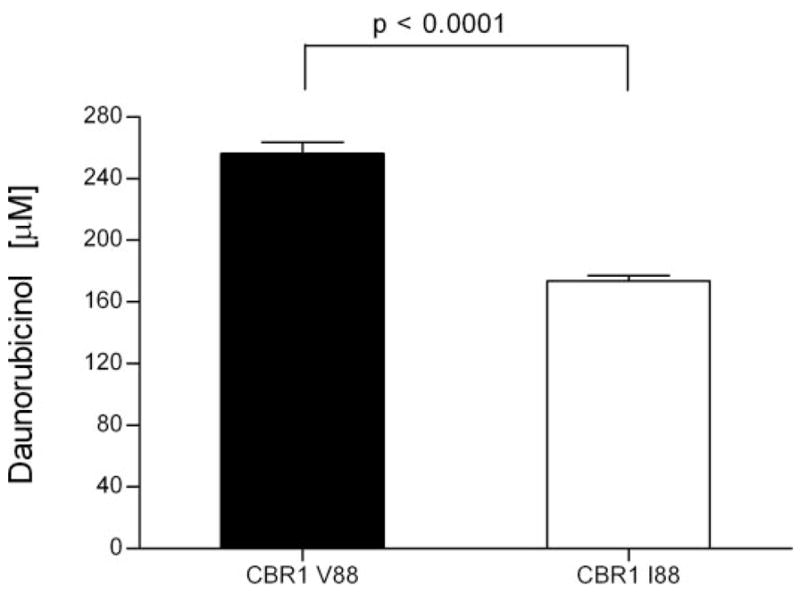
Synthesis of daunorubicinol by CBR1 V88 and CBR1 I88. Each bar represents the average ± S.D. of two independent experiments performed in duplicate.
Kinetic studies with varying concentrations of NADPH (range 10–200 μM) and menadione (250 μM) showed a relatively lower Km (p = 0.095) and a higher Vmax (p = 0.096) for CBR1 V88 as compared with CBR1 I88 (Table 3). Further comparison of CBR1 V88 and CBR1 I88 data sets showed that the overall Michaelis-Menten kinetics for NADPH in the presence of menadione differed significantly between both isoforms (p < 0.001).
CBR1 V88 and CBR1 I88 Inhibition by Rutin
The development of specific CBR inhibitors has been proposed as a potentially useful therapeutic strategy to minimize the incidence of anthracycline-related cardiotoxicity during cancer treatment (Olson et al., 2003). Some flavonoids such as rutin, quercetin, and kaempferol inhibit CBR1 activity with IC50 values in the micromolar range (Wermuth, 1981). Therefore, we tested whether the CBR1 V88 and CBR1 I88 isoforms were distinctively inhibited by rutin. CBR1 V88 and CBR1 I88 displayed different levels of dose-dependent inhibition by rutin (Fig. 4). Interestingly, the rutin IC50 value for CBR1 I88 was 3.6-fold lower than the IC50 value obtained for CBR1 V88 (CBR1 I88 IC50 = 15.0 ± 0.1 μM versus CBR1 V88 IC50 = 54.0 ± 0.4 μM, p < 0.001).
Fig. 4.
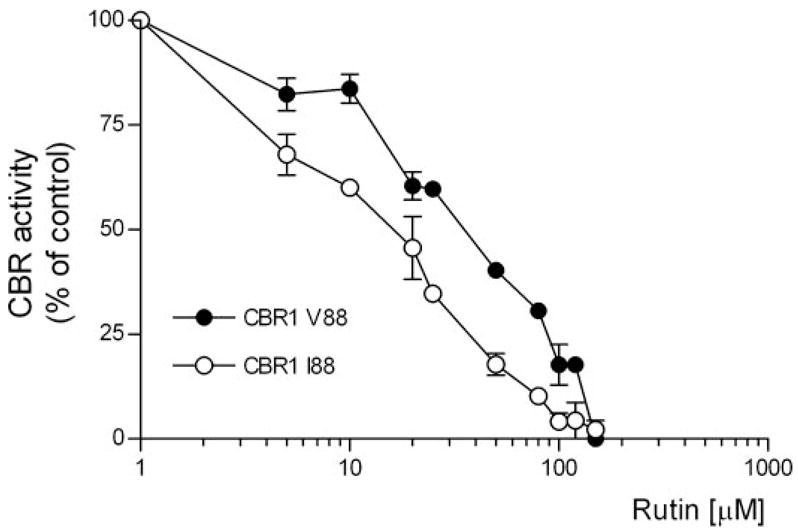
CBR1 V88 and CBR1 I88 inhibition by rutin (substrate = 40 μM menadione). Each point represents the average ± S.D. of two experiments performed in duplicate. Differences between inhibition curves were statistically significant at p < 0.001.
CBR1 V88 and CBR1 I88 Thermolability
Some functional SNPs result in protein isoforms that show differential activity profiles after heat treatment (Yamada et al., 2001; Li et al., 2005). Consequently, we analyzed the thermolability of CBR1 V88 and CBR1 I88 by determining enzymatic activities at different temperatures with the substrates menadione and daunorubicin. CBR1 V88 and CBR I88 retained 100% of activity after heating to 46°C. Incubations at 50°C resulted in similar reductions (≈13%) on the activities of CBR1 V88 and CBR1 I88. Both isoforms were rendered inactive after heating to 60°C (Fig. 5). In conclusion, CBR1 V88 and CBR1 I88 showed similar thermolability profiles.
Fig. 5.
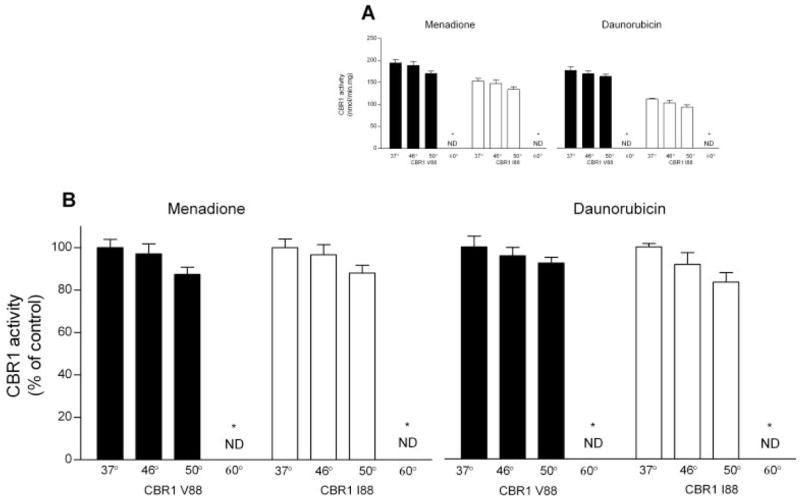
Thermolability profiles of CBR1 V88 and CBR1 I88. A, CBR1 V88 and CBR1 I88 maximal activities at different temperatures for the substrates menadione (left) and daunorubicin (right). B, CBR1 V88 and CBR1 I88 maximal activities relative to control incubations at 37°C (100%). Each value represents the average ± S.D. of two experiments performed in duplicate (ND, nondetectable).
CBR1 V88 and CBR1 I88 NADPH Binding Affinities
We performed isothermal titration calorimetric analyses to further explore the basis of the distinctive catalytic properties of CBR1 V88 and CBR1 I88. The NADPH titration profiles of CBR1 V88 and CBR1 I88 differed significantly (p < 0.001; Fig. 6, A and B). Further integration analyses demonstrated that both isoforms presented essentially identical values for enthalpy (ΔH), free Gibbs’ energy (ΔG), and entropy (ΔS), respectively (Fig. 6, C and D; Table 4). Notably, CBR1 V88 and CBR1 I88 showed different NADPH dissociation constants (Kd; Table 4). The Kd of CBR1 I88 was 1.7-fold higher than the Kd of CBR1 V88. Therefore, the valine-to-isoleucine substitution encoded by the variant CBR1 I88 allele results in a CBR1 isoform (I88) with lower affinity for the NADPH cofactor compared with the “wild-type” protein (V88).
Fig. 6.
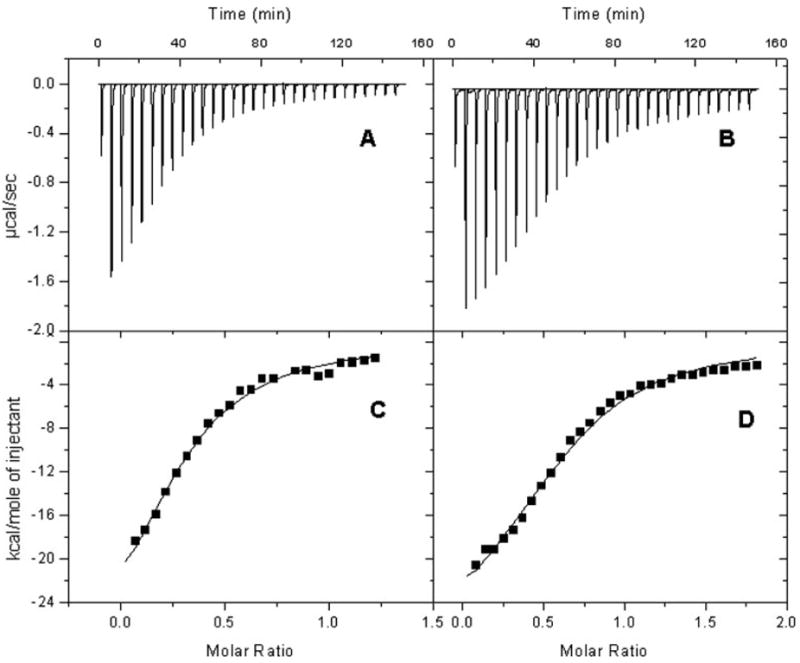
Isothermal titration calorimetry profiles of CBR1 V88 and CBR1 I88 with NADPH. Top, each peak shows the rate of heat release (μcal/s) after the interaction of CBR1 V88 (A) and CBR1 I88 (B) with the NADPH cofactor. Differences in titration profiles were significant at p < 0.001. Bottom, integration analysis for the titration profiles of CBR1 V88 (C) and CBR1 I88 (D). The solid line represents the best fit with a one-binding site model.
TABLE 4.
Thermodynamic constants of CBR1 V88 and CBR1 I88 derived from microcalorimetry titration analyses with NADPH
| CBR1 V88 | CBR1 I88 | |
|---|---|---|
| Kda (μM) | 3.8 ± 0.5 | 6.3 ± 0.6 |
| Kaa × 105 M−1 | 2.6 ± 0.2 | 1.6 ± 0.2 |
| ΔH (kcal/mol) | −29 ± 1 | −29 ± 1 |
| ΔG (kcal/mol) | −52 ± 1 | −51 ± 1 |
| TΔS (kcal/mol) | −20 ± 1 | −21 ± 1 |
Differences between CBR1 variant isoforms were significant at p < 0.001.
Docking and Modeling Analysis
A three-dimensional atomic model of the polymorphic human CBR1-NADP-PGE2 ternary complex was generated by docking a substrate molecule to the active site using the crystal structure of the human CBR1-NADP complex (Tanaka et al., 2005). Figure 7 is a close-up view of the active site showing the bound NADP molecule (pink), the docked substrate PGE2 (yellow), and the location of the polymorphic V88I site. The polymorphic V88I site is situated behind the NADP molecule. The catalytic residues Y194, K198, and S140 are shown in blue. The I88 side chain has no direct contact either with NADP (distance ~9 Å) or with PGE2. In the docked orientation, the 9C=O group of PGE2 makes a hydrogen bond with the catalytic Y194 side chain in the correct stereochemical orientation for the hydride and proton transfer for the reduction of PGE2 to PGE2α (with a H9αC–OH functional group). Furthermore, this orientation of the 9C=O group satisfies the mechanistic requirement that the direction of approach of the Pro-S hydride (from the 4C position of the reduced nicotinamide ring) is roughly normal to the carbonyl plane. The carboxylic moiety of PGE2 makes a salt bridge with the R145 side chain, an interaction that may be important for binding and stabilization of the substrate.
Fig. 7.
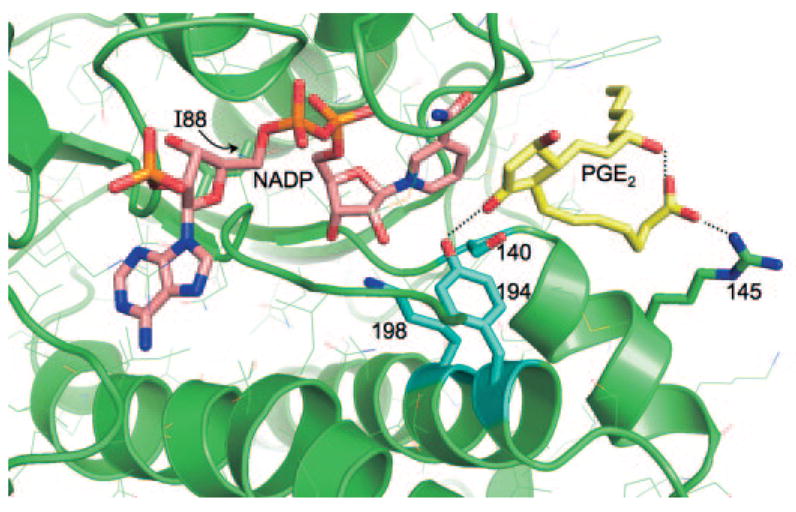
Three-dimensional model of the active site of polymorphic human CBR1 V88I with the substrate PGE2. In this view, the polymorphic V88I residue is located behind the NADP molecule (pink). The catalytic residues Y194, K198, and S140 are shown in light blue, and PGE2 is shown in yellow. Other side chains are shown in thin lines.
Discussion
Human CBR1 catalyzes the reduction of a wide variety of endogenous and xenobiotic compounds including the fever mediator PGE2 and the anticancer anthracycline daunorubicin. CBR activity varies widely among individuals, and the reasons for such disparities remain to be elucidated. It is possible that genetic polymorphism may in part dictate variable CBR activity. Therefore, we conducted this study to identify and characterize the functional impact of SNPs in the coding region of CBR1. We detected three synonymous SNPs and one non-synonymous SNP on CBR1 (CBR1 V88I).
Our functional characterization studies with prototypical CBR1 substrates showed that CBR1 V88I encodes for CBR1 isoforms with distinctive kinetic and thermodynamic properties. Interestingly, the CBR1 V88 isoform presented higher Vmax values for PGE2 (51%) and daunorubicin (50%) than did CBR1 I88. In agreement, CBR1 V88 synthesized higher levels (47%) of the cardiotoxic C-13 alcohol metabolite daunorubicinol than did CBR1 I88. Olson et al. (2003) reported that the plasma levels of C-13 alcohol metabolites 4 h after the administration of doxorubicin (20 mg/kg i.p.) were 75% higher in mice with two active Cbr1 alleles (Cbr1+/+) than in heterozygous Cbr1+/− animals. The authors also observed that the overall 40 to 50% decrease in Cbr1 protein levels among Cbr1+/− mice was sufficient to confer significant protection against anthracycline-related cardiotoxicity. As a result, the percentages of dead animals 18 days after doxorubicin treatment were 91% (Cbr1+/+) versus 18% (Cbr1+/−). In this context, it is tempting to speculate that individuals with homozygous CBR1 I88 genotype may exhibit a slower rate of synthesis of C-13 alcohol metabolites that may consequently have an impact on the risk of cardiotoxicity associated with the use of anthracyclines for cancer therapy.
CBR1 V88 also showed higher Vmax (31%) for the small quinone substrate menadione (vitamin K3), as compared with CBR1 I88. Together, our results suggest that the functional impact of CBR1 V88I appears not to be substrate-specific. Therefore, it is possible that CBR1 V88 and CBR1 I88 would also differ in their ability to catalyze the reduction of other relevant CBR1 substrates such as the antipsychotic haloperidol or the potent carcinogen N-methyl-N-(4-oxo-4-pyridin-3-yl-butyl)nitrous amide (Someya et al., 1992; Kudo and Ishizaki, 1999; Forrest and Gonzalez, 2000).
CBR1 V88 and CBR1 I88 showed different Michaelis-Menten kinetics for the NADPH cofactor in the presence of the substrate menadione. In line with this finding, our titration calorimetry studies indicated that the NADPH dissociation constants (Kd) of CBR1 V88 and CBR1 I88 differed significantly (CBR1 V88 = 3.8 ± 0.5 μM versus CBR1 I88 = 6.3 ± 0.6 μM). Thus, the distinct catalytic activities of CBR1 V88 and CBR1 I88 toward different substrates are due to the fact that both isoforms bind the NADPH cofactor with different affinities. We performed molecular modeling together with substrate docking simulation using the crystal structure of human CBR1 to further define the molecular basis underlying the functional impact of CBR1 V88I. The docking of a PGE2 molecule in the active site provided additional insights into the enzyme-substrate interplays during the catalytic reduction of PGE2 by CBR1, in spatial relation to the V88I substitution. Figure 7 shows the CBR1-PGE2 complex at the enzyme’s active site. The model indicates that the 9C=O group of PGE2 accepts a proton from the Y194 hydroxyl group in a precise stereochemical orientation that allows the hydride and proton transfer during the reduction of the substrate. The 1C carboxyl group of PGE2 binds to the side chain of R145 through a salt bridge that stabilizes the enzyme-substrate complex during catalysis. The side chains inside and bordering the substrate, and cofactor-binding cavities including the catalytic residues K198, S194, and Y140 interact directly with NADP and the substrate (PGE2). The model also shows that the V88I substitution is located behind the ribose moiety of the NADP molecule, roughly 9 Å away from it. At this polymorphic site, V88 packs against I20 in a hydrophobic pocket with restricted although sufficient space to accommodate the extra methyl group from variant I88. The methyl group from I88 increases the nonpolar surface area (V, 135 Å2 versus I, 155 Å2), and augments the overall hydrophobicity of the pocket. Thus, the altered packing between I20 and I88 may in turn have an impact on NADPH binding due to the proximity of the cofactor. This effect is probably reflected in the docking simulation result showing that the ternary CBR1-NADP-PGE2 structure with the V88 side chain is energetically more favorable (−5.9 kcal/mol) than that with I88 (−3.6 kcal/mol). Together, these facts provide the structural rationale for the experimental differences between the NADPH dissociation constants of both isoforms.
Our genotypic survey suggests that the CBR1 V88I polymorphism may be confined to individuals with African ancestry. Africa is the continent with the highest incidence of malaria and, for example, in Uganda, the number of cases per 1000 individuals is as high as 478 (http://www.globalhealthfacts.org). Recent studies suggest that elevated plasma levels of PGE2 in healthy children infected with malarial parasites may protect against severe clinical forms of malaria (Perkins et al., 2001, 2005). In humans, malaria is the most powerful known source of genetic selective pressure, and the frequency of polymorphic changes with malaria-protective effects is expected to be higher in affected populations (Kwiatkowski, 2005). Therefore, we hypothesize that CBR1 V88I may constitute a protective polymorphism against severe malaria because the slower CBR1 I88 isoform would result in higher levels of PGE2 among carriers of the variant CBR1 I88 allele. Genetic association studies on selected human groups are needed to test this hypothesis. In conclusion, we have identified and characterized the first genetic determinant of human CBR1 activity.
Acknowledgments
The excellent assistance of Jennifer Griswold and Darowan Akajagbor is gratefully acknowledged.
This work was supported by National Institutes of Health/National Institute of General Medical Sciences Grant R01GM73646 to J.G.B., and by National Institutes of Health/National Institute of General Medical Sciences Grant U01GM61393 and National Institutes of Health/National Institute of General Medical Sciences Pharmacogenetics Research Network and Database Grant U01GM61374 (http://pharmgkb.org). V.G.-C. is a Merck fellow.
ABBREVIATIONS
- CBR
carbonyl reductase
- SNP
single nucleotide polymorphism
- PCR
polymerase chain reaction
- PGE2
prostaglandin E2
- HPLC
high-performance liquid chromatography
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
References
- Bohren KM, von Wartburg JP, Wermuth B. Kinetics of carbonyl reductase from human brain. Biochem J. 1987;244:165–171. doi: 10.1042/bj2440165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias VG, Lakhman SS, Forrest A, Relling MV, Blanco JG. Higher activity of polymorphic NAD(P)H:quinone oxidoreductase in liver cytosols from blacks compared to whites. Toxicol Lett. 2006;164:249–258. doi: 10.1016/j.toxlet.2006.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowens JW, Creaven PJ, Greco WR, Brenner DE, Tung Y, Ostro M, Pilkiewicz F, Ginsberg R, Petrelli N. Initial clinical (phase I) trial of TLC D-99 (doxorubicin encapsulated in liposomes) Cancer Res. 1993;53:2796–2802. [PubMed] [Google Scholar]
- Forrest GL, Gonzalez B. Carbonyl reductase. Chem-Biol Interact. 2000;129:21–40. doi: 10.1016/s0009-2797(00)00196-4. [DOI] [PubMed] [Google Scholar]
- Forrest GL, Gonzalez B, Tseng W, Li X, Mann J. Human carbonyl reductase overexpression in the heart advances the development of doxorubicin-induced cardiotoxicity in transgenic mice. Cancer Res. 2000;60:5158–5164. [PubMed] [Google Scholar]
- Ivanov AI, Romanovsky AA. Prostaglandin E2 as a mediator of fever: synthesis and catabolism. Front Biosci. 2004;9:1977–1993. doi: 10.2741/1383. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Scheck AC, Romanovsky AA. Expression of genes controlling transport and catabolism of prostaglandin E2 in lipopolysaccharide fever. Am J Physiol. 2003;284:R698–R706. doi: 10.1152/ajpregu.00570.2002. [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Nickerson DA. Variation is the spice of life. Nat Genet. 2001;27:234–236. doi: 10.1038/85776. [DOI] [PubMed] [Google Scholar]
- Kudo S, Ishizaki T. Pharmacokinetics of haloperidol: an update. Clin Pharmacokinet. 1999;37:435–456. doi: 10.2165/00003088-199937060-00001. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005;77:171–192. doi: 10.1086/432519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhman SS, Ghosh D, Blanco JG. Functional significance of a natural allelic variant of human carbonyl reductase 3 (CBR3) Drug Metab Dispos. 2005;33:254–257. doi: 10.1124/dmd.104.002006. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang X, van Breemen RB, Bolton JL. Characterization of two new variants of human catechol O-methyltransferase in vitro. Cancer Lett. 2005;230:81–89. doi: 10.1016/j.canlet.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- Mordente A, Meucci E, Martorana GE, Giardina B, Minotti G. Human heart cytosolic reductases and anthracycline cardiotoxicity. IUBMB Life. 2001;52:83–88. doi: 10.1080/15216540252774829. [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Tobe VO, Taylor SL. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara H, Miyabe Y, Deyashiki Y, Matsuura K, Hara A. Reduction of drug ketones by dihydrodiol dehydrogenases, carbonyl reductase and aldehyde reductase of human liver. Biochem Pharmacol. 1995;50:221–227. doi: 10.1016/0006-2952(95)00124-i. [DOI] [PubMed] [Google Scholar]
- Olson LE, Bedja D, Alvey SJ, Cardounel AJ, Gabrielson KL, Reeves RH. Protection from doxorubicin-induced cardiac toxicity in mice with a null allele of carbonyl reductase 1. Cancer Res. 2003;63:6602–6606. [PubMed] [Google Scholar]
- Perkins DJ, Hittner JB, Mwaikambo ED, Granger DL, Weinberg JB, Anstey NM. Impaired systemic production of prostaglandin E2 in children with cerebral malaria. J Infect Dis. 2005;191:1548–1557. doi: 10.1086/429332. [DOI] [PubMed] [Google Scholar]
- Perkins DJ, Kremsner PG, Weinberg JB. Inverse relationship of plasma prostaglandin E2 and blood mononuclear cell cyclooxygenase-2 with disease severity in children with Plasmodium falciparum malaria. J Infect Dis. 2001;183:113–118. doi: 10.1086/317660. [DOI] [PubMed] [Google Scholar]
- Pey AL, Martinez A. The activity of wild-type and mutant phenylalanine hydroxylase and its regulation by phenylalanine and tetrahydrobiopterin at physiological and pathological concentrations: an isothermal titration calorimetry study. Mol Genet Metab. 2005;86(Suppl 1):S43–S53. doi: 10.1016/j.ymgme.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Rosemond MJ, Walsh JS. Human carbonyl reduction pathways and a strategy for their study in vitro. Drug Metab Rev. 2004;36:335–361. doi: 10.1081/dmr-120034154. [DOI] [PubMed] [Google Scholar]
- Someya T, Shibasaki M, Noguchi T, Takahashi S, Inaba T. Haloperidol metabolism in psychiatric patients: importance of glucuronidation and carbonyl reduction. J Clin Psychopharmacol. 1992;12:169–174. [PubMed] [Google Scholar]
- Tanaka M, Bateman R, Rauh D, Vaisberg E, Ramachandani S, Zhang C, Hansen KC, Burlingame AL, Trautman JK, Shokat KM, et al. An unbiased cell morphology-based screen for new, biologically active small molecules. PLoS Biol. 2005;3:e128. doi: 10.1371/journal.pbio.0030128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg IS, Jacques PF, Selhub J, Bostom AG, Chen Z, Curtis Ellison R, Eckfeldt JH, Rozen R. The 1298A→C polymorphism in methylenetetrahydrofolate reductase (MTHFR): in vitro expression and association with homocysteine. Atherosclerosis. 2001;156:409–415. doi: 10.1016/s0021-9150(00)00671-7. [DOI] [PubMed] [Google Scholar]
- Wermuth B. Purification and properties of an NADPH-dependent carbonyl reductase from human brain. Relationship to prostaglandin 9-ketoreductase and xenobiotic ketone reductase. J Biol Chem. 1981;256:1206–1213. [PubMed] [Google Scholar]
- Wermuth B, Platts KL, Seidel A, Oesch F. Carbonyl reductase provides the enzymatic basis of quinone detoxication in man. Biochem Pharmacol. 1986;35:1277–1282. doi: 10.1016/0006-2952(86)90271-6. [DOI] [PubMed] [Google Scholar]
- Wouters KA, Kremer LC, Miller TL, Herman EH, Lipshultz SE. Protecting against anthracycline-induced myocardial damage: a review of the most promising strategies. Br J Haematol. 2005;131:561–578. doi: 10.1111/j.1365-2141.2005.05759.x. [DOI] [PubMed] [Google Scholar]
- Yamada K, Chen Z, Rozen R, Matthews RG. Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc Natl Acad Sci USA. 2001;98:14853–14858. doi: 10.1073/pnas.261469998. [DOI] [PMC free article] [PubMed] [Google Scholar]