

Abstract
This report describes the synthesis and evaluation of glycosylated polyacrylate nanoparticles that have covalently-bound antibiotics within their framework. The requisite glycosylated drug monomers were prepared from one of three known antibiotics, an N-sec-butylthio β-lactam, ciprofloxacin, and a penicillin, by acylation with 3-O-acryloyl-1,2-O-isopropylidene-5,6 bis((chlorosuccinyl)oxy)-D-glucofuranose (7) or 6-O-acetyl-3-O-acryloyl-1,2-O-isopropylidene-5-(chlorosuccinyl)oxy-α-D-glucofuranose (10). These acrylated monomers were subjected to emulsion polymerization in a 7:3 (w:w) mixture of butyl acrylate-styrene in the presence of sodium dodecyl sulfate as surfactant (3 weight %) and potassium persulfate as a radical initiator (1 weight %). The resulting nanoparticle emulsions were characterized by dynamic light scattering and found to have similar diameters (~40 nm) and size distributions to those of our previously studied systems. Microbiological testing showed that the N-sec-butylthio β-lactam and ciprofloxacin nanoparticles both have powerful in vitro activities against methicillin-resistant Staphylococcus aureus and Bacillus anthracis, while the penicillin-bound nanoparticles have no antimicrobial activity. This indicates the need for matching a suitable antibiotic with the nanoparticle carrier. Overall, the study shows that even relatively large, polar acrylate monomers (MW>1000 amu) can be efficiently incorporated into the nanoparticle matrix by emulsion polymerization, providing opportunities for further advances in nanomedicine.
Keywords: carbohydrates, emulsion polymerization, nanoparticle, MRSA, anthrax, drug delivery
1. Introduction
Recent advances in biotechnology have accelerated the discovery of new drug molecules. However, many of these compounds suffer from sensitivity to degradative processes in the blood, or have limited water solubility that provides for minimal dissolution in the gastrointestinal tract and low bioavailability. There has been a renewed effort to find strategies for protecting and delivering such drugs, including the use of liposomes and various nanoparticles. These drug delivery vehicles improve bioavailability, efficacy, and specificity of some pharmaceutical compounds.1,2 Recently, our laboratory has investigated the properties of polyacrylate emulsions that contain nanoparticles measuring 30–50 nm in diameter, and onto which an antibiotic molecule can be covalently attached.3
These nanoparticles can be prepared easily in water by radical-induced emulsion polymerization using an acrylated derivative of the antibiotic as a co-monomer in a warmed mixture of butyl acrylate and styrene. Sodium dodecyl sulfate (3 weight %) and sodium persulfate (1 weight %) are used as surfactant and radical initiator, respectively. The resulting emulsions are stable indefinitely up to at least 80°C. Also by extending this emulsion polymerization procedure, we established that penicillin-containing polyacrylate nanoparticles can be easily prepared and are effective at rejuvenating the in vitro antibacterial activity of penicillin drugs against β-lactamase-producing microbes such as methicillin-resistant Staphylococcus aureus (MRSA).4 More recently, we described the application of this polymerization methodology to the synthesis of carbohydrated poly(ethyl acrylate) nanoparticles which contain a variety of O-protected and even unprotected monosaccharides within the polymeric framework.5 These carbohydrated nanoparticles appear to be the first examples of polymeric glyconanoparticles reported in the literature, and required that relatively bulky, water-soluble glycosylated acrylates be used as monomers in the emulsion polymerization process. Glycosylated nanoparticles have been employed to study carbohydrate interactions in the field of chemical glycobiology, but their potential in drug delivery is relatively unexplored particularly in the context of antibacterials research.6 Consequently, a number of issues regarding this capability need to be better investigated, and the first thing we wanted to explore was whether the glycosylated nanoparticles could be made to have antibacterial activity. Thus, the goal of the present work was to adapt our previous studies on antibiotic-conjugated nanoparticles and carbohydrated nanoparticles to construct an amalgamated system we refer to as glyconanoparticle antibiotics, or glyconanobiotics for short, and to evaluate their in vitro antibacterial capabilities against two dangerous pathogenic microbes, MRSA and B. anthracis. A graphic of what this nanoparticle is comprised of is depicted in Scheme 1. Secondly, we also were interested in whether the relatively large monomeric acrylates required for the formation of these nanoparticles could even be successfully employed, since this would then signal the potential use of other surrogates needed for biomedical applications. At the outset of this study, it was not obvious that this could in fact be achieved, since until now only small (MW<250) lipophilic monomers have been successfully introduced as polyacrylates by emulsion polymerization. Therefore, our study is focused on three representative antibiotic agents having entirely different biological modes of action, an N-thiolated β-lactam, a fluoroquinolone, and a penicillin. To prepare the nanoparticles (designated NP1–3), it was necessary to first convert each of the antibiotics to their glycosylated monomers 1–3, where the critical acrylate moiety is located at the 3’ center of the furanose ring (Fig. 1). Acrylate monomer 1 contains two appended N-thiolated β-lactams, while monomers 2 and 3 bear a single ciprofloxacin or 6-aminopencillinanic acid moiety, respectively. The latter acrylate was used as a control, since our prior investigation on penicillin-bound nanoparticles identified it as being devoid of antibacterial activity against MRSA, with a broth minimum inhibitory concentration of >256 ug/mL.4
Scheme 1.
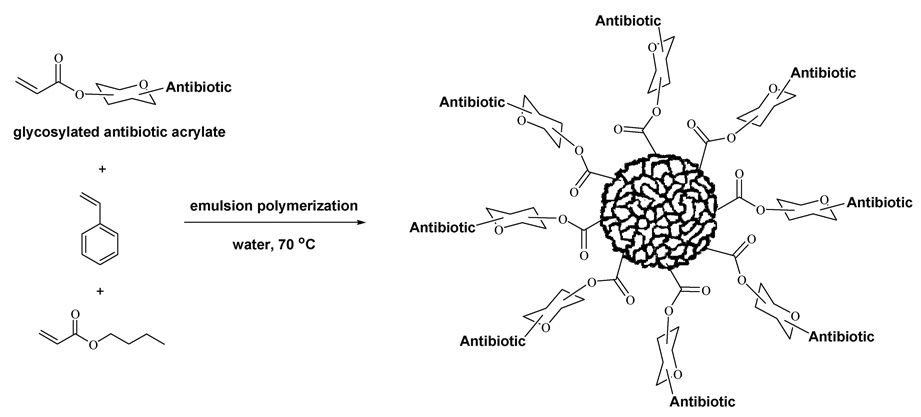
Synthesis of glyconanobiotics by emulsion polymerization
Figure 1.

Glycosylated antibiotic acrylates used for the formation of glyconanobiotics: bis-N-sec-butylthio β-lactam acrylate (1), ciprofloxacin acrylate (2), 6-aminopenicillanic acid acrylate (3).
2. Results and discussion
To construct acrylates 1–3, it was first necessary to synthesize the two acyl chlorides 7 and 10 from the protected glucofuranose 4 as shown in Scheme 2. Subsequently, the antibiotics were attached by direct acylation according to the procedures described below.
Scheme 2.
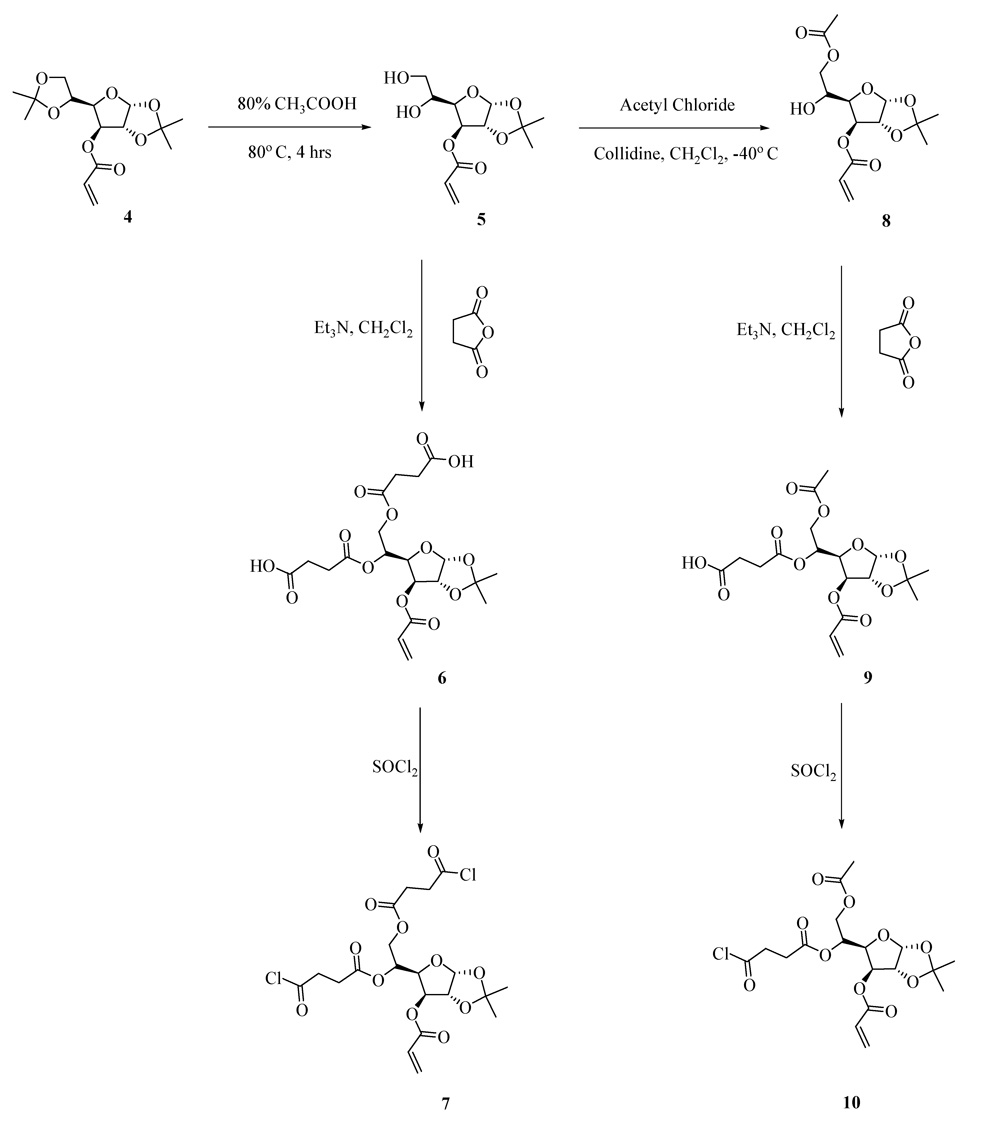
Synthesis of 3-O-acryloyl-1,2-O-isopropylidene-5,6 bis((chlorosuccinyl)oxy)-D-glucofuranose (7) and 6-O-acetyl-3-O-acryloyl-1,2-O-isopropylidene-5-(chlorosuccinyl)oxy-α-D-glucofuranose (10).
2.1. Synthesis of acyl chlorides 7 and 10
3-O-acryloyl-1,2-O-isopropylidene-5,6-bis((chlorosuccinyl)oxy)-D-glucofuranose (7) and 6-O-acetyl-3-O-acryloyl- 1,2-O-isopropylidene-5-(chlorosuccinyl)oxy-α-D-glucofuranose (10) were both synthesized from 1,2:5,6-di-O-isopropylidine-α-D-glucofuranose-3-acrylate (4) as shown in Scheme 2. Partial deacetonation of 4 with 80% acetic acid at 80°C afforded product 5 in 62% isolated yield. Diol 5 was diacylated with succinic anhydride in the presence of Et3N in CH2Cl2 to give di-acid 6 in 65% yield. Thionyl chloride was used to convert compound 6 to the corresponding di-acyl chloride 7 in 98% yield. Acyl chloride 10 was obtained similarly by selective acylation7 of the primary hydroxyl group of diol 5 with acetyl chloride in dichloromethane in the presence of 2,4,6-collidine at −40°C to afford compound 8 in 72% yield. Following succinic acid acylation of alcohol 8 to afford carboxylic acid 9, conversion to the mono-acyl chloride 10 with thionyl chloride proceeded in 65% yield for the two steps. A significant feature revealed in this synthesis is the stability of the endo-cyclic acetonide of the monosaccharide during the reaction of thionyl chloride (generation of HCl).
2.2. Synthesis of antibiotic-conjugated acrylates 1–3
With the two key acyl chlorides in hand, we evaluated their use for acylating the three selected antibiotic drugs to prepare monomers 1–3 (Scheme 3–Scheme 5). N-Thiolated β-lactams discovered in our group are a new family of antibacterial agents active against Staphylococcus bacteria, including methicillin-resistant strains of Staphylococcus aureus (MRSA)8,9, and Bacillus anthracis10 which is the causative agent of anthrax infections. For the goal of preparing glyconanoparticle variants of this class of antibacterial agents, bis-N-thiolated β-lactam glucose acrylate 1 was prepared. Monomer 1 was synthesized in 62% yield from N-sec-butylthio β-lactam derivative 1111 by coupling with di-acyl chloride 7 and Et3N in CH2Cl2 (Scheme 3). As it was expected that similar bisciprofloxacin and bis-penicillin glucose acrylates would be less lipophilic due to the presence of their carboxylic acid moieties, which in turn could cause problems in the emulsion polymerization due to poor solubility in the butyl acrylate-styrene mixture, we decided to instead try to prepare the mono-antibiotic acrylate forms. Monociprofloxacin acrylate 2 was successfully synthesized in 56% yield from ciprofloxacin by coupling with acyl chloride 10 and Et3N in CH2Cl2 (Scheme 4) whereas the mono-penicillin acrylate 3 was synthesized4 in 51% yield from 6-aminopenicillanic acid (13) via its trimethylsilyl ester 14, which was then reacted with acyl chloride 10 (Scheme 5). The structures of the carbohydrated monomers 1 and 2 were determined by 1H and 13C NMR, and mass spectrometry.12
Scheme 3.
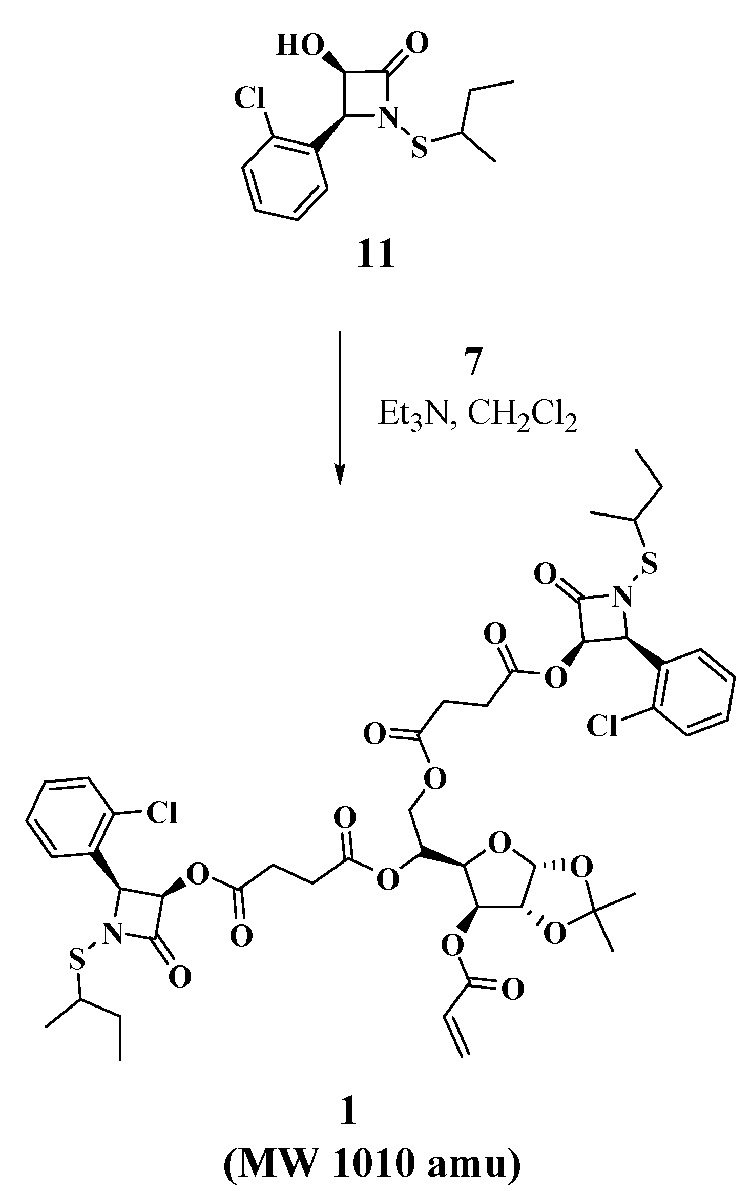
Synthesis of bis-N-sec-butylthio β-lactam acrylate (1)
Scheme 5.
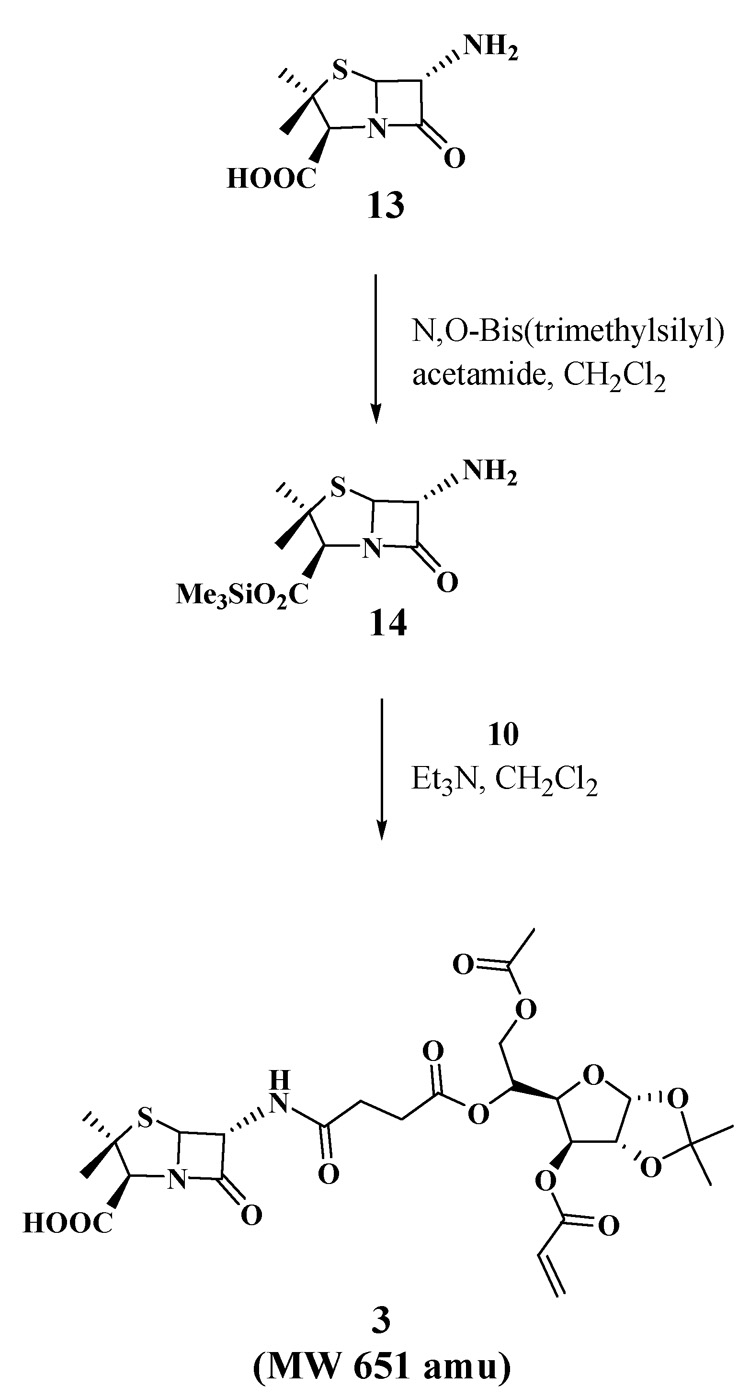
Synthesis of 6-aminopenicillinic acid acrylate (3).
Scheme 4.
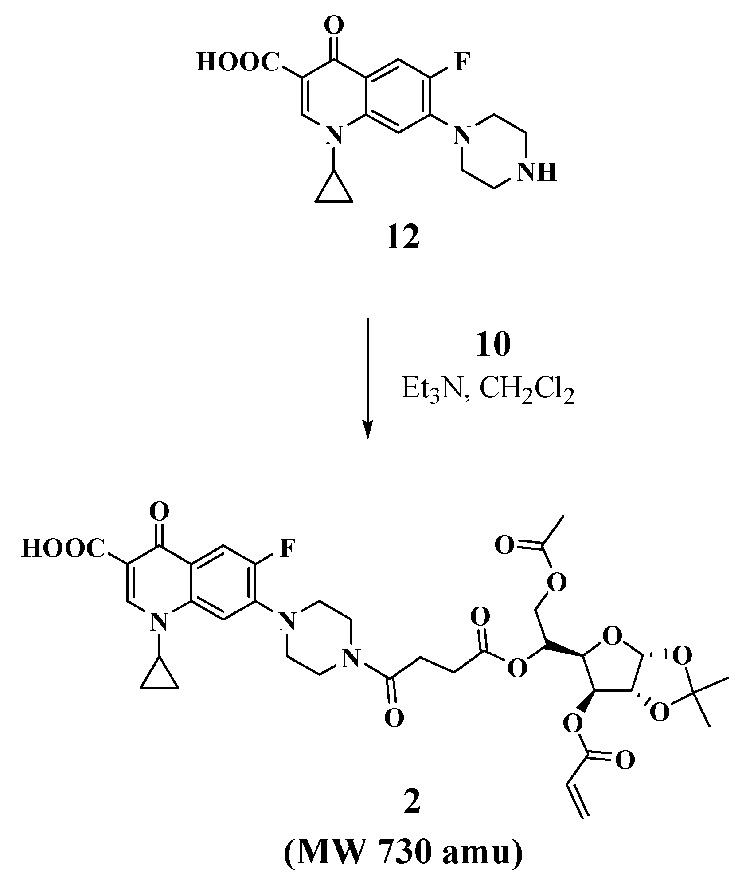
Synthesis of ciprofloxacin acrylate (2).
2.3 Preparation of glyconanobiotics
The carbohydrate-conjugated antibiotic monomers (1–3) prepared from N-sec-butylthio β-lactam, ciprofloxacin, and 6-aminopenicillanic acid were found to be readily soluble in a warm 7:3 (w:w) mixture of butyl acrylate and styrene, and undergo emulsion polymerization with ease using the prescribed protocol. In this way, the nanoparticles bearing one of the three antibiotics could be prepared by emulsion polymerization of antibiotic monomers 1–3 (2 weight %) in a 7:3 (w:w) blend of butyl acrylate-styrene, upon stirring in water for 6 hours at 70°C. Sodium dodecyl sulfate (3 weight %) and potassium persulfate (1 weight %) were used as surfactant and radical initiator, respectively. Dynamic light scattering (DLS) analysis of the final emulsion revealed that the average diameter of the glyconanoparticles is uniformly about 40 nm with narrow size distributions spanning only from about 30–50 nm, as has been previously reported for similar polyacrylate nanoparticles.4
2.4 Biological activity
The glycosylated nanoparticle antibiotics NP1–3 were evaluated for in vitro antibacterial activity against three pathogenic bacteria, namely Staphylococcus aureus, a strain of methicillin-resistant Staphylococcus aureus (MRSA), and Bacillus anthracis. Additionally, the acrylated monomers 1–3 were also examined for comparison. Activity was assessed by determining the minimum inhibitory concentration (MIC) values by broth serial dilution, and is summarized in Table 1. The concentration of antibiotic in the nanoparticle was assumed to be equivalent to that used for the polymerization, as expressed in µg of drug per mL of undiluted emulsion. The results show that the glyconanoparticles containing N-thiolated β-lactam and ciprofloxacin exhibited significant antibacterial activity, while the penicillin-bound glyconanoparticles and non-drug containing nanoparticles made of just butyl acrylate-styrene (7:3) (controls) were completely inactive against these microbes, with MIC values of >256 ug/mL. The bioactivities are similar to or better than that of the corresponding drug monomers. The finding that the N-thiolated β-lactam and cipro nanoparticles are active, while the penicillin variant is not, suggests that the type of antibiotic and the comparative activity of the free drug being released (presumably by ester or amide cleavage) are both critical. Our laboratories’ attempts to measure the rate of drug release from the nanoparticles has not yet been successful, and are ongoing, as are experiments to identify the mechanism of how these nanoparticle antibiotics may exert their effects on bacterial cells.
Table 1.
Antibacterial activity of glycosylated monomers 1–3 and their nanoparticle emulsions NP 1–3 against Staphylococcus aureus (ATCC 25923), MRSA (ATCC 43300) and Bacillus anthracis, as determined by broth dilution MIC assaysa
| Sample | Microbe |
||
|---|---|---|---|
| S. aureus (µg/mL) | MRSA (µg/mL) | B. anthracis (µg/mL) | |
| Penicillin G (control) | 0.012 | 16 | n/a |
| Lactam monomer 1 | 128 | 128 | 64 |
| Lactam NP 1 | 64 | 64 | 64 |
| Cipro monomer 2 | 32 | 32 | 16 |
| Cipro NP 2 | 32 | 32 | 16 |
| Penicillin monomer 3 | >256 | >256 | >256 |
| Penicillin NP 3 | >256 | >256 | >256 |
MIC values refer to the lowest concentration of the bound drug (µg/mL) required to completely inhibit bacterial growth for 24 h in culture.
3. Experimental
Solvents and chemicals were purchased from Acros Organics and used without further purification. 1,2:5,6-Di-O-isopropylidine-α-D-glucofuranose-3-acrylate (4)13 was prepared following the cited literature procedure. 1H NMR and 13C NMR spectra were recorded on Bruker DPX-250 and Varian Inova 400 spectrometers. Mass spectra were obtained with on an ESI LC MS system (Agilent Technologies).
3.1 Synthesis
3.1.1 Synthesis of 3-O-acryloyl-1,2-O-isopropylidene-5,6 bis((chlorosuccinyl)oxy)-D-glucofuranose (7)
1,2:5,6-Di-O-isopropylidine-α-D-glucofuranose-3-acrylate (4) (0.5g, 1.59 mmol) was dissolved in 4 mL of 80% acetic acid and stirred 4 hours at 80°C. The solvents were removed in vacuo and the residue was purified by column chromatography (silica gel, hexane-EtOAc, 1:1) to afford compound 5 (0.32 g, 62%) as a syrup. This monoacetonide product (0.3g, 1.09 mmol) and succinic anhydride (0.65g, 6.54 mmol) were stirred with Et3N (0.76 mL, 5.45 mmol) in CH2Cl2 (10 mL) at rt for 5h. The reaction mixture was washed with 5% KHSO4, then water. The organic layer was dried over Na2SO4, filtered and evaporated to dryness. The brown oil obtained was chromatographed on silica gel (EtOAc) and a solid material was obtained that contained ammonium salt and succinic acid, which could easily be removed by centrifugation in 10 mL of CH2Cl2. After evaporation, compound 6 (0.34g, 65%) was obtained as a colorless syrup. This di-acid (0.3g, 0.80 mmol) was dissolved in 3 mL of thionyl chloride, stirred 45 min. at room temperature, and the solvents were removed in vacuo to afford compound 7 (0.31g, 98%) as a syrup. 1H NMR (400 MHz, CDCl3): δ 6.39 (m, 1H), 6.04 (m, 1H), 5.87 (m, 2H), 5.28 (m, 2H), 4.50 (m, 3H), 4.17 (m, 1H), 3.13 (m, 4H), 2.62 (m, 4H) 1.48 (s, 3H), 1.27 (s, 3H);13C NMR (100 MHz, CDCl3): δ 173.2, 172.9, 171.0, 170.1, 169.8, 165.7, 164.7, 132.5, 127.5, 112.8, 105.3, 83.3, 76.6, 75.2, 68.5, 63.8, 41.8, 29.3, 28.6, 26.9.
3.1.2 Synthesis of 6-O-acetyl-3-O-acryloyl-1,2-O-isopropylidene-5-(chlorosuccinyl)oxy-α-D-glucofuranose (10)
3-O-acryloyl-1,2-O-isopropylidene-α-D-glucofuranoside (5) (0.4 g, 1.52 mmol) was dissolved in 10 mL of CH2Cl2 and cooled to −40 °C. Collidine (0.36 g, 3.04 mmol) was added to the solution, then acetyl chloride (0.14g, 1.82 mmol), and the mixture was stirred at −40 °C for 3 h and at rt for an additional hour. The mixture was washed with water and extracted into CH2Cl2 (3×25 mL). The solvents were evaporated and the residue was purified by column chromatography to afford compound 8 (0.33 g, 72 %) as a syrup. This intermediate product (0.3 g, 0.98 mmol) and succinic anhydride (0.29 g, 2.96 mmol) were stirred with Et3N (0.34 mL, 2.45mmol) in CH2Cl2 (10 mL) at rt for 5h. The reaction mixture was washed with 5% KHSO4, then water. The organic layer was dried over Na2SO4, filtered and evaporated to dryness. The brown oil obtained was chromatographed on silica gel (EtOAc), and a solid material was obtained that contained ammonium salt and succinic acid, which could easily be removed by centrifugation in 10 mL of CH2Cl2. After evaporation, compound 9 (0.26 g, 65%) was obtained as a syrup. Subsequently, this acid was dissolved in 3 mL of thionyl chloride, stirred 45 min. at room temperature, and the solvents were removed in vacuo to afford compound 10 (0.34 g, 98%) as a syrup. 1H NMR (400 MHz, CDCl3): δ 6.35 (d, 1H, J=17.2 Hz), 6.04 (dd, 1H, J =10.4, 17.2 Hz), 5.87 (m, 2H), 5.33 (d, 1H, J=2.8 Hz), 5.17 (m, 1H), 4.52 (m, 3H), 4.08 (dd, 1H, J =5.6, 12.4 Hz), 3.10 (m, 3H), 2.57 (m, 3H), 2.00 (s, 3H), 1.49 (s, 3H), 1.27 (s, 3H);13C NMR (100 MHz, CDCl3): δ 172.8, 170.8, 169.8, 164.8, 132.7, 127.3, 112.7, 105.3, 83.3, 76.8, 75.2, 68.6, 63.2, 41.7, 29.4, 28.6, 26.3, 20.8.
3.1.3 Synthesis of bis-N-sec-butylthio β-lactam acrylate (1)
To a stirred solution of N-sec-butylthio β-lactam derivative 1111 (0.2 g, 0.7 mmol) and triethylamine (0.29 mL, 2.1 mmol) in CH2Cl2 (5 mL) at 0 °C was added dropwise diacyl chloride 7 (0.173g, 0.42 mmol in 0.5 mL of CH2Cl2). The solution was stirred at rt for 10 h, then treated with water (10 mL) and extracted with CH2Cl2 (3×15 mL). The combined organic layers were washed with water (2×50 mL), dried (MgSO4) and evaporated under reduced pressure. The residue was purified by column chromatography (hexane-EtOAc, 2:1) to afford compound 1 (0.22 g, 62%) as a syrup. 1H NMR (400 MHz, CDCl3): δ 7.35 (m, 8H), 6.35 (d, 1H, J=17.2 Hz), 6.14 (m, 2H), 6.03 (dd, 1H, J =10.8, 17.6 Hz), 5.84 (m, 2H), 5.47 (m, 2H), 5.25 (s, 1H), 5.09 (m, 1H), 4.45 (m, 2H), 4.33 (m, 1H), 4.05 (m, 1H), 3.04 (m, 2H), 2.17 (m, 8H), 1.56 (m, 4H), 1.47 (s, 3H), 1.26 (m, 9H), 0.96 (m, 6H);13C NMR (100 MHz, CDCl3): δ 171.2, 170.3, 169.8, 169.0, 164.7, 134.6, 123.6, 130.7, 130.1, 130.0, 128.9, 127.3, 126.9, 112.7, 105.3, 83.3, 75.3, 68.0, 63.7, 63.6, 48.6, 28.5, 28.3, 26.9, 26.4, 19.2, 18.9, 11.3. ESI-MS calcd for [M+NH4]+, 1028.0; found, 1028.3.
3.1.4 Synthesis of ciprofloxacin acrylate (2)
To a stirred solution of ciprofloxacin hydrochloride (0.2g, 0.6 mmol) and triethylamine (0.5 mL, 3.6 mmol) in CH2Cl2 (8 mL) at 0 °C was added dropwise acyl chloride 10 (0.31g, 0.66 mmol in 0.5 mL of CH2Cl2). The solution was stirred at rt for 10 h then treated with water (10 mL) and extracted with CH2Cl2 (3×15 mL). The combined organic layers were washed with water (2×50 mL), dried (MgSO4) and evaporated under reduced pressure. The residue was purified by column chromatography (EtOAc) to afford compound 2 (0.23 g, 56%) as a syrup. 1H NMR (400 MHz, CDCl3): δ 8.64(s, 1H), 7.90 (d, 1H, J=12.4 Hz), 7.32 (d, 1H, J=6.8 Hz), 6.39 (d, 1H, J=17.2 Hz), 6.05 (dd, 1H, J =10.4, 17.2 Hz), 5.88 (d, 1H, J=3.6 Hz), 5.86 (d, 1H, J=10.4 Hz), 5.32 (d, 1H, J=2.8 Hz), 5.24(m, 1H), 4.52(m, 3H), 4.12 (dd, 1H, J =5.2, 12.0 Hz), 3.79(m, 2H), 3.68(m, 2H), 3.53(m, 1H), 3.33(m, 2H), 3.25(m, 2H), 2.59(m, 4H), 2.02(s, 3H), 1.49(s, 3H), 1.37(m, 2H), 1.27(s, 3H), 1.18(m, 2H);13C NMR (100 MHz, CDCl3): δ 177.1, 171.8, 170.8, 169.6, 166.9, 164.8, 155.0, 152.5, 147.6, 145.6, 145.5, 139.1, 132.3, 127.7, 120.2, 112.7, 112.4, 108.2, 105.3, 105.2, 83.3, 75.5, 68.0, 63.3, 53.6, 50.2, 49.5, 45.2, 41.5, 35.5, 29.4, 28.1, 26.9, 26.4, 20.9, 8.4. ESI-MS calcd for [M+Na]+, 752.6; found, 752.2.
3.1.5 Synthesis of 6-aminopenicillanic acid acrylate (3)12
6-Aminopenicillanic acid (2 g, 0.924 mmol) and N,O-bis(trimethylsilyl)acetamide (0.21 g, 1.02 mmol) were added to CH2Cl2 (10 mL) and stirred at rt overnight. Triethylamine (0.19 mL, 1.38 mmol) was added and cooled on ice. Acyl chloride 10 (0.52 g, 1.11 mmol in 0.5 mL of CH2Cl2) was added dropwise and stirred at room temperature for 10 h. The reaction mixture was treated with water (10 mL) and extracted into CH2Cl2 (3×15 mL). The combined organic layers were washed with water (2×50 mL), dried (MgSO4) and evaporated under reduced pressure. The residue was purified by column chromatography (EtOAc) to afford compound 312 (0.23 g, 56%) as a syrup. 1H NMR (400 MHz, CDCl3): δ 6.55 (d, 1H, J=8.8 Hz), 6.39 (d, 1H, J=17.2 Hz), 6.02 (m, 1H), 5.88 (m, 2H), 5.63 (dd, 1H, J =4.0, 8.8 Hz), 5.48 (d, 1H, J=4.0 Hz), 5.31 (m, 1H), 5.19 (m, 1H), 4.48 (m, 3H), 4.04 (m, 1H), 2.55 (m, 4H), 2.01 (s, 3H), 1.62 (s, 3H), 1.54 (s, 3H), 1.48 (s, 3H), 1.27 (s, 3H).
3.2 Emulsion polymerization
The general procedure is as previously reported.3,4 The drug monomer (2 weight %) was pre-dissolved in a 7:3 (w:w) mixture of butyl acrylate-styrene, and sodium dodecyl sulfate in purified water was added to pre-emulsify the mixture with stirring under a nitrogen atmosphere. The amounts of each component added to the mixture are given in Table 2. The polymerized nanoparticles were prepared by adding potassium persulfate to a rapidly stirred (magnetic stirrer) mixture at 70°C for 6 hours, at which time heating and stirring were stopped and the sample could be transferred to a vial for storage or biological testing.
Table 2.
Formulation parameters used for emulsion polymerization
| Components | Amounts | Solid Content |
|---|---|---|
| Drug monomer | 20 mg | 2% |
| Butyl acrylate | 658 mg | 66% |
| Styrene | 282 mg | 28% |
| Sodium dodecyl sulfate | 30 mg | 3% |
| Potassium persulfate | 10 mg | 1% |
| Water | 4 mL |
3.3 Dynamic light scattering analysis
Sample size and distribution of the emulsions was examined by dynamic light scattering using a UPA 150 Honeywell MicroTrac DLS instrument equipped with a laser beam at 780 nm. The sample was pre-diluted with deionized distilled water (0.05 mL of emulsion in 9.95 mL of water, polymer concentration 0.1% wt) and sonicated for 3 minutes for the analysis. The value is expressed in weight-averaged scales as unimode at a scattering angle of 180° (backscatter) at a temperature of 25 °C.
4. Conclusions
In summary, these studies have demonstrated the preparation and in vitro evaluation of polyacrylate glyconanoparticles that bear a covalently-attached antibiotic. With this methodology, even large monomers (MW > 1000 amu) can be efficiently incorporated into the polyacrylate nanoparticle matrix up to 2 weight % of the total solid content. The antibiotic-conjugated acrylate monomers needed for the polymerization are readily prepared by a simple one-step procedure, from acylation of the antibiotic with the corresponding carbohydrated acyl chloride. These structurally-complex acrylate drug monomers show excellent solubility in the butyl acrylate-styrene mixture required for radical-induced emulsion polymerization in aqueous media. The resulting nanoparticles measure around 40 nm in diameter according to dynamic light scattering experiments, with narrow size distributions (30–50 nm), and possess equivalent or improved in vitro antibacterial activity to that of the corresponding acrylated antibiotic monomer. Further investigations in our laboratory are attempting to fully assess the underlying reasons for antibiotic effects of these nanoparticles, including how, where and at what rate the drug is being released, and their in vivo properties as potential treatment of life-threatening bacterial infections.
Acknowledgements
We sincerely thank Gil Brubaker (University of Florida Particle Engineering Research Center) for providing assistance with the particle analyses, and generous financial support from the National Institutes of Health (Grant AI05135) and National Science Foundation (NSF 0620572).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alle’mann E, Gurny R, Doelker E. Eur. J. Pharm. Biopharm. 1993;39:173–191. [Google Scholar]
- 2.Couvreur P, Dubernet C, Puisieux F. Eur. J. Pharm. Biopharm. 1995;41:2–13. [Google Scholar]
- 3.Turos E, Shim YJ, Wang Y, Greenhalgh K, Reddy GSK, Dickey S, Lim DV. Bioorg. Med. Chem. Lett. 2007;17:53–56. doi: 10.1016/j.bmcl.2006.09.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turos E, Reddy GSK, Greenhalgh K, Ramaraju P, Abeylath SC, Jang S, Dickey S, Lim DV. Bioorg. Med. Chem. Lett. 2007;17:3468–3472. doi: 10.1016/j.bmcl.2007.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abeylath SC, Turos E. Carbohydr. Polymer. 2007;70:32–37. doi: 10.1016/j.carbpol.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuente JM, Penadés S. Biochim. Biophys. Acta. 2006;1760:636–651. doi: 10.1016/j.bbagen.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Ishihara K, Kurihara H, Yamamoto H. J. Org. Chem. 1993;58:3791–3793. [Google Scholar]
- 8.Turos E, Konaklieva MI, Ren RXF, Shi H, Gonzalez J, Dickey S, Lim D. Tetrahedron. 2002;56:5571–5578. [Google Scholar]
- 9.Turos E, Long TE, Konaklieva MI, Coates C, Shim JY, Dickey S, Lim DV, Cannons A. Bioorg. Med. Chem. Lett. 2002;12:2229–2231. doi: 10.1016/s0960-894x(02)00343-8. [DOI] [PubMed] [Google Scholar]
- 10.Turos E, Long TE, Heldreth B, Leslie JM, Reddy SK, Wang Y, Coates C, Konaklieva M, Dickey S, Lim DV, Alonso E, Gonzalez J. Bioorg. Med. Chem. Lett. 2006;16:2084–2090. doi: 10.1016/j.bmcl.2006.01.070. [DOI] [PubMed] [Google Scholar]
- 11.Heldreth B, Long TE, Jang S, Reddy SK, Turos E, Dickey S, Lim DV. Bioorg. Med. Chem. 2006;14:3773–3784. doi: 10.1016/j.bmc.2006.01.029. [DOI] [PubMed] [Google Scholar]
- 12.A small amount of carboxylic acid 9 mixed with penicillin acrylate 3 showed up in the 1H NMR spectrum after eluting with ethyl acetate, and this mixture was used for the emulsion polymerization as a mixture.
- 13.Krishna PR, Kannan V, Ilangovan A, Sharma GVM. Tetrahedron: Asymmetry. 2001;12:829–837. [Google Scholar]