

Abstract
Until recently, parallel genotypic adaptation was considered unlikely because phenotypic differences were thought to be controlled by many genes. There is increasing evidence, however, that phenotypic variation sometimes has a simple genetic basis and that parallel adaptation at the genotypic level may be more frequent than previously believed. Here, we review evidence for parallel genotypic adaptation derived from a survey of the experimental evolution, phylogenetic, and quantitative genetic literature. The most convincing evidence of parallel genotypic adaptation comes from artificial selection experiments involving microbial populations. In some experiments, up to half of the nucleotide substitutions found in independent lineages under uniform selection are the same. Phylogenetic studies provide a means for studying parallel genotypic adaptation in non-experimental systems, but conclusive evidence may be difficult to obtain because homoplasy can arise for other reasons. Nonetheless, phylogenetic approaches have provided evidence of parallel genotypic adaptation across all taxonomic levels, not just microbes. Quantitative genetic approaches also suggest parallel genotypic evolution across both closely and distantly related taxa, but it is important to note that this approach cannot distinguish between parallel changes at homologous loci versus convergent changes at closely linked non-homologous loci. The finding that parallel genotypic adaptation appears to be frequent and occurs at all taxonomic levels has important implications for phylogenetic and evolutionary studies. With respect to phylogenetic analyses, parallel genotypic changes, if common, may result in faulty estimates of phylogenetic relationships. From an evolutionary perspective, the occurrence of parallel genotypic adaptation provides increasing support for determinism in evolution and may provide a partial explanation for how species with low levels of gene flow are held together.
Keywords: adaptation, artificial selection, convergent evolution, experimental evolution, homoplasy, natural selection, parallel evolution
Introduction
Homoplasy, or the recurrence of similarity in distinct evolutionary lineages, occurs frequently in nature. Such similarities have been documented at practically every level of biological organization, from nucleotide/amino acid sequences (Stewart, Schilling & Wilson 1987) to large scale deletions (Downie & Palmer, 1992), whole genome duplications (Soltis & Soltis, 1991), and the acquisition of complex phenotypic characters such as succulent, spiny stems in the Euphorbiaceae and Cactaceae. There is even evidence of the repeated origin of animal and plant species (Soltis & Soltis, 1991; Rundle et al., 2000; reviewed in Levin, 2001). This list includes examples of both molecular and morphological homoplasy, which are generally thought to be the result of distinct evolutionary processes. Because it is unlikely that complex phenotypes would arise repeatedly via a stochastic process, morphological homoplasy is widely regarded to be the result of selection. In contrast, nucleotide sequences are limited in the number of ways that they can evolve, thus most instances of molecular homoplasy have been interpreted as the chance fixation of independently arising variants in diverging lineages (Doolittle, 1994; Wells, 1996).
Although morphological homoplasy is generally viewed as being driven by natural selection, many evolutionary biologists assume that the phenotypes of interest result from unique genetic changes. In some cases, they are clearly right: The evolution of spines in euphorbs and cacti results from the modification of non-homologous structures. In cases where homology is plausible, this view is perhaps best explained by the traditional acceptance of Fisher’s infinitesimal model, in which quantitative traits are assumed to be controlled by an effectively infinite number of genes, each of very small effect (Fisher, 1930). Under this view, there should be numerous paths from any one phenotype to another. Thus, the likelihood that two lineages would independently accumulate changes at the same subset of underlying loci would be low. It has become increasingly clear, however, that continuous patterns of variation may sometimes be explained by the existence of a few major quantitative trait loci (QTLs) (Tanksley, 1993). Under this so-called oligogenic model of inheritance, the number of pathways from one phenotype to another is considerably more limited, increasing the likelihood that parallel phenotypic changes have a common genetic basis.
In organisms where connections between genotype and phenotype have been made, there is emerging evidence that molecular homoplasy is sometimes driven by natural selection. Unfortunately, our understanding of the genetic basis of all but the simplest traits in the simplest organisms is woefully incomplete. Thus, it is difficult to say with any certainty whether or not some of the more complex instances of morphological homoplasy have a common genetic basis. Here, we review the best examples of selection driving different lineages to the same phenotype through the fixation of independent changes at homologous loci. This pattern of evolution has several important implications. With respect to phylogeny reconstruction, it is widely recognized that homoplasy, regardless of the cause, can lead to inaccurate conclusions regarding the evolutionary history of taxa. Parallel selection responses at the genotypic level also suggest that adaptation may be a more deterministic process than previously believed, with genetic background effects and historical contingency playing a lesser role. If parallel changes prove to be common, they may provide a mechanism by which populations of a species can evolve collectively. Furthermore, such changes may increase the likelihood of the recurrent origin of taxa by allowing geographically isolated populations of the same species to independently invade a novel, unoccupied habitat.
Definitions
Historically, taxonomists have divided phenotypic homoplasy into two categories, parallelism and convergence. Parallel evolution is defined as ‘the independent occurrence of similar changes in groups with a common ancestry and because they had a common ancestry’ (Simpson, 1961, p. 103). In contrast, ‘convergence is the development of similar characteristics separately in two or more lineages without a common ancestry pertinent to the similarity but involving adaptation to similar ecological status’ (Simpson, 1961, pp. 78–79). As noted above, selection is believed to be the primary evolutionary force causing the recurrence in both situations.
The advent of DNA and protein sequencing necessitated a more precise definition of these terms. Molecular evolutionary biologists use parallelism and convergence in an analogous yet distinct manner. Nucleotide or protein sequence changes from the same ancestral state to the same derived state are called parallel changes, whereas changes from different ancestral states to a common derived state are considered convergent changes (Zhang & Kumar, 1997; Figure 1). Because our goal is to make an explicit connection between evolution at the phenotypic and genotypic levels, we need an operational definition that bridges the phenotypic and molecular views. Thus, we define parallel genotypic adaptation as the independent evolution of homologous loci to fulfill the same function in two or more lineages. Note that these changes need not be identical, just functionally equivalent. Under this definition, changes at non-homologous loci resulting in the same phenotype would be considered convergent (e.g., Chen Devries & Cheng, 1997), and fall outside the scope of this review.
Figure 1.
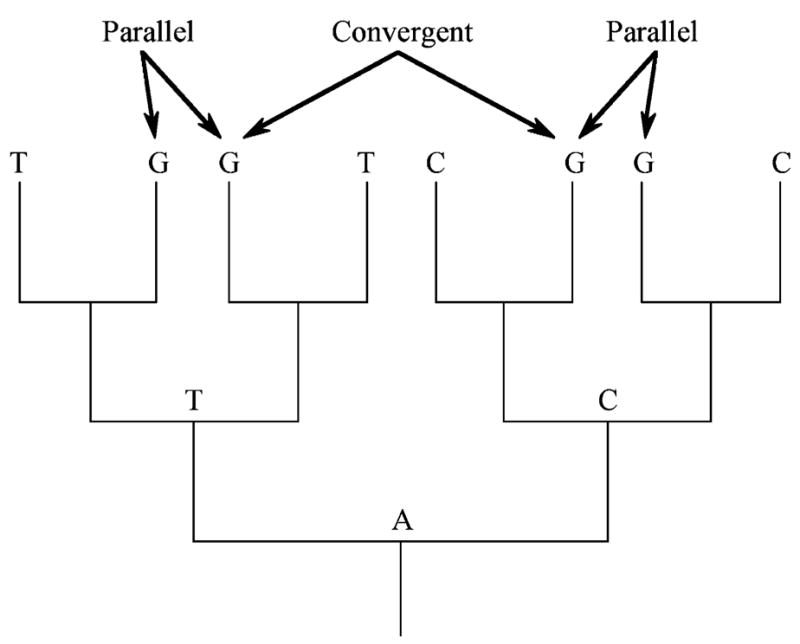
Parallelism versus convergence in molecular evolution. Character states at a single, homoplastic nucleotide site are mapped onto a gene tree. Parallelism refers to the independent evolution of the same derived state from a common ancestral state (the two Gs from T, or the two Gs from C). In contrast, convergence involves the evolution of the same derived state from different ancestral states (G derived independently from T and C). (After Zhang & Kumar, 1997)
Another possibility involves the independent duplication of a homologous, ancestral locus (A) to yield two descendant loci (B) (Figure 2). In this case, the two independently derived loci are not technically homologous. However, because the two loci are direct descendants of true homologues, we consider cases in which such loci evolve to fulfill a common function to be examples of parallel genotypic evolution. The growing body of genomic data suggests that gene duplication is a common phenomenon (Lynch & Conery, 2000), and its importance in generating the raw material for adaptive evolution has been widely recognized (e.g., Haldane, 1932; Ohno, 1970). Thus, future analyses may reveal this process to be a common mode of parallel evolution.
Figure 2.
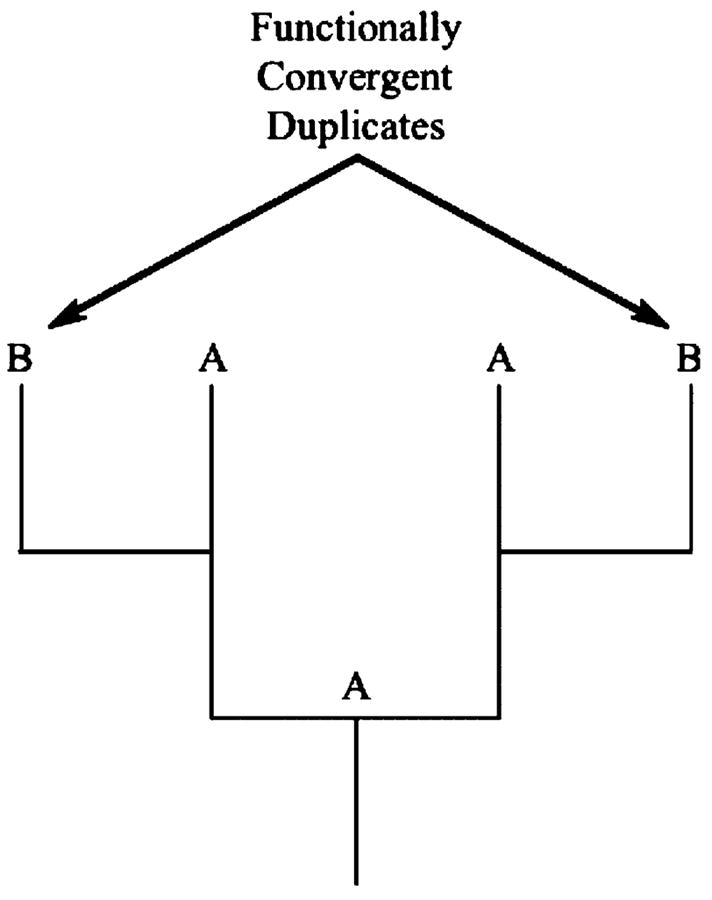
Convergent evolution of gene duplicates. The lateral branches leading to functional state B represent independent duplications of a homologous gene that fulfills function A. Functional state B evolved independently from changes in the duplicate copies. See text for details.
Empirical evidence
Experimental evolution studies
The clearest evidence of parallel genotypic adaptation comes from artificial selection experiments in the lab or greenhouse (Table 1, Section A). The strength of this approach lies in the fact that researchers control both the relevant selective pressures acting upon and the evolutionary histories of the populations under study. The short generation time and relative ease of characterizing genetic variation in certain microbes makes them ideal organisms in which to study the genotypic response to uniform selective pressures. In general, these studies have revealed that selection pressures such as temperature or host shifts commonly lead to parallel genotypic adaptation (Table 1). Moreover, there is evidence that these phenotypic shifts often result from minor sequence changes; in some cases, one or a few nucleotide substitutions at a single locus accounted for the entire response to selection (Liao, Mckenzie & Hageman, 1986; Cunningham et al., 1997; Crill, Wichman & Bull, 2000). While these studies are intriguing, they have an obvious shortcoming – the dynamics of selection in these simple organisms might not be representative of adaptation in more complex organisms. In taxa with larger and more complex genomes, selective constraints due to genetic background effects or antagonistic pleiotropy may play a more important role.
Table 1.
List of studies documenting parallel genotypic adaptation. The upper, middle, and lower panels include laboratory or greenhouse selection experiments, phylogeny-based studies, and genetic analyses of controlled crosses, respectively. See Appendix 1 for a summary of each study
| Taxonomic Group(s) | Phenotype | Type of evidence | Reference |
|---|---|---|---|
| Maize & Cocklebur | Herbicide resistance | Amino acid substitution | Bernasconi et al., 1995 |
| Human influenza A | Virulence | Amino acid substitution | Brown et al., 2001 |
| Bacteriophage ΦX 174 | Thermotolerance | Nucleotide substitution | Bull et al., 1997 |
| Bacteriophage ΦX 174 | Host shift | Amino acid substitution | Crill et al., 2001 |
| Bacteriophage T7 | Fitness | Deletion/nucleotide Substitution | Cunningham et al., 1997 |
| Escherichia coli | Drug resistance | Amino acid substitution | Levin et al., 2000 |
| Bacillus subtilis KNTase | Thermostability | Amino acid substitution | Liao et al., 1986 |
| Annual Sunflower spp. | Fertility | Genome composition | Rieseberg et al., 1996 |
| Arabidopsis thaliana | Fitness | Genome composition | Ungerer, 2000 |
| Bacteriophage ΦX 174 | Thermotolerance/host shift | Nucleotide substitution | Wichman, 1999 |
| Flour Beetle | Pesticide resistance | Amino acid substitution | Andreev et al., 1999 |
| Nematodes & Fungi | Pesticide resistance | Amino acid substitution | Elard et al., 1996 |
| Coleopterans, Dipterans & Dictyopterans | Pesticide resistance | Amino acid substitution | ffrench-Constant, 1994 |
| Arabidopsis thaliana | Flowering time | Deletion | Johanson et al., 2001 |
| Wild Mice spp. | Immune response | Amino acid substitution | Jouvin-Marche et al., 1988 |
| Human & Non-Human Primates | Blood groups | Nucleotide substitution | Kermarrec et al., 1999 |
| Human & Old/New World Monkeys | Immune response | Nucleotide substitution | Kriener, 2000 |
| Escherichia coli | Drug resistance | Nucleotide substitution | Low et al., 2001 |
| Potato Virus X | Virulence | See Appendix 1 | Malcuit et al., 2000 |
| Human Immunodeficiency Virus (HIV) | Drug resistance | Amino acid substitution | Molla et al., 1996 |
| Primates & Squid | Visual pigments | Amino acid substitution | Morris et al., 1993 |
| Chimpanzee & Gorilla | Blood groups | Amino acid substitution | O’h Uigin et al., 1997 |
| Human & Sooty Mangabey | Disease resistance | Deletion | Palacios et al., 1998 |
| Escherichia coli | Virulence | Horizontal transfer | Reid et al., 2000 |
| Escherichia coli | Thermotolerance | Duplication/deletion | Riehle et al., 2001 |
| Cetaceans & Pinnipeds | Respiration | Amino acid substitution | Romero-Herrera et al., 1978 |
| Human & Pea | Enzyme function | Amino acid substitution | Shafqat et al., 1996 |
| Human, Marmoset & Squirrel Monkey | Visual pigments | Amino acid substitution | Shyue et al., 1995 |
| Colobine Monkey, Ruminants & Hoatzin | Enzyme function | Amino acid substitution | Stewart et al., 1987; Zhang and Kumar, 1997 |
| Human & Blind Cave Fish | Visual pigments | Amino acid substitution | Yokoyama and Yokoyama, 1990 |
| Cowpea & Mung Bean | Seed weight | Comparative QTL mapping | Fatokun et al., 1992 |
| Maize, Rice & Sorghum | Seed mass and dispersal | Comparative QTL mapping | Paterson et al., 1995 |
| Silene vulgaris | Metal tolerance | Complementation test | Schat et al., 1996 |
Although our understanding of the molecular basis of selection response in higher organisms is incomplete, several studies in Table 1 document parallel evolution in eukaryotes. The best experimental evidence comes from a comparison of resistance to acetolactate synthase inhibitors in naturally occurring cocklebur and two mutagenized maize lines (Bernasconi et al., 1995). Given that resistance in this case is based on a single enzyme, this result may not be predictive of the types of changes that underlie parallel phenotypic evolution in more complex traits. While there are very few studies that bear on this issue, Ungerer (2000) found that the frequency of QTL alleles governing life history traits responded uniformly to viability selection in replicate Arabidopsis populations, even when genetic background was varied. Similarly, working in sunflower, Rieseberg et al. (1996) showed that experimental hybrid lineages subjected to strong fertility selection converged on a common genomic composition. Because this fertility selection was primarily the result of selection for the recovery of viable gametes in interspecific hybrids, the underlying adaptive process is mechanistically distinct from classical examples of adaptation involving allelic substitution at a targeted locus. However, this study clearly demonstrates that parallel selection among lineages can yield remarkably similar genotypic responses. One weakness of conclusions drawn from these two studies is that they did not provide the necessary resolution to conclude that selection is acting on variation at homologous loci across populations. In addition, both of these studies relied on variation generated in crosses between different lineages, rather than on novel variation. They do, however, show that selection response at the genotypic level is repeatable across populations. Thus, given the appropriate genetic variation, we might expect the evolution of complex traits to mirror the findings from genetically simpler traits.
Phylogenetic studies
While experimental studies allow researchers to control the branching pattern of lineages and monitor their response to selection, parallel genotypic adaptation can be assessed in non-experimental systems as well. One approach is to use phylogenetic methods to infer the evolutionary history of the organisms of interest. This phylogeny can then be used to reconstruct the historical sequence of mutational changes in a nucleotide or protein sequence with known function. The advantage of this approach is that it can be applied to virtually any organism; thus, parallel evolution can be studied across vast taxonomic distances and in organisms that are not amenable to experimental manipulation. The main difficulty is that, in order to show that homoplasy is adaptive in origin rather than the result of chance fixation, the functional effects of a sequence change must be known, or at least inferred (Doolittle, 1994).
Once a relationship between genotype and phenotype has been established, the basic challenge is to demonstrate that shared sequence similarities are not simply the result of common ancestry. Because sequences that have evolved in parallel will show phylogenetic affinity, the detection of parallel genotypic adaptation can be problematic. Of course, if the adaptive change results from relatively few nucleotide substitutions, homoplasy may have only minor effects on phylogenetic inference. In other cases, where the ratio of informative sites to selectively advantageous substitutions is relatively low, the framework for these analyses should be based on independent phylogenetic data. Assuming that the structure of the resulting tree represents the true evolutionary history of the organisms, detecting homoplasy is as simple as mapping character states onto this tree (Figure 1). The phylogenetic approach can also be used within taxa to examine the pattern of evolution of a gene in a geographic context. For example, Andreev et al. (1999) used a phylogeny of alleles of Resistance to dieldrin to demonstrate that the same point mutation arose on multiple occasions in different populations of the red flour beetle, Tribolium castaneum
The middle panel of Table 1 lists examples of parallel genotypic adaptation documented with phylogenetic methods. Although this set of studies includes examples from microorganisms, the taxonomic diversity represented clearly demonstrates that parallel genotypic adaptation occurs at all taxonomic levels. Once again, many of these examples involve minor sequence changes. In fact, parallel adaptation in four of these studies was based on a single amino acid substitution (Morris, Bowmaker & Hunt, 1993; Elard, Comes & Humbert, 1996; ffrench-Constant, 1996; Andreev et al., 1999).
While many of the traits listed would generally be viewed as complex, what the studies in Table 1 say about parallel evolution in simple versus complex traits is unclear. Part of the problem here stems from the definition of traits. For example, the spectral properties of visual pigments represent one aspect of color vision, which is clearly a complex trait (Yokoyama & Yokoyama, 1990; Morris, Bowmaker & Hunt, 1993; Shyue et al., 1995). Thus, parallel evolution of the genes encoding these pigments could be viewed as the parallel evolution of a highly complex trait. If, on the other hand, the trait is defined to be spectral tuning, then the trait of interest is Mendelian, no different from herbicide resistance in cocklebur and maize. The difficulty here lies in the fact that, from an evolutionary perspective, traits should be defined by what selection sees, not what the researcher sees. For example, if selection acts to increase the height of a hypothetical organism, parallel genotypic responses may be less likely than if selection acts on a specific component of height, such as cell number or cell size.
A number of the studies included in this section demonstrate sequence homoplasy for loci that have a known adaptive function, but the parallel changes themselves have not been demonstrated to be under selection. Thus, although an adaptive role for these changes is plausible, their functional significance has not been directly assessed (e.g., Romero-Herrera et al., 1978; Jouvin-Marche et al., 1988). Moreover, only two of the examples in this section (Stewart, Schilling & Wilson, 1987; Zhang & Kumar, 1997; Kriener, 2000) have been evaluated statistically. Unfortunately, the statistical model used to evaluate the role of selection in parallel sequence changes (Zhang & Kumar, 1997) is, out of necessity, naive to protein function. Because it uses a general evolutionary model to ascribe probabilities to changes between sequence states, this approach can lead to false positives. For example, if a given amino acid site is constrained on the basis of charge, it is free to evolve, but in a more limited number of ways. Therefore, the number of possible states can be far fewer than the model allows. In such cases, the test will be biased toward detecting significant parallelisms even though the changes may have occurred by chance. Ultimately, sequence changes need to be linked to a change in function to demonstrate unequivocally parallel genotypic adaptation.
Quantitative genetic studies
Another approach to detecting parallel genotypic adaptation in non-experimental systems involves quantitative genetic analysis. The most direct method is a complementation test, in which two lineages are crossed and the segregation patterns of their hybrid offspring are analyzed. If a shared, yet independently derived character state has a common genotypic basis, it will not segregate in the second (or later) generation(s). In contrast, if the character is determined by non-homologous loci, the hybrid progeny should exhibit significant phenotypic variation. An example of this approach is the work of Schat, Voour & Kuiper, (1996; Table 1), who demonstrated that metal tolerance in genetically isolated populations of Silene results from changes at homologous loci.
Comparative QTL mapping can also yield evidence for parallel genotypic responses. In this case, molecular markers are used to identify chromsomal regions underlying the trait(s) of interest in a segregating population (see Mauricio, 2001 for a review). In cases where homologous markers are shared across mapping populations, QTL positions can be compared between taxa. When QTLs map to the same marker intervals, the results are consistent with parallel genotypic adaptation. Although QTL methods have been applied to a wide variety of study organisms, there are only three good examples of parallel adaptation identified through this approach (Fatokun et al., 1992; Paterson et al., 1995; Hu et al., 2003; Table 1).
In all three of these cases, it is important to note that the effects of closely linked, but non-homologous loci cannot be discounted. Thus, like the map-based studies of Ungerer (2000) and Rieseberg et al. (1996) detailed above, conclusions regarding homology of the changes are premature. In addition, all three of the studies focus on domestication traits. Like the examples listed under experimental evolution above, these traits have evolved in response to strong artificial selection. Because artificially selected lineages are generally maintained in a controlled environment (e.g., lab, greenhouse, or agricultural setting), they are not necessarily subject to the same pleiotropic constraints as naturally evolving populations. Therefore, the relevance of these studies to the evolution of traits in the wild is tenuous (Coyne & Lande, 1985).
Evolutionary implications
Each of the studies reviewed here provides at least circumstantial evidence that parallel genotypic adaptation occurs at all taxonomic levels. This finding stands in stark contrast to the traditional view that parallel phenotypic evolution results from unique genetic changes. Given that a number of the traits listed above are simple (i.e., Mendelian), this result should not be surprising. After all, if a trait is controlled by a single gene, phenotypic evolution can involve changes in only that gene. As the complexity of an adaptation increases, the likelihood of its parallel recurrence should decrease. In other words, if there are numerous pathways connecting two phenotypic states, it is relatively unlikely that evolution will follow the same path twice. As stated above, however, there is a growing body of evidence that many quantitative traits are controlled oligogenically (Tanksley, 1993). In addition, apparently complex traits can often be decomposed into their component parts (e.g., color vision versus visual pigments; Morris, Bowmaker & Hunt, 1993; Shyue et al., 1995; Yokoyama & Yokoyama, 1990). If selection acts on these parts, rather than on their sum, the number of potential pathways will be fewer, which makes parallel genotypic adaptation even more likely. Finally, if the genetic variance–covariance matrices are similar across populations or taxa, then populations may be predisposed to adaptation along the path of least resistance, thereby leading to parallel genotypic adaptation (Endler, 1986; Schluter, 1996).
From a practical standpoint, perhaps one of the greatest concerns regarding homoplasy is the confounding effect it can have on phylogeny reconstruction. Because phylogenetic algorithms are designed to minimize homoplasy, shared character states that truly arose multiple times may be grouped together erroneously (Forey et al., 1992). However, a number of the studies reviewed here suggest that selection often targets only one or a few sites in a sequence (e.g., Andreev et al., 1999). Thus, even if a gene responds identically to selective pressures in evolutionarily distinct lineages, the majority of the sequence will track the branching patterns of the taxa. That is, if the selectively important changes are rare relative to the number of phylogenetically informative sites, the gene tree may still track the species tree. On the other hand, if the sequence changes represent a larger proportion of the informative sites, the resulting tree may be incongruent with the true phylogeny. For example, Kriener et al. (2000) examined sequence variation in certain alleles of the DRB gene family in monkeys and humans. Similarities among coding sequences were strong enough to cause a conflict between the exon-based tree and true organismal relationships. Because systematists are increasingly using multiple gene sequences to reconstruct phylogenies, these sorts of conflicts are less likely to lead to incorrect phylogenetic inferences.
From an evolutionary perspective, the occurrence of parallel genotypic adaptation suggests that adaptive evolution may be a more deterministic process than previously believed. Although some authors have argued that the most likely outcome of parallel selection in isolated populations is divergence (e.g., Wade & Goodnight, 1998; Goodnight, 2000; Levin, 2000), two studies in particular suggest that selection response at the genotypic level is repeatable across populations (Rieseberg et al., 1996; Ungerer, 2000). These studies, therefore, suggest that the effects of genetic background on selection response may have been overemphasized. If this turns out to be generally true, then parallel genotypic adaptation might provide a mechanism for both the collective evolution of populations within a species (Lande, 1983; Templeton, 1989) and the recurrent origin of taxa (reviewed in Levin, 2001).
Classical studies of gene flow have suggested that migration rates are too low to account for the apparent integration of species across their ranges (e.g., Ehrlich & Raven, 1969; Grant, 1980). If this were true, species would not be different from higher taxa, mere aggregates of the actual units of evolution (local populations or metapopulations). Recent work has revealed that the joint effects of selection and migration are, in general, sufficient to account for the integration of populations across a species range (Rieseberg & Burke, 2000). The studies reviewed above take this idea further, suggesting that local populations of a species subjected to similar selective pressures may arrive at the same genetical solutions. Another type of evidence supporting this idea comes from experimental selection studies in which populations subjected to parallel selection maintained reproductive compatibility, whereas those subjected to divergent selection often evolved incompatibilities (Rice & Hostert, 1993). The importance of parallel genotypic adaptation in species cohesion will vary with the relative rates of mutation and migration; in cases where gene flow is limiting, parallel genotypic adaptation would be expected to play a more central role. In this context, it is interesting to note that many of the characters used to differentiate plant species are governed by one or two genes (Gottlieb, 1984; Hilu, 1983). Thus, traits used in species identification may be especially likely to evolve in parallel.
Just as parallel genotypic adaptation can help maintain species cohesion, the potential for recurrent evolution of key adaptations makes the repeated origin of taxa plausible. In general terms, this evolutionary process could allow local populations to independently invade a similar habitat. Because these lineages would share a common solution to a unique ecological challenge, they would be demographically exchangeable (sensu Templeton, 1989) for the same genetic reasons. Indeed, more and more evolutionary biologists are recognizing the importance of ecology in speciation (Schluter, 2001). Because different habitat types are often interspersed across the range of a species, the requisite ecological opportunities may occur frequently. An example of this process, albeit at the infraspecific level, would be metal tolerance in Silene (Schat, Voour & Kuiper 1996; Table 1). Given enough time, these independently derived populations may ascend to species status. Though not yet characterized genetically, threespine stickleback fishes are another possible example of recurrent divergence due to parallel genotypic adaptation (Rundle et al., 2000).
Taken together, the studies reviewed here provide evidence that parallel genotypic adaptation can occur in organisms ranging from microbes to plants to primates. Although the relevance of studies in microorganisms to adaptation in general has been questioned, this body of data suggests that Jacques Monod may have been right when he suggested that ‘What is true for E. coli is true for elephants, only more so.’ In some cases, the parallelisms spanned remarkably wide taxonomic distances – e.g., the independent evolution of ethanol-active ADH in pea plants and humans (Shafqat et al., 1996). Given that the genetic basis of most adaptations is still unknown, our understanding of the prevalence of parallel genotypic adaptation is still in its infancy. The advent of functional genomics should lead to a wealth of data connecting genotype to phenotype, allowing researchers to identify and compare the genetic mechanisms underlying adaptive traits in a variety of organisms.
Acknowledgments
We thank Jeff Demuth and Mark Welch for useful discussions of various aspects of this work. Our research on the genetics of adaptive evolution has been supported by grants from the National Institutes of Health, National Science Foundation, and United States Department of Agriculture.
Appendix 1
Brief summaries of studies
|
References
- Andreev D, Kreitman M, Phillips TW, Beeman RW, ffrench-Constant RH. Multiple origins of cyclodiene insecticide resistance in Tribolium castaneum (Coleoptera: Tenbrionidae) J Mol Evol. 1999;48:615–624. doi: 10.1007/pl00006504. [DOI] [PubMed] [Google Scholar]
- Barlow M, Hall BG. Predicting evolutionary potential: in vitro evolution accurately reproduces natural evolution of the TEM β-lactamase. Genetics. 2002;160:823–832. doi: 10.1093/genetics/160.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow M, Hall BG. Experimental prediction of the natural evolution of antibiotic resistance. Genetics. 2003;163:1237–1241. doi: 10.1093/genetics/163.4.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi P, Woodworth AR, Rosen BA, Subramanian MV, Siehl DL. A naturally occurring point mutation confers broad range tolerance to herbicides that target acetolactate synthase. J Biol Chem. 1995;270:17381–17385. doi: 10.1074/jbc.270.29.17381. [DOI] [PubMed] [Google Scholar]
- Brown EG, Liu H, Chang Kit L, Baird S, Nesrallah M. Pattern of mutation in the genome of influenza A virus on adaptation to increased virulence in the mouse lung: identification of functional themes. Proc Natl Acad Sci USA. 2001;98:6883–6888. doi: 10.1073/pnas.111165798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ, Badgett MR, Wichman HA, Huelsenbeck JP, Hillis DM, Gulati A, Ho C, Molineux IJ. Exceptional convergent evolution in a virus. Genetics. 1997;147:1497–1507. doi: 10.1093/genetics/147.4.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, DeVries AL, Cheng CHC. Convergent evolution of antifreeze glycoproteins in Antarctic notothenioid fish & Arctic cod. Proc Natl Acad Sci USA. 1997;94:3817–3822. doi: 10.1073/pnas.94.8.3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA, Lande R. The genetic basis of species differences in plants. Am Nat. 1985;126:141–145. [Google Scholar]
- Crill WD, Wichman HA, Bull JJ. Evolutionary reversals during viral adaptation to alternating hosts. Genetics. 2000;154:27–37. doi: 10.1093/genetics/154.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CW, Jeng K, Husti J, Badgett M, Molineux IJ, Hillis DM, Bull JJ. Parallel molecular evolution of deletions & nonsense mutations in bacteriophage T7. Mol Biol Evol. 1997;14:113–116. doi: 10.1093/oxfordjournals.molbev.a025697. [DOI] [PubMed] [Google Scholar]
- Doolittle RF. Convergent evolution: the need to be explicit. Trends Biochem Sci. 1994;19:15–18. doi: 10.1016/0968-0004(94)90167-8. [DOI] [PubMed] [Google Scholar]
- Downie SR, Palmer JD. Use of chloroplast DNA rearrangements in reconstructing plant phylogeny. In: Soltis PS, Soltis DE, Doyle JJ, editors. Molecular Systematics of Plants. Chapman & Hall; N.Y.: 1992. pp. 14–35. [Google Scholar]
- Elard L, Comes AM, Humbert JF. Sequences of B-tubulin cDNA from benzimidazole-susceptible and -resistant strains of Teladorsagia cirumcincta, a nematode parasite of small ruminants. Mol Biochem Parasitol. 1996;79:249–253. doi: 10.1016/0166-6851(96)02664-3. [DOI] [PubMed] [Google Scholar]
- Endler JA. Natural Seleciton in the Wild. Princeton University Press; Princeton: 1986. [Google Scholar]
- Ehrlich PR, Raven PH. Differentiation of populations. Science. 1969;165:1228–1232. doi: 10.1126/science.165.3899.1228. [DOI] [PubMed] [Google Scholar]
- Fatokun CA, Menancio-Hautea DI, Danesh D, Young ND. Evidence for orthologous seed weight genes in cowpea & mung bean based on RFLP mapping. Genetics. 1992;132:841–846. doi: 10.1093/genetics/132.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ffrench-Constant RH. The molecular & population genetics of cyclodiene insecticide resistance. Insect Bicochem Mol Biol. 1994;24:335–345. doi: 10.1016/0965-1748(94)90026-4. [DOI] [PubMed] [Google Scholar]
- Forey PL, Humphries CJ, Kitching IL, Scotland RW, Siebert DJ, Williams DM. Cladistics. Oxford University Press; NY: 1992. [Google Scholar]
- Goodnight CJ. Quantitative trait loci & gene interaction: the quantitative genetics of metapopulations. Heredity. 2000;84:587–598. doi: 10.1046/j.1365-2540.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- Gottlieb LD. Genetics & morphological evolution in plants. Am Nat. 1984;123:681–709. [Google Scholar]
- Grant V. Gene flow and the homogeneity of species populations. Biol Zentralbl. 1980;99:157–169. [Google Scholar]
- Haldane JBS. The Causes of Evolution. Longmans Green; London: 1932. [Google Scholar]
- Hilu KW. The role of single-gene mutations in the evolution of flowering plants. Evol Biol. 1983;36:97–128. [Google Scholar]
- Hu FY, Tao DY, Sacks E, Fu BY, Xu P, Li J, Yang Y, McNally K, Khush GS, Paterson AH, Li ZK. Convergent evolution of perenniality in rice & sorghum. Proc Natl Acad Sci USA. 2003;100:4050–4054. doi: 10.1073/pnas.0630531100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson U, West J, Lister C, Michaels S, Amasino R, Dean C. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science. 2001;290:344–347. doi: 10.1126/science.290.5490.344. [DOI] [PubMed] [Google Scholar]
- Jouvin-Marche E, Cuddihy A, Butler S, Hansen JN, Fitch WM, Rudikoff S. Modern Evolution of a single-copy gene: the immunoglobulin Ck locus in wild mice. Mol Biol Evol. 1988;5:500–511. doi: 10.1093/oxfordjournals.molbev.a040514. [DOI] [PubMed] [Google Scholar]
- Kermarrec N, Roubinet F, Apoil PA, Blancher A. Comparison of allele O sequences of the human & non-human primate ABO system. Immunogenetics. 1999;49:517–526. doi: 10.1007/s002510050529. [DOI] [PubMed] [Google Scholar]
- Kriener K, O’hUigin C, Tichy H, Klein J. Convergent evolution of major histocompatibility complex molecules in humans & New World monkeys. Immunogenetics. 2000;51:169–178. doi: 10.1007/s002510050028. [DOI] [PubMed] [Google Scholar]
- Lande R. The response to selection on major & minor mutations affecting a metrical trait. Heredity. 1983;50:47–65. [Google Scholar]
- Levin BR, Perrot V, Walker N. Compensatory mutations, antibiotic resistance & the population genetics of adaptive evolution in bacteria. Genetics. 2000;154:985–997. doi: 10.1093/genetics/154.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin DA. The Origin, Expansion, & Demise of Plant Species. Oxford University Press; NY: 2000. [Google Scholar]
- Levin DA. The recurrent origin of plant races & species. Syst Bot. 2001;26:197–204. [Google Scholar]
- Liao H, McKenzie T, Hageman R. Isolation of a thermostable enzyme variant by cloning & selection in a thermophile. Proc Natl Acad Sci USA. 1986;83:576–580. doi: 10.1073/pnas.83.3.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low AS, MacKenzie FM, Gould IM, Booth IR. Protected environments allow parallel evolution of a bacterial pathogen in a patient subjected to long-term antibiotic therapy. Mol Microbiol. 2001;42:619–630. doi: 10.1046/j.1365-2958.2001.02647.x. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate & consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Malcuit I, De Jong W, Baulcombe DC, Shields DC, Kavanagh TA. Acquisition of multiple virulence/avirulence determinants by potato virus X (PVX) has occurred through convergent evolution rather than through recombination. Virus Genes. 2000;20:165–172. doi: 10.1023/a:1008178800366. [DOI] [PubMed] [Google Scholar]
- Mauricio R. Mapping quantitative trait loci in plants: uses & caveats for evolutionary biology. Nat Rev Genet. 2001;2:370–381. doi: 10.1038/35072085. [DOI] [PubMed] [Google Scholar]
- Molla A, Korneyeva M, Gao Q, Vasavanonda S, Schipper PJ, et al. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat Med. 1996;2:760–766. doi: 10.1038/nm0796-760. [DOI] [PubMed] [Google Scholar]
- Morris A, Bowmaker JK, Hunt DM. The molecular basis of a spectral shift in the rhodopsins of two species of squid from different photic environments. Proc R Soc Lond Ser B Biol Sci. 1993;254:233–240. doi: 10.1098/rspb.1993.0151. [DOI] [PubMed] [Google Scholar]
- Ohno S. Evolution by Gene Duplication. Springer; Berlin: 1970. [Google Scholar]
- O’hUigin C, Sato A, Klein J. Evidence for convergent evolution of A & B blood group antigens in primates. Hum Genet. 1997;101:141–148. doi: 10.1007/s004390050603. [DOI] [PubMed] [Google Scholar]
- Palacios E, Digilio L, McClure HM, Chen Z, Marx PA, Goldsmith MA, Grant RM. Parallel evolution of CCR5-null phenotypes in humans & in a natural host of simian immunodeficiency virus. Curr Biol. 1998;8:943–946. doi: 10.1016/s0960-9822(07)00378-8. [DOI] [PubMed] [Google Scholar]
- Paterson AH, Lin YR, Li Z, Schertz KF, Doebley JF, Pinson SRM, Liu SC, Stansel JW, Irivine JE. Convergent domestication of cereal crops by independent mutations at corresponding genetic loci. Science. 1995;269:1714–1718. doi: 10.1126/science.269.5231.1714. [DOI] [PubMed] [Google Scholar]
- Reid SD, Herbelin CJ, Bumbaugh AC, Selander RK, Whittam TS. Parallel evolution of virulence in pathogenic Escherichia coli. Nature. 2000;406:64–67. doi: 10.1038/35017546. [DOI] [PubMed] [Google Scholar]
- Rice WR, Hostert EE. Laboratory experiments on speciation: what have we learned in 40 years? Evolution. 1993;47:1637–1653. doi: 10.1111/j.1558-5646.1993.tb01257.x. [DOI] [PubMed] [Google Scholar]
- Riehle MM, Bennett AF, Long AD. Genetic architecture of thermal adaptation in Escherichia coli. Proc Natl Acad Sci USA. 2001;98:525–530. doi: 10.1073/pnas.021448998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH, Sinervo B, Linder CR, Ungerer MC, Arias DM. Role of gene interactions in hybrid speciation: evidence from ancient & experimental hybrids. Science. 1996;272:741–745. doi: 10.1126/science.272.5262.741. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Burke JM. The biological reality of species: gene flow, selection, & collective evolution. Taxon. 2001;50:47–67. [Google Scholar]
- Romero-Herrera AE, Lehmann H, Josey KA, Friday AE. On the evolution of myoglobin. Philos Trans R Soc Lond Ser B Biol Sci. 1978;283:61–163. doi: 10.1098/rstb.1978.0018. [DOI] [PubMed] [Google Scholar]
- Rundle HD, Nagel L, Boughman JW, Schluter D. Natural selection & parallel speciation in sympatric sticklebacks. Scinece. 2000;287:306–308. doi: 10.1126/science.287.5451.306. [DOI] [PubMed] [Google Scholar]
- Saitou N, Yamamoto F. Evolution of primate ABO blood group genes & their homologous genes. Mol Biol Evol. 1997;14:399–411. doi: 10.1093/oxfordjournals.molbev.a025776. [DOI] [PubMed] [Google Scholar]
- Schat H, Voous R, Kuiper E. Identical major gene loci for heavy metal tolerances that have independently evolved in different local populations & subspecies of Silene vulagaris. Evolution. 1996;50:1888–1895. doi: 10.1111/j.1558-5646.1996.tb03576.x. [DOI] [PubMed] [Google Scholar]
- Schluter D. Adaptive radiation along genetic lines of least resistance. Evolution. 1996;50:1766–1774. doi: 10.1111/j.1558-5646.1996.tb03563.x. [DOI] [PubMed] [Google Scholar]
- Schluter D. Ecology & the origin of species. Trends Ecol Evol. 2001;16:372–380. doi: 10.1016/s0169-5347(01)02198-x. [DOI] [PubMed] [Google Scholar]
- Shafqat J, El-Ahmad M, Danielsson O, Martinez MC, Persson B, Pares X, Jornvall H. Pea formaldehyde-active class III alcohol dehydrogenase: common derivation of the plant & animal forms but not of the corresponding ethanol-active forms (classes I & P) Proc Natl Acad Sci USA. 1996;93:5595–5599. doi: 10.1073/pnas.93.11.5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyue SK, Hewett-Emmett D, Sperling HG, Hunt DM, Bowmaker JK, Mollon JD, Li WH. Adaptive evolution of color vision genes in higher primates. Science. 1995;269:1265–1267. doi: 10.1126/science.7652574. [DOI] [PubMed] [Google Scholar]
- Simpson GG. Principles of Animal Taxonomy. New York: Columbia University Press; 1961. [DOI] [PubMed] [Google Scholar]
- Soltis PS, Soltis DS. Multiple origins of the allotetraploid Tragopogon mirus (Compositae): rDNA evidence. Syst Bot. 1991;16:407–413. [Google Scholar]
- Stewart CB, Schilling JW, Wilson AC. Adaptive evolution in the stomach lysozymes of foregut fermenters. Nature. 1987;330:401–404. doi: 10.1038/330401a0. [DOI] [PubMed] [Google Scholar]
- Sucena E, Delon I, Jones I, Payre F, Stern DL. Regulatory evolution of shavenbaby/ovo underlies multiple cases of morphological parallelism. Nature. 2003;424:935–938. doi: 10.1038/nature01768. [DOI] [PubMed] [Google Scholar]
- Tanksley SD. Mapping polygenes. Annual Rev Genet. 1993;27:205–233. doi: 10.1146/annurev.ge.27.120193.001225. [DOI] [PubMed] [Google Scholar]
- Templeton AR. The meaning of species & speciation: a genetic perspective. In: Otte D, Endler JA, editors. Speciation & Its Consequences. Sunderland, MA: 1989. pp. 3–27. [Google Scholar]
- Ungerer M. PhD dissert. Indiana Univ.; Bloomington: 2000. Selection, genetic environment & adaptive evolution: an analysis of microevolutionary dynamics in experimental plant populations. [Google Scholar]
- Wade MJ, Goodnight CJ. Perspective: the theories of Fisher & Wright in the context of metapopulations: when nature does many small experiments. Evolution. 1998;52:1537–1553. doi: 10.1111/j.1558-5646.1998.tb02235.x. [DOI] [PubMed] [Google Scholar]
- Wells RS. Excessive homoplasy in an evolutionarily constrained protein. Proc R Soc Lond Ser B Biol Sci. 1996;263:393–400. doi: 10.1098/rspb.1996.0060. [DOI] [PubMed] [Google Scholar]
- Wichman HA, Badgett MR, Scott LA, Boulianne CM, Bull JJ. Different trajectories of parallel evolution during viral adaptation. Science. 2001;285:422–424. doi: 10.1126/science.285.5426.422. [DOI] [PubMed] [Google Scholar]
- Yokoyama R, Yokoyama S. Convergent evolution of the red- & green-like visual pigment genes in fish, Astyanax fasciatus & human. Proc Natl Acad Sci USA. 1990;87:9315–9318. doi: 10.1073/pnas.87.23.9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kumar S. Detection of convergent & parallel evolution at the amino acid sequence level. Mol Biol Evol. 1997;14:527–536. doi: 10.1093/oxfordjournals.molbev.a025789. [DOI] [PubMed] [Google Scholar]