

Hypertrophic cardiomyopathy (HCM) is a common hereditary disease, with a prevalence of 1:500 in the general population.1,2 HCM is clinically diagnosed by the presence of – often asymmetrical – left ventricular (LV) hypertrophy in the absence of a disease likely to cause this hypertrophy, such as diabetes mellitus, hypertension and/or aortic valve stenosis.3
Hereditary HCM has a Mendelian pattern of inheritance and is mainly caused by mutations in genes that encode for sarcomeric proteins.4 To date, over 430 mutations have been associated with the development of HCM.5 Histologically, HCM is characterised by myocyte disarray and interstitial fibrosis.6
The clinical course of HCM has a large inter- and intrafamilial variability, ranging from the onset of sudden cardiac death at a young age to the development of mild symptoms of heart failure late in life. Therefore, risk estimation of asymptomatic family members of HCM patients for the development of HCM is important to enable timely initiation of therapy.
If a mutation is found in an HCM patient, asymptomatic family members at risk can be identified by screening for carriership of this mutation. Unfortunately, mutations are only found in 63% of HCM patients.7 In the remaining 37%, identification of asymptomatic family members at risk is confined to the use of echocardiography and electrocardiography, by which subtle diastolic dysfunction and/or electrocardiographic abnormalities can be detected.8,9 However, no specific structural abnormalities have been detected in human HCM mutation carriers before frank hypertrophy has developed.
Cardiac magnetic resonance imaging (CMR) has excellent tomographical qualities and can be used to detect focal fibrosis with delayed contrast enhancement (DCE) imaging techniques. Consequently, we performed a study to evaluate if structural abnormalities could be discerned with CMR in HCM mutation carriers before LV hypertrophy has developed.10
In brief, we selected 28 HCM mutation carriers from 15 different families, who had an LV wall thickness ≤13 mm measured by 2D echocardiography in the year prior to the study. The HCM mutation carriers either had a 2373 insG mutation in the gene encoding for cardiac myosin binding protein C, or a Glu62Gln missense mutation in the gene encoding for 〈-tropomyosin.11,12 The structure and global function of the LV of the HCM mutation carriers were evaluated with CMR and echocardiography and compared with 16 age- and gender-matched healthy volunteers. Also, a standard twelve-lead ECG was performed. LV hypertrophy was evaluated using the Romhilt-Estes criteria. ST segments were defined as normal or abnormal, and an R wave in V1 >3 mm was considered abnormal.
We found that global LV and left atrial dimensions of the HCM mutation carriers were comparable with those of the healthy volunteers. In one HCM carrier, CMR showed inferoseptal basal hypertrophy which was not detected by echocardiography. In the remaining 27 HCM mutation carriers, maximum septal wall thickness, maximal lateral wall thickness, LV mass and LA dimensions were within normal limits. In 82% of HCM mutation carriers (23/28), only CMR revealed an abnormal structure of the myocardium consisting of profound crypts in the basal and mid segments of the inferoseptal LV myocardium, at the junction of the right and left ventricle (figure 1). Best visualisation was obtained in enddiastole on a dedicated long-axis slice, slightly modified from the two-chamber view, cutting through the inferior septum (figure 1B). All healthy volunteers had normal LV myocardium without structural abnormalities. Patchy DCE was found in two HCM mutation carriers within the septal wall (figure 2). In these HCM mutation carriers, maximum septal wall thickness was 17 and 12 mm respectively. None of the HCM mutation carriers met the Romhilt-Estes ECG criteria for left ventricular hypertrophy. In 74% of the crypt-positive carriers (17/23), the ECG was considered abnormal, based on abnormal ST segments, a tall R wave in V1, and/or prominent q waves in the inferior limb leads. One crypt-negative female HCM mutation carrier had an abnormal ECG. To date, the number of HCM mutation carriers included in this study has increased, and still approximately 80% of the HCM mutation carriers exhibit crypts, particularly in the inferoseptum.
Figure 1.
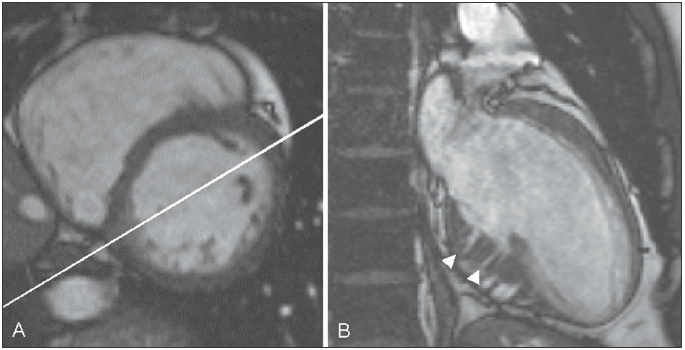
End-diastolic short-axis (A) and dedicated long-axis (B) cine images of a hypertrophic cardiomyopathy mutation carrier. Note the bright triangular spot in the inferoseptum (A) through which the dedicated long-axis image is planned. The white arrowheads indicate crypts in the inferoseptum of the left ventricle (B).
Figure 2.
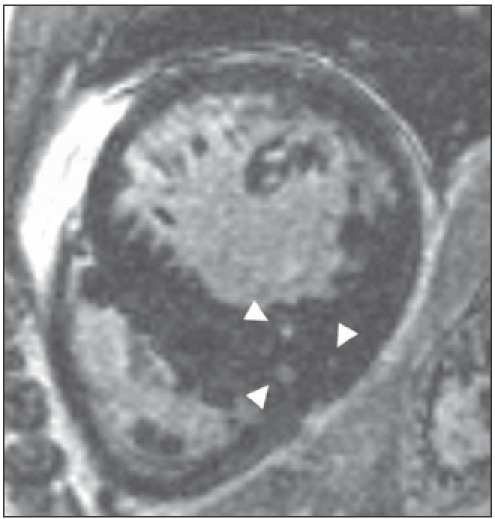
Short-axis contrast enhanced image of a hypertrophic cardiomyopathy mutation carrier with regional inferoseptal hypertrophy. Note the patchy contrast enhancement within the inferoseptum of the left ventricle, as indicated by the three white arrowheads.
The pathophysiological background of the crypts is currently unknown, but the presence of crypts contradicts the hypothesis that HCM is the result of a disease process that directly induces myocyte hypertrophy. The crypts may be the macroscopic representation of myocyte disarray and a precursor stage of a disease process that ultimately leads to hypertrophy and focal fibrosis.13
To further investigate the role of the crypts in the development of HCM, we set up a multicentre, long-term follow-up study, in which HCM mutation carriers will undergo a biannual CMR examination in addition to the regular ambulatory ECG monitoring, echocardiogram and exercise testing. The clinical and CMR data will be registered within the national database for genetically identified cardiomyopathies (GENCOR).
Parallel to the follow-up study with human HCM mutation carriers, an experimental study with an HCM mouse model has been set up to determine the histological background of the crypts.14 For this purpose, transgenic mice will undergo serial CMR examinations at the live mouse MRI facility in Utrecht. If the crypts are visualised with CMR, histological examination will be performed.
This study illustrated that structural abnormalities can be detected with CMR in HCM mutation carriers at a very early stage of disease.10 Therefore, CMR may serve as an additional tool to screen for asymptomatic family members at risk of developing HCM. This unique finding challenges current concepts on the development of HCM and merits further research.
Acknowledgement
Tjeerd Germans is supported by the Netherlands Heart Foundation, grant no. 2006B213, Arthur A.M. Wilde and Yigal M. Pinto are supported by the Netherlands Heart Foundation, grant no. 2003T302.
References
- 1.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995; 92:785-9. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ, Spirito P, Roman MJ, Paranicas M, Okin PM, Best LG, et al. Prevalence of hypertrophic cardiomyopathy in a population-based sample of American Indians aged 51 to 77 years (the Strong Heart Study). Am J Cardiol 2004;93:1510-4. [DOI] [PubMed] [Google Scholar]
- 3.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002;287:1308-20. [DOI] [PubMed] [Google Scholar]
- 4.Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev 2002;82:945-80. [DOI] [PubMed] [Google Scholar]
- 5.NHLBI Program for Genomic Applications HMS. Genomics of Cardiovascular Development, Adaptation, and Remodeling. URL: http://www.cardiogenomics.org, accessed 01-2007. [Google Scholar]
- 6.Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, et al. A mouse model of familial hypertrophic cardiomyopathy. Science 1996;272:731-4. [DOI] [PubMed] [Google Scholar]
- 7.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003;107:2227-32. [DOI] [PubMed] [Google Scholar]
- 8.McKenna WJ, Spirito P, Desnos M, Dubourg O, Komajda M. Experience from clinical genetics in hypertrophic cardiomyopathy: proposal for new diagnostic criteria in adult members of affected families. Heart 1997; 77:130-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagueh SF, McFalls J, Meyer D, Hill R, Zoghbi WA, Tam JW, et al. Tissue Doppler imaging predicts the development of hypertrophic cardiomyopathy in subjects with subclinical disease. Circulation 2003;108:395-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Germans T, Wilde AA, Dijkmans PA, Chai W, Kamp O, Pinto YM, et al. Structural abnormalities of the inferoseptal left ventricular wall detected by cardiac magnetic resonance imaging in carriers of hypertrophic cardiomyopathy mutations. J Am Coll Cardiol 2006;48:2518-23. [DOI] [PubMed] [Google Scholar]
- 11.Alders M, Jongbloed R, Deelen W, van den WA, Doevendans PA, Ten Cate FJ, et al. The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur Heart J 2003;24:1848-53. [DOI] [PubMed] [Google Scholar]
- 12.Jongbloed RJ, Marcelis CL, Doevendans PA, Schmeitz-Mulkens JM, Van Dockum WG, Geraedts JP, et al. Variable clinical manifestation of a novel missense mutation in the alpha-tropomyosin (TPM1) gene in familial hypertrophic cardiomyopathy. J Am Coll Cardiol 2003;41:981-6. [DOI] [PubMed] [Google Scholar]
- 13.Varnava AM, Elliott PM, Sharma S, McKenna WJ, Davies MJ. Hypertrophic cardiomyopathy: the interrelation of disarray, fibrosis, and small vessel disease. Heart 2000;84:476-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrier L, Knoll R, Vignier N, Keller DI, Bausero P, Prudhon B, et al. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res 2004;63:293-304. [DOI] [PubMed] [Google Scholar]