

Abstract
We hypothesized that myelin breakdown in vulnerable late-myelinating regions releases oligodendrocyte- and myelin-associated iron that promotes amyloid beta (Aβ) oligomerization, its associated toxicity, and the deposition of oligomerized Aβ and iron in neuritic plaques observed in Alzheimer’s disease (AD). The model was tested by using published maps of cortical myelination from 1901 and recent in vivo imaging maps of Aβ deposits in humans. The data show that in AD, radiolabeled ligands detect Aβ deposition in a distribution that matches the map of late-myelinating regions. Furthermore, the strikingly lower ability of this imaging ligand to bind Aβ in animal models is consistent with the much lower levels of myelin and associated iron levels in rodents when compared with humans. The hypotheses derived from the “myelin model” are testable with current imaging methods and have important implications for therapeutic interventions that should be expanded to include novel targets such as oligodendrocytes, myelin, and brain iron.
Keywords: Myelin, Oligodendrocyte, White matter, Amyloid, Iron, Alzheimer’s disease, PIB, Degeneration, Dementia, Aging, Medications, Treatment, Prevention
1. Introduction
The extensive scope of myelination is the single-most unique aspect in which the human brain differs from that of other species [1–3]. In this myelin model of human evolution and development, our brain’s extensive myelination accounts for the high processing speeds and precise temporal coding underlying higher cognitive and behavioral functions [4,5]. Myelin and the oligodendrocytes that produce it are extremely vulnerable to a variety of insults including those caused by amyloid beta (Aβ) oligomers and fibrils [4,6,7]. The vulnerabilities of oligodendrocytes and myelin, especially later-developed myelin, are directly pertinent to many uniquely human degenerative disorders such as Alzheimer’s disease (AD) [3,4,7] in which white matter damage has been directly associated with brain parenchyma Aβ load [6,8].
Myelination produces a continuum of increasingly vulnerable oligodendrocytes as later-differentiating oligodendrocytes that populate later-myelinating association regions ensheath increasing numbers of axons with smaller axon diameters [9,10]. Thus, during development, the myelination process produces a roughly bilaterally symmetrical continuum of oligodendrocyte vulnerability (Figure 1B, D) [3,7]. Under the influence of multiple risk factors such as age, apolipoprotein E genotype, and increasing Aβ and iron levels, these thinner myelin sheaths are differentially lost with age (27% to 45% reductions) in a pattern of bilaterally progressive myelin breakdown [4,7,11–13]. The myelin breakdown process thus recapitulates the developmental process of myelination in reverse [14] and is hypothesized to underlie the progressive, bilaterally symmetrical spread of the pathognomonic lesions of AD (neuritic plaques and neurofibrillary tangles) from late-myelinating regions toward earlier-myelinating regions [3,4] (Figure 1A, C). A similar process of developmental recapitulation has been described clinically as a progression of the cognitive, functional, and neurologic declines that accompany AD [15].
Fig 1.
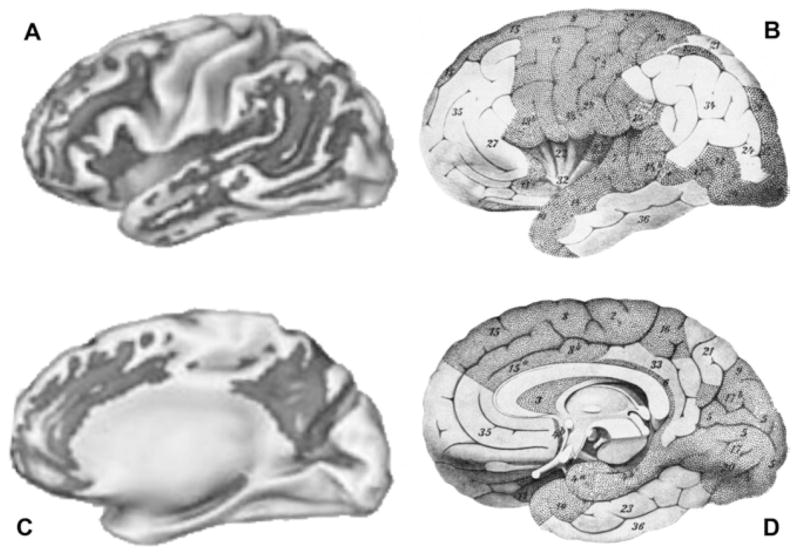
(A) Amyloid deposition in dark grey, with lighter areas within the dark grey representing progressively more significant changes, as imaged with 11C Pittsburgh Compound-B, displayed in standard space lateral view. Reprinted from Buckner et al [35] with permission. Copyright 2005 by the Society for Neuroscience. (B) Late-myelinating regions in white; lateral brain view corresponds to positron emission tomography (PET)–derived image on the left. Reprinted from Meyer [37] with permission. (C) Amyloid deposition in dark grey, with lighter areas within the dark grey representing progressively more significant changes, as imaged with 11C Pittsburgh Compound-B, displayed in standard space medial view. Reprinted from Buckner et al [35] with permission. Copyright 2005 by the Society for Neuroscience. (D) Late-myelinating regions in white; medial brain view corresponds to PET-derived image on the left. Note: This image is “tilted” to expose the underside of the frontal and temporal lobe regions, which is not the case for the PET image on the left (C), and the exposed underside of those lobes should be ignored when comparing (C) and (D). Reprinted from Meyer [37] with permission.
Oligodendrocytes and myelin have the highest levels of iron of any brain cells [16–19]. Several lines of circumstantial evidence support the possibility that brain iron levels might be a risk factor for age-related neurodegenerative diseases such as AD [20,21]. Post mortem and in vivo studies have established that brain iron levels increase with age [21–25], and iron levels are abnormally elevated in age-related neurodegenerative diseases, suggesting that increased iron levels might contribute to their age risk factor [3,4,26]. Men have higher brain iron levels than women [21], and men also have a younger age at onset of AD [27,28]. In large representative American populations after statistically controlling for the risk associated with the apolipoprotein E (APOE) genotype, men have a peak risk for AD onset that is 5 years younger than in women [29,30]. Thus, the peak probability of AD is reached 5 years earlier for men than women: age 78 and 83 years for APOE e4/e4, age 91 and 96 for APOE eX/eX, and age 92 and 97 for APOE e4/eX, respectively [29]. Furthermore, in patients with Down syndrome, who have an elevated risk for developing AD on a genetic basis, men develop AD at an earlier age than women [31]. In women with Down syndrome, an earlier menopause, which increases peripheral iron levels [32], is associated with earlier age at onset of AD [33,34].
2. Methods, Results, and Discussion
Recent in vivo methods of imaging Aβ deposits in humans by using Pittsburgh Compound-B (PIB) [35] (Figure 1A, C) are strikingly consistent with a myelin-based model of human brain development and degeneration [3,4]. The PIB data show that in AD, Aβ deposition is also observed predominantly in late-myelinating regions [35–37] (Figure 1A, C). We hypothesized that myelin breakdown in vulnerable late-myelinating regions (Figure 1B, D) releases oligodendrocyte- and myelin-associated iron [19,21,28,38,39], thus promoting Aβ oligomerization, its associated toxicity, and deposition of oligomerized Aβ and iron in neuritic plaques [7,28,40–43] (Figure 1A, C). This hypothesis has been supported in transgenic mouse models that demonstrated increased vulnerability of oligodendrocytes to toxicity [44], age-related white matter volume reductions [45], and age-related iron deposition in amyloid plaques [42]. Human studies have likewise confirmed age-related myelin breakdown that is exacerbated in healthy APOE e4 carriers and AD subjects [7,43] and age-related increases in brain iron [21] that are exacerbated in AD subjects [28].
The unique myelination of the human brain might also help explain the conundrum of why in vivo Aβ labels have high retention in human brain amyloid and very low retention in rodent amyloid [46,47]. Transgenic mouse models as well as human studies have demonstrated that a strong association exists between tissue iron and the production of senile plaques [28,40–42]. Myelin and oligodendrocytes have the highest iron content of brain tissues. The percentage of dry brain weight in humans accounted for by myelin is 30% higher than in rodents [48], and it is thus not surprising that rodents have much lower levels of brain iron (up to an order of magnitude) than humans [24,49,50]. Iron levels increase with age in both species [21,23,24,42,50]. These age-related increases might contribute to both the age-related increase in amyloid plaques observed in both species [14,41,42] and the correlation between tissue iron and amyloid deposit load observed in late-myelinating regions [14,42] that are most vulnerable to myelin breakdown and the release of iron [3,7,21,28,43].
The estimated 30% higher proportion of myelin in human versus rodent brain [48] is disproportionately higher in late-myelinating brain regions (Figure 1A to D). These regions contain predominantly thinly myelinated fibers with smaller axons and thus contain an increased proportion of myelin [9,43]. The age-related loss of myelin from primarily these thinly myelinated small axons (estimated at 10% per decade [13]) would take a disproportionate toll in late-myelinating regions and make much higher iron levels available for interaction with Aβ and deposition in amyloid plaques in these regions [41,42].
The histologic gold standard for labeling Aβ deposits in tissue is thioflavine-S [41], and PIB is a thioflavine derivative [46,47]. In animal models thioflavine-Aβ reactivity has been associated with the presence of metals in the deposits of Aβ [41,51]. We therefore suggest that the striking similarity between published maps of human in vivo Aβ labels and maps of later-myelinating cortical regions (Figure 1A to D) [35,36] supports the myelin model of AD [3,4,7]. The myelin model and known species differences in myelination and iron levels might also explain the human/rodent differences in Aβ label retention, with high iron levels in humans acting as a necessary “scaffolding” for the PIB label to bind to the amyloid plaques [46,47] in which Aβ and iron colonize [41,42,52,53].
The hypotheses derived from the myelin model are eminently testable with currently available imaging methods and animal models. These hypotheses have important implications for therapeutic interventions that could include novel primary prevention measures focused on targets such as oligodendrocytes, myelin, and brain iron [7,20,21,28,54,55].
Acknowledgments
Part of this work was supported by NIA Alzheimer’s Disease Center Grant (AG16570), NIMH grant (MH 0266029), Research and Psychiatry Services of the Department of Veterans Affairs.
References
- 1.Semendeferi K, Lu A, Schenker N, Damasio H. Humans and great apes share a large frontal cortex. Nat Neurosci. 2002;5:272–6. doi: 10.1038/nn814. [DOI] [PubMed] [Google Scholar]
- 2.Schoenemann PT, Sheehan MJ, Glotzer LD. Prefrontal white matter volume is disproportionately larger in humans than in other primates. Nat Neurosci. 2005;8:242–52. doi: 10.1038/nn1394. [DOI] [PubMed] [Google Scholar]
- 3.Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging. 2004;25:5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Bartzokis G. Quadratic trajectories of brain myelin content: unifying construct for neuropsychiatric disorders. Neurobiol Aging. 2004;25:49–62. [Google Scholar]
- 5.Bartzokis G. Brain myelination in prevalent neuropsychiatric developmental disorders: Primary and comorbid addiction. Adolescent Psychiatry. 2005;29:55–96. [PMC free article] [PubMed] [Google Scholar]
- 6.Jantaratnotai N, Ryu JK, Kim SU, McLarnon JG. Amyloid beta peptide-induced corpus callosum damage and glial activation in vivo. Neuroreport. 2003;14:1429–33. doi: 10.1097/00001756-200308060-00005. [DOI] [PubMed] [Google Scholar]
- 7.Bartzokis G, Lu PH, Geschwind DH, Edwards N, Mintz J, Cummings JL. Apolipoprotein E genotype and age-related myelin breakdown in healthy individuals: implications for cognitive decline and dementia. Arch Gen Psychiatry. 2006;63:63–72. doi: 10.1001/archpsyc.63.1.63. [DOI] [PubMed] [Google Scholar]
- 8.Chalmers K, Wilcock G, Love S. Contributors to white matter damage in the frontal lobe in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2005;31:623–31. doi: 10.1111/j.1365-2990.2005.00678.x. [DOI] [PubMed] [Google Scholar]
- 9.Lamantia AS, Rakic P. Cytological and quantitative characteristics of four cerebral commissures in the rhesus monkey. J Comp Neurol. 1990;291:520–37. doi: 10.1002/cne.902910404. [DOI] [PubMed] [Google Scholar]
- 10.Pandya DN, Seltzer B. The topography of commissural fibers, two hemispheres-one brain: functions of the corpus callosum. New York: Alan R. Liss, Inc; 1986. pp. 47–73. [Google Scholar]
- 11.Kemper T. Neuroanatomical and neuropathological changes during aging and dementia. In: Albert M, Knoefel J, editors. Clinical neurology of aging. 2. New York: Oxford University Press; 1994. pp. 3–67. [Google Scholar]
- 12.Tang Y, Nyengaard JR, Pakkenberg B, Gundersen HJ. Age-induced white matter changes in the human brain: a stereological investigation. Neurobiol Aging. 1997;18:609–15. doi: 10.1016/s0197-4580(97)00155-3. [DOI] [PubMed] [Google Scholar]
- 13.Marner L, Nyengaard JR, Tang Y, Pakkenberg B. Marked loss of myelinated nerve fibers in the human brain with age. J Comp Neurol. 2003;462:144–52. doi: 10.1002/cne.10714. [DOI] [PubMed] [Google Scholar]
- 14.Braak H, Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol (Berl) 1996;92:197–201. doi: 10.1007/s004010050508. [DOI] [PubMed] [Google Scholar]
- 15.Reisberg B, Franssen EH, Souren LE, Auer SR, Akram I, Kenowsky S. Evidence and mechanisms of retrogenesis in Alzheimer’s and other dementias: management and treatment import. Am J Alzheimers Dis Other Demen. 2002;17:202–12. doi: 10.1177/153331750201700411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. 1996;17:83–93. doi: 10.1002/(SICI)1098-1136(199606)17:2<83::AID-GLIA1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 17.Erb GL, Osterbur DL, LeVine SM. The distribution of iron in the brain: a phylogenetic analysis using iron histochemistry. Brain Res Dev Brain Res. 1996;93:120–8. doi: 10.1016/0165-3806(96)00020-x. [DOI] [PubMed] [Google Scholar]
- 18.Griffiths PD, Dobson BR, Jones GR, Clarke DT. Iron in the basal ganglia in Parkinson’s disease: an in vitro study using extended X-ray absorption fine structure and cryo-electron microscopy. Brain. 1999;122:667–73. doi: 10.1093/brain/122.4.667. [DOI] [PubMed] [Google Scholar]
- 19.Quintana C, Bellefqih S, Laval JY, Guerquin-Kern JL, Wu TD, Avila J, et al. Study of the localization of iron, ferritin, and hemosiderin in Alzheimer’s disease hippocampus by analytical microscopy at the subcellular level. J Struct Biol. 2006;153:42–54. doi: 10.1016/j.jsb.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–73. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 21.Bartzokis G, Tishler TA, Lu PH, Villablanca P, Altshuler LL, Carter M, et al. Brain ferritin iron may influence age- and gender-related risks of neurodegeneration. Neurobiol Aging. 2006;28:414–23. doi: 10.1016/j.neurobiolaging.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 22.Bartzokis G, Beckson M, Hance DB, Marx P, Foster JA, Marder SR. MR evaluation of age-related increase of brain iron in young adult and older normal males. Magn Reson Imaging. 1997;15:29–35. doi: 10.1016/s0730-725x(96)00234-2. [DOI] [PubMed] [Google Scholar]
- 23.Bartzokis G, Mintz J, Sultzer D, Marx P, Herzberg JS, Phelan CK, et al. In vivo MR evaluation of age-related increases in brain iron. AJNR Am J Neuroradiol. 1994;15:1129–38. [PMC free article] [PubMed] [Google Scholar]
- 24.Hallgren B, Sourander P. The effect of age on the non-haemin iron in the human brain. J Neurochem. 1958;3:41–51. doi: 10.1111/j.1471-4159.1958.tb12607.x. [DOI] [PubMed] [Google Scholar]
- 25.Klintworth GK. Huntington’s chorea: morphologic contributions of a century. In: Barbeau A, Paulson GW, Chase TN, editors. Advances in neurology, vol 1: Huntington’s chorea, 1872–1972. New York: Raven Press; 1973. pp. 353–68. [Google Scholar]
- 26.Bartzokis G, Aravagiri M, Oldendorf WH, Mintz J, Marder SR. Field dependent transverse relaxation rate increase may be a specific measure of tissue iron stores. Magn Reson Med. 1993;29:459–64. doi: 10.1002/mrm.1910290406. [DOI] [PubMed] [Google Scholar]
- 27.Bartzokis G, Cummings JL, Markham CH, Marmarelis PZ, Treciokas LJ, Tishler TA, et al. MRI evaluation of brain iron in earlier- and later-onset Parkinson’s disease and normal subjects. Magn Reson Imaging. 1999;17:213–22. doi: 10.1016/s0730-725x(98)00155-6. [DOI] [PubMed] [Google Scholar]
- 28.Bartzokis G, Tishler TA, Shin I-S, Lu PH, Cummings JL. Brain ferritin iron as a risk factor for age at onset in neurodegenerative diseases. In: LeVine S, Connor J, Schipper H, editors. Redox-active metals in neurological disorders. Vol. 1012. New York: Ann N Y Acad Sci; 2004. pp. 224–36. [DOI] [PubMed] [Google Scholar]
- 29.Miech RA, Breitner JC, Zandi PP, Khachaturian AS, Anthony JC, Mayer L. Incidence of AD may decline in the early 90s for men, later for women: the Cache County study. Neurology. 2002;58:209–18. doi: 10.1212/wnl.58.2.209. [DOI] [PubMed] [Google Scholar]
- 30.Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 2004;25:641–50. doi: 10.1016/j.neurobiolaging.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 31.Schupf N, Kapell D, Nightingale B, Rodriguez A, Tycko B, Mayeux R. Earlier onset of Alzheimer’s disease in men with Down syndrome. Neurology. 1998;50:991–5. doi: 10.1212/wnl.50.4.991. [DOI] [PubMed] [Google Scholar]
- 32.Whitfield JB, Treloar S, Zhu G, Powell LW, Martin NG. Relative importance of female-specific and non-female-specific effects on variation in iron stores between women. Br J Haematol. 2003;120:860–6. doi: 10.1046/j.1365-2141.2003.04224.x. [DOI] [PubMed] [Google Scholar]
- 33.Schupf N, Pang D, Patel BN, Silverman W, Schubert R, Lai F, et al. Onset of dementia is associated with age at menopause in women with Down’s syndrome. Ann Neurol. 2003;54:433–8. doi: 10.1002/ana.10677. [DOI] [PubMed] [Google Scholar]
- 34.Prasher VP, Gosling P, Blair J. Role of iron in Alzheimer-type dementia in Down syndrome. Int J Geriatr Psychiatry. 1998;13:818–9. doi: 10.1002/(sici)1099-1166(1998110)13:11<818::aid-gps864>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 35.Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–17. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flechsig P. Developmental (myelogenetic) localisation of the cerebral cortex in the human subject. Lancet. 1901:1027–9. [Google Scholar]
- 37.Meyer A. Paul Flechsig’s system of myelogenetic cortical localization in the light of recent research in neuroanatomy and neurophysiology: part I. Can J Neurol Sci. 1982;8:1–6. doi: 10.1017/s031716710004275x. [DOI] [PubMed] [Google Scholar]
- 38.de los Monteros AE, Korsak RA, Tran T, Vu D, de Vellis J, Edmond J. Dietary iron and the integrity of the developing rat brain: a study with the artificially-reared rat pup. Cell Mol Biol (Noisy-le-grand) 2000;46:501–15. [PubMed] [Google Scholar]
- 39.Francois C, Nguyen-Legros J, Percheron G. Topographical and cytological localization of iron in rat and monkey brains. Brain Res. 1981;215:317–22. doi: 10.1016/0006-8993(81)90510-2. [DOI] [PubMed] [Google Scholar]
- 40.Falangola MF, Lee SP, Nixon RA, Duff K, Helpern JA. Histological co-localization of iron in Abeta plaques of PS/APP transgenic mice. Neurochem Res. 2005;30:201–5. doi: 10.1007/s11064-004-2442-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jack CR, Jr, Wengenack TM, Reyes DA, Garwood M, Curran GL, Borowski BJ, et al. In vivo magnetic resonance microimaging of individual amyloid plaques in Alzheimer’s transgenic mice. J Neurosci. 2005;25:10041–8. doi: 10.1523/JNEUROSCI.2588-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El Tannir El Tayara N, Delatour B, Le Cudennec C, Guegan M, Volk A, Dhenain M. Age-related evolution of amyloid burden, iron load, and MR relaxation times in a transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2005;22:199–208. doi: 10.1016/j.nbd.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 43.Bartzokis G, Sultzer D, Lu PH, Nuechterlein KH, Mintz J, Cummings J. Heterogeneous age-related breakdown of white matter structural integrity: implications for cortical “disconnection” in aging and Alzheimer’s disease. Neurobiol Aging. 2004;25:843–51. doi: 10.1016/j.neurobiolaging.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 44.Pak K, Chan SL, Mattson MP. Presenilin-1 mutation sensitizes oligodendrocytes to glutamate and amyloid toxicities, and exacerbates white matter damage and memory impairment in mice. Neuromolecular Med. 2003;3:53–64. doi: 10.1385/NMM:3:1:53. [DOI] [PubMed] [Google Scholar]
- 45.Delatour B, Guegan M, Volk A, Dhenain M. In vivo MRI and histological evaluation of brain atrophy in APP/PS1 transgenic mice. Neurobiol Aging. 2005;27:835–47. doi: 10.1016/j.neurobiolaging.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 46.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 47.Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, Abrahamson EE, et al. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Norton WT. Formation, structure, and biochemistry of myelin. In: Siegel GJ, Albers RW, Agranoff BW, Katzman R, editors. Basic neurochemistry. 3. Boston: Little, Brown and Co; 1981. pp. 63–92. [Google Scholar]
- 49.Sastry S, Arendash GW. Time-dependent changes in iron levels and associated neuronal loss within the substantia nigra following lesions within the neostriatum/globus pallidus complex. Neuroscience. 1995;67:649–66. doi: 10.1016/0306-4522(94)00618-f. [DOI] [PubMed] [Google Scholar]
- 50.Maynard CJ, Cappai R, Volitakis I, Cherny RA, White AR, Beyreuther K, et al. Overexpression of Alzheimer’s disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J Biol Chem. 2002;277:44670–6. doi: 10.1074/jbc.M204379200. [DOI] [PubMed] [Google Scholar]
- 51.Larry Sparks D. Cholesterol, copper, and accumulation of thioflavine S-reactive Alzheimer’s-like amyloid beta in rabbit brain. J Mol Neurosci. 2004;24:97–104. doi: 10.1385/jmn:24:1:097. [DOI] [PubMed] [Google Scholar]
- 52.Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Abeta peptides. J Biol Inorg Chem. 2004;9:954–60. doi: 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 53.Ghribi O, Golovko MY, Larsen B, Schrag M, Murphy EJ. Deposition of iron and beta-amyloid plaques is associated with cortical cellular damage in rabbits fed with long-term cholesterol-enriched diets. J Neurochem. 2006;99:438–49. doi: 10.1111/j.1471-4159.2006.04079.x. [DOI] [PubMed] [Google Scholar]
- 54.Doraiswamy PM, Finefrock AE. Metals in our minds: therapeutic implications for neurodegenerative disorders. Lancet Neurol. 2004;3:431–4. doi: 10.1016/S1474-4422(04)00809-9. [DOI] [PubMed] [Google Scholar]
- 55.Ke Y, Ming Qian Z. Iron misregulation in the brain: a primary cause of neurodegenerative disorders. Lancet Neurol. 2003;2:246–53. doi: 10.1016/s1474-4422(03)00353-3. [DOI] [PubMed] [Google Scholar]