

Abstract
Homoploid hybrid speciation has been recognized for its potential rapid completion, an idea that has received support from experimental and modeling studies. Following initial hybridization, the genomes of parental species recombine and junctions between chromosomal blocks of different parental origin leave a record of recombination and the time period before homogenization of the derived genome. We use detailed genetic maps of three hybrid species of sunflowers and models to estimate the time required for the stabilization of the new hybrid genome. In contrast to previous estimates of 60 or fewer generations, we find that the genomes of three hybrid sunflower species were not stabilized for hundreds of generations. These results are reconciled with previous research by recognizing that the stabilization of a hybrid species’ genome is not synonymous with hybrid speciation. Segregating factors that contribute to initial ecological or intrinsic genetic isolation may become stabilized quickly. The remainder of the genome likely becomes stabilized over a longer time interval, with recombination and drift dictating the contributions of the parental genomes. Our modeling of genome stabilization provides an upper bound for the time interval for reproductive isolation to be established and confirms the rapid nature of homoploid hybrid speciation.
Keywords: Admixture, Helianthus, hybrid speciation, junction theory, linkage disequilibrium, recombination
Homoploid hybrid speciation is the process by which an independent lineage arises through hybridization and the combination of parental genomes, but without an increase in ploidy. This form of speciation has been hypothesized for the origin of various species and has been confirmed in a small number of plant and animal taxa (Rieseberg 1997; Gross and Rieseberg 2005), including recent studies in plants (Howarth and Baum 2005; Mir et al. 2006), insects (Schwarz et al. 2005; Gompert et al. 2006; Mavárez et al. 2006), and fish (Meyer et al. 2006). Hybrid speciation is thought to have the potential to be achieved in a relatively short period of time. This is because the derived lineage may become isolated rapidly from gene flow from parental taxa by the invasion of a novel ecological setting and the generation of novel combinations of parental incompatibility factors (e.g., chromosomal rearrangements). A few generations of recombination of genes or chromosomal regions that contribute to isolation may be sufficient for the origin of a lineage with high fitness and reproductive isolation, and that may then increase in abundance (Buerkle et al. 2000). Given that our understanding of the process of homoploid hybrid speciation predicts that it has the potential to be rapid, it is desirable to test whether these predictions hold in species of known hybrid origin. Estimates of the rate of hybrid speciation may lead to refinement of our understanding of the process, and allow contrasts with the relatively slow accumulation of isolating barriers in some other modes of speciation.
Three diploid species of sunflowers are known to have resulted from hybridization between the same two parental species, Helianthus annuus and H. petiolaris (Rieseberg 1991, 1997). The three hybrid species (H. anomalus, H. deserticola, and H. paradoxus) provide a basis for independent tests of the prediction of rapid speciation. An earlier study of the rate of speciation in H. anomalus estimated that the hybrid species likely arose in fewer than 60 generations (Ungerer et al. 1998). There were three reasons to revisit the question of the rate of speciation in Helianthus: (1) relevant genetic data are now available for all three hybrid species in the genus (independent speciation events and tests of predictions), (2) the genetic map now available for H. anomalus is of much higher resolution than the map used in Ungerer et al. (1998), and (3) we wanted to refine aspects of the modeling and analysis of the genetic data.
One measure of the rate of homoploid hybrid speciation is the amount of time that passed between the initial hybridization and the time at which the genomic composition of the derived species became stabilized. For some time after hybridization and the establishment of an isolated lineage, the derived population will be segregating for genomic contributions of the parental species. During this period, recombination in portions of the genome that are not identical-by-descent will decay the size of parental chromosomal blocks. The genetic maps for each of the hybrid species of sunflowers give estimates for the sizes of, and number of transitions between, chromosomal blocks inherited from each of the parental species. With these maps and appropriate theory for the relationship between time and size of chromosomal blocks, we can estimate the amount of time that passed during the transitional period between initial hybridization and stabilization of the genomes of the three hybrid sunflower species.
The time interval for genome stabilization is of interest because it is a function of the rate at which reproductive isolation of the derived lineage was established. Lack of isolation of a hybrid lineage would inhibit genome stabilization as parental chromosomal blocks would be continually introduced. In the origin of a homoploid hybrid species, after reproductive isolation is established, genome stabilization would proceed for some additional time. Therefore any estimate of the time frame for genome stabilization sets an upper bound for the time interval for reproductive isolation to be established.
Theory of Junctions
DETERMINISTIC MODEL
In the process of hybrid speciation, hybrid individuals give rise to a novel lineage that is largely isolated from the parental species. Recurrent backcrossing would inhibit the establishment of an independent lineage (Buerkle et al. 2000). Thus, we assume that the hybrid lineage is equivalent to an isolated, founding population and apply theory for recombination between parental chromosomes and the generation of junctions.
Fisher (1954) made the initial observation that the full genetic makeup of an individual can be described on the basis of junctions between chromosomal regions of different ancestry. In this approach, the ancestry of chromosomal blocks is tracked, rather than the parental origin of alleles at individual loci. In the case of a founding population of hybrids, each chromosome is derived from the parental species and recombination in heterozygous individuals will reduce the size of chromosomal blocks. Fisher (1954) referred to the points at which chromosomal blocks of different ancestry meet as junctions. Junction formation depends on the presence of heterozygous individuals in the population and on recombination in heterozygous chromosomal regions (Barton 1983; Baird 1995). Thus, as heterozygosity declines, the rate of introduction of junctions will decline.
This theoretical relationship between heterozygosity and the expected number of junctions (E[J]) in a given generation (t) has been analyzed fully (Stam 1980; Chapman and Thompson 2002, 2003; MacLeod et al. 2005) and is simple
| (1) |
for heterozygosity H, and map length of L Morgans (eq. 5 in Chapman and Thompson 2002). For a randomly mating population, the heterozygosity remaining in a given generation is a function of population size (N) and the heterozygosity of the founding population (H 0; H 0 = 0.5 herein), (Crow and Kimura 1970). After some rearrangement of terms, a simple, deterministic expression for the expected number of junctions is 2N (H 0 − Ht)L , which will approach a limit of 2NH 0 L as heterozygosity (Ht) goes to zero (MacLeod et al. 2005). Similarly, for an obligately outcrossing species, such as the sunflowers considered here, the limit is given by J∞ = 2(N + 1)L, when H 0 = 1 (Stam 1980; MacLeod et al. 2005). Thus, restricting the mating to outcrossing introduces just one junction per Morgan relative to the random mating model and the predicted difference will be small for N of interest (N > 20, see below). Finally, the number of generations over which heterozygosity declines from H 0 to Ht is given by
An analytical approximation of the variance of the expected number of junctions has been derived for a randomly mating population (Chapman and Thompson 2002). The approximation is complicated to calculate and was shown to be an underestimate of the variance observed in simulations of a randomly mating population (Chapman and Thompson 2002). Additionally, we require an estimate of the variance for an obligately outcrossing system, which is expected to have a slightly higher variance, so our estimates of dispersion come from simulation.
SIMULATION MODEL
As an independent verification of the above analytical models and as a means to obtain variance estimates for the expected number of junctions, we used a simulation model of the process of junction formation in finite populations. Additionally, the simulation model allows the assessment of how selection (underdominance) may affect the rate of junction formation.
In the simulation two parental species were mixed in equal proportions to form an ancestral, admixed population of fixed size. The parental species were assumed to be highly diverged, such that chromosomal blocks can be assigned to the parental species of origin and initial heterozygosity (H 0) is 0.5. As in similar simulation models (Baird 1995; Ungerer et al. 1998; Chapman and Thompson 2002; MacLeod et al. 2005), in each generation mating occurred by randomly choosing two parents and forming gametes (obligate, random outcrossing). The simulation was of hermaphroditic, annual organisms, with nonoverlapping generations. Recombination was modeled as a Poisson process, with a Poisson distribution for the number of crossovers and a mean of one crossover per chromosome pair (i.e., each chromosome is assumed to be 1 Morgan in length). The location of crossovers was uniformly distributed and there was no interference. Junctions were tracked for 17 homologous chromosome pairs in each individual to match the Helianthus species of interest. Modeling multiple chromosome pairs is not necessary to estimate the number of junctions, because the estimate could simply be scaled by some factor for map length (see L above). However, the sampling variance for the number of junctions within a genome will be appropriately reduced because chromosomes will have independent histories of recombination. The computer model was written in C.
The simple approach to modeling population size (constant) and structure (equal admixture of parental species) is obviously an abstraction. It was also used in an earlier analysis of the rate of speciation in Helianthus (Ungerer et al. 1998), which facilitates comparisons with the earlier results. Because new junctions can be formed only in regions of the genome that are heterozygous (not identical-by-descent), the longer heterozygosity is maintained in a population (a function of population size), the larger the number of junctions will be at end of the process. Thus, inferences about the length of time before the genome was stabilized in hybrid species are also inferences about the effective population size during establishment. We also consider more complex population size histories in the analysis below.
Because the goal was to estimate the number of generations until the genome of the hybrid species was stabilized, simulations continued until the population was very near fixation for all chromosomes. The simulation was halted in the generation in which heterozygosity had declined below 0.001 (operational criterion for homogenization). Population heterozygosity was determined by quantifying the fraction of the genome that was heterozygous, across all chromosomes in the population. At the end of each simulation, summary statistics on the genome composition were calculated, including the density of junctions.
The effect of selection on the rate of junction formation and stabilization of the genome of the hybrid species was investigated by allowing fertility to be a function of heterozygosity. Heterozygous combinations of parental chromosomal blocks are likely to be underdominant (e.g., pollen fertility of F 1 between H. annuus and H. petiolaris averages 4.8%; Rieseberg 2000). Thus, the probability of producing a viable gamete was modeled as 1 − hs, where h is the heterozygosity of the individual and s is the magnitude of selection against a completely heterozygous individual.
Genetic Maps of Hybrid Species
Genetic maps of H. anomalus, H. deserticola, and H. paradoxus were published previously (Rieseberg et al. 2003). These maps are for microsatellite, RAPD, and AFLP markers (total number of markers: 1019 for H. anomalus, 672 for H. deserticola, 771 for H. paradoxus). It was possible to determine the parental species of origin for species-specific random amplified polymorphic DNA (RAPD) and amplified fragment length polymorphism (AFLP) markers (Fig. 1; 427, 290, and 325 markers), whereas the remaining markers improved estimates of marker position and map length (see table S4 in Rieseberg et al. 2003). Transitions between chromosomal blocks that were inferred to be of different parental origin were counted as junctions. In cases for which markers of different parental origin mapped to identical locations on a linkage group (18, 3, and 5 markers per species), the state of adjacent markers was compared. If one or more adjacent markers had the same inferred parental origin as a marker at the shared location, they were assumed to be part of the same parental, chromosomal block (i.e., parsimony).
Figure 1.

Genetic linkage maps for each of three hybrid species of Helianthus (data from Rieseberg et al. 2003). For a species’ map, the 17 linkage groups are plotted along with their designations. Portions of a linkage group that are white were inferred to have originated in H. annuus, and those that are black to have originated in H. petiolaris. Hash marks to the right of each linkage group indicate the position of informative markers (inferred parentage: H. annuus—gray, and H. petiolaris—black). Regions that were inferred to harbor junctions are indicated by transitions from black to white (abrupt or grayscale transition).
The analyses involve only that portion of the map that was covered by markers for which parental origin could be inferred. Thus, for some linkage groups, the first and last utilized markers are not at the end of the full map (i.e., a few cM from the end) and this portion was removed in all analyses (96–97% of each species’ map retained). This is because junctions can be detected only between markers of known origin. The density of markers of known origin is roughly one marker every 4 cM (Table 1).
Table 1.
Descriptive statistics for the genetic maps of three species of Helianthus, including the observed junction density and the inferred point estimate of the underlying junction density, and lower and upper limits of the 95% confidence interval for underlying junction density.
| Species | Map size (cM) | Marker density | Junction density (junctions/cM)
|
|||
|---|---|---|---|---|---|---|
| Observed | Lower | Point estimate | Upper | |||
| H. anomalus | 1850.1 | 0.23 | 0.086 | 0.44 | 0.64 | 1.32 |
| H. deserticola | 1189.1 | 0.24 | 0.083 | 0.34 | 0.45 | 0.76 |
| H. paradoxus | 1362.8 | 0.24 | 0.098 | 0.48 | 0.73 | — |
Mapped markers allow the number of junctions to be inferred only imperfectly, because when the density of junctions is higher than the marker density, junctions will be missed (see MacLeod et al. 2005). To assess the sensitivity of our marker distributions to underlying junction density, we used a simple stochastic model. For a given number of junctions, we generated random uniform distributions for the locations of all junctions. For each simulation of genomic composition and junctions, the parental origin at the mapped marker locations was determined, and the number of detected junctions was counted. The distribution of detected junction density for the 500 replicates was determined initially for a broad range of true junction density between 0.1 and 2.2 junctions/cM (500 replicates at 20 points between 0.1 and 2.2) and for each of the species’ genetic maps (10,000 random maps for each species; Fig. 2). To make higher precision estimates of the expectation and 95% confidence intervals, 100 replicates at each 0.01 junction/cM increment were simulated for each species (H. anomalus: 0.3–1.5, 12,100 maps; H. deserticola: 0.28–1.2, 9300 maps; H. paradoxus 0.4–1, 6100 maps). The smallest simulated junction density for which the median of the distribution of detected density exceeded the junction density observed on the species’ map was taken as the point estimate (Fig. 2). Lower and upper confidence limits were based on the simulated density at which 2.5% and 97.5% quantiles of the distribution of detected density exceeded the observed junction density.
Figure 2.
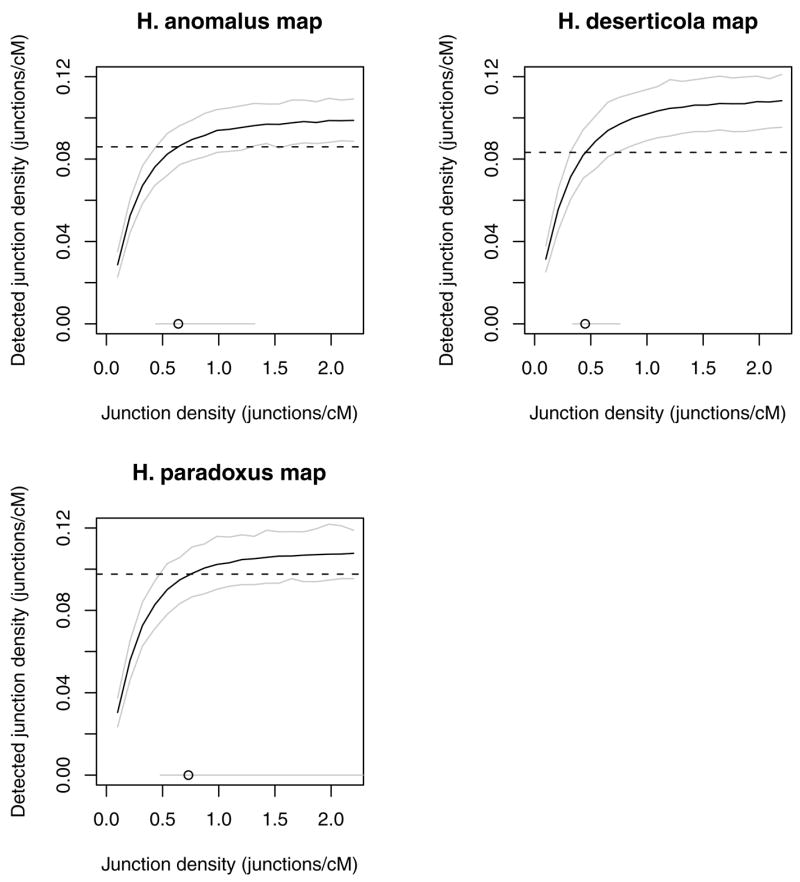
Simulations quantify how the distribution and density of informative markers on the species’ maps limit the detection of underlying junctions. The median detected density increases with simulated junction densities (0.1–2.2 junctions/cM), but saturates (black line, 95% confidence interval—gray lines). The intersection of these curves with the junction density observed on the maps of the hybrid species (dashed line), gives rise to a point estimate and confidence interval for the underlying junction density (circle and gray line at bottom).
The maps of the three species vary in their sensitivity to true junction density (Fig. 2). Therefore, whereas the observed junction densities for H. anomalus and H. deserticola are very similar (0.086 and 0.083 junctions/cM), they imply different true junction densities (0.64 vs. 0.45 junctions/cM; Table 1). Point estimates and confidence limits were established in all cases except for the upper confidence limit for the junction density in H. paradoxus. For this species’ map, the 95% confidence interval overlaps with the observed density for all values of simulated junction density above the point estimate (Fig. 2).
Finally, there would have been no reason to proceed with an analysis of the size of parental chromosomal blocks in the hybrid species unless there were evidence for the existence of parental chromosomal blocks. In other words, a prerequisite step was to determine whether the length of parental chromosomal blocks exceeded the length that would be expected on the basis of chance ordering of parentage at marker locations. A null distribution of block sizes, or the inverse, the density of junctions, was modeled simply by permuting the parental origin of all markers on the map. In this way, the map distances and distribution of markers on linkage groups were preserved but the state variable (parental origin) was randomized 1000 times for each map. For each species’ map, the observed junction density was much smaller (i.e., block size larger) than the densities in the simulations (P < 0.001 for H. anomalus and H. deserticola, and P = 0.009 for H. paradoxus). Thus, there was an indication of chromosomal blocks that were inherited intact from the parental species and the possibility that some signal of time to stabilization of the genome would be discernable.
Analysis
Given the inferred junction densities on each of the species’ maps, the main objective of this analysis was to estimate the number of generations that would be required to generate that density of junctions. The deterministic and simulation models resulted in indistinguishable expectations for junction density as a function of time (generations). The simulations added the stochastic component of genetic drift in finite populations and gave rise to appropriate estimates of variance around the expectation.
A linear model for time as a function of junction density was used to summarize the simulations and estimate the sampling variance in the time parameter (Fig. 3A). The regression is a summary of 4000 simulations with no selection, and with fixed population size sampled uniformly between 25 and 150. The regression equation for the simulations was used in combination with the estimated junction densities for each species (Table 1) to infer the number of generations to stabilization of the hybrid species’ genomes (Table 2): 795 (H. anomalus), 558 (H. deserticola), and 907 (H. paradoxus). The uncertainty in these estimates that comes from inferring the true, underlying junction density on a map was much more substantial than the sampling error due to genetic drift (Fig. 3B–D; Table 2A). Across all three species, the minimum time estimate for a species to reach a heterozygosity of 0.001 and a junction density consistent with the species’ map was 311 generations (H. deserticola). The equivalent regression of population size as a function of junction density gave estimates of effective population sizes that are consistent with the estimated junction densities (range of point estimates: 48–78 inds.; Table 2B). Each of these results depends on the assumption of 1 Morgan per chromosome. Consistent, genome-wide departures from this rate would simply change the slope of the relationship between junction density and generations.
Figure 3.
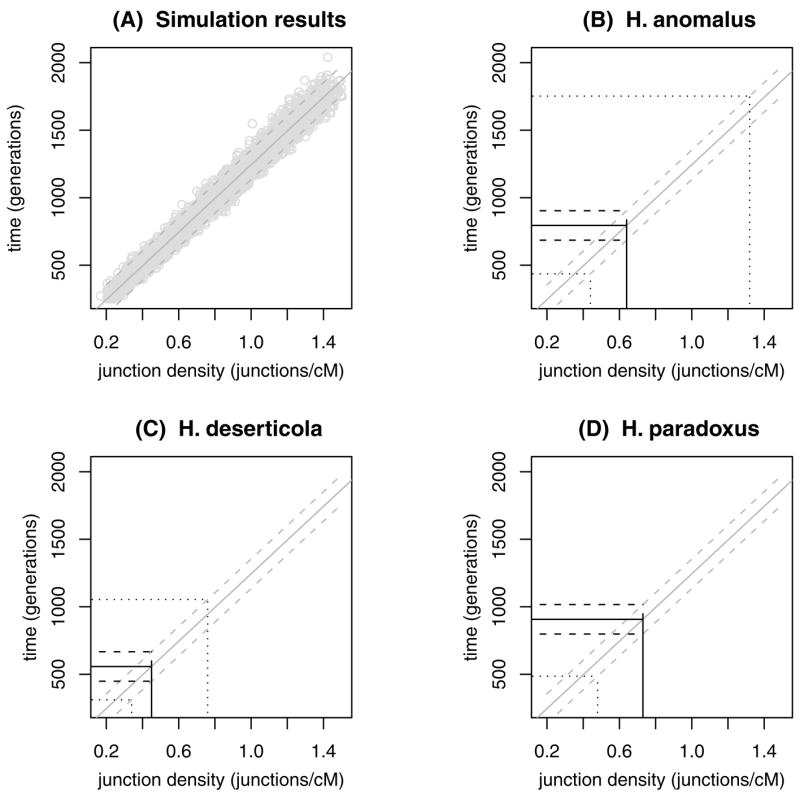
The time to stabilization of the hybrid genome (heterozygosity less than 0.001) is predicted by junction density with a relatively small variance introduced by genetic drift. (A) The results of 4000 simulations, with no selection and constant population sizes between 25 and 150, can be fit by a linear model (solid line, y = 1248x − 4). Residual variance around the regression allows the estimation of a 95% confidence interval for observations (dashed line). (B–D) Point estimates of junction density predict times to stabilization of the hybrid genomes (solid black lines). Variance due to genetic drift introduces some uncertainty in estimates (dashed black lines). Uncertainty due to the sensitivity of maps to underlying junction density is substantially larger (dotted black lines).
Table 2.
Simulations and the inferred junction densities (Table 1) allowed the number of generations between initial hybridization and stabilization of genomic composition of diploid hybrid species (Ht = 0.001) to be estimated (A) Similarly, the effective population sizes associated with simulations that fit the inferred junction densities were estimated. (B) For each species, a point estimate is given, along with confidence limits (CL) on the basis of residual variance in the regression (see Fig. 3 for prediction of generations). The minimum and maximum estimates are based on the inferred interval for underlying junction density (uncertainty in the map), as well as the variance around the regression.
| Species | Min. | Low CL | Point estimate | High CL | Max. |
|---|---|---|---|---|---|
| (A) Number of generations | |||||
| H. anomalus | 436 | 686 | 795 | 904 | 1753 |
| H. deserticola | 311 | 448 | 558 | 667 | 1054 |
| H. paradoxus | 486 | 798 | 907 | 1016 | — |
| (B) Effective population size | |||||
| H. anomalus | 39 | 60 | 68 | 76 | 147 |
| H. deserticola | 29 | 40 | 48 | 56 | 88 |
| H. paradoxus | 44 | 70 | 78 | 85 | — |
The above time estimates are on the basis of the criterion that heterozygosity in the population declined to 0.001 or less. It may be reasonable to allow some segregation of parental polymorphisms and still refer to genome stabilization as being complete. For example, it might be that heterozygosity will be favored for some chromosomal segments because of deleterious recessive alleles. When the criterion for “fixation” is relaxed to a heterozygosity (Ht) of 0.01 or 0.05, the point estimates for the number of generations to produce the observed junction densities are: 507, 358, and 577 for Ht ≤ 0.01, and 326, 229, and 372 for Ht ≤0.05 (for H. anomalus, H. deserticola, and H. paradoxus, respectively).
The effect of selection on the rate of speciation was investigated with selection parameters up to 0.9. A regression of time on junction density was performed for 400 replicate simulations with population sizes between 25 and 150 (as above). The inferred number of generations to speciation decreased only 5–8 generations (788, 553, and 899) and the variance around the regression was essentially unchanged (residual standard error 55.65 for s = 0 and 55.34 for s = 0.9).
The estimates of time to stabilization of the genome and the associated junction density depend on the length of time that heterozygosity persists, which is a function of population size and drift. In the modeling above we considered only fixed population sizes. Previous deterministic modeling has shown that linear and exponential growth in populations greatly affect the accumulation of junctions in populations that are initially small (Chapman and Thompson 2002, 2003). Numerical results of the deterministic model from Chapman and Thompson (2002) show that the principal consequence of population growth is to reduce the effects of drift and to extend the time to reach any given threshold for fixation (e.g., h = 0.001, as above). For a given initial population size, the time to fixation for a population of constant size is the minimum. Thus, the above estimates of time to stabilization are minimum estimates. The accumulation of junctions is affected much less by population growth, because most of the junction formation occurs early in the population history, when the size is still relatively small, and before most of the heterozygosity is lost to drift. Without independent evidence, the history of population size cannot be inferred, but the time estimates for stabilization of the genome can be considered minimum estimates.
Our modeling of junctions involved the assumption that there was no backcrossing to parental species. However, because immigration of parental chromosomes would simply slow the decline of heterozygosity in the hybrid population, the models make clear the outcome of relaxing the assumption of no backcrossing. If rates of gene flow due to backcrossing to parental species outpace the loss of heterozygosity due to drift, the time to fixation will increase. However, substantial backcrossing would also lead to the nonindependence of the derived lineage and would prevent speciation. Also, a model with backcrossing, but with smaller population size, would result in the same decline in heterozygosity and consequently in the same junction density and time estimate for stabilization. Different rates of backcrossing would be balanced by different population sizes to result in the same junction density at stabilization. Conversely, larger populations than were estimated to fit the data and more backcrossing would preserve heterozygosity over a larger number of generations and would lead to more junctions than were observed on the sunflower maps.
Discussion
Genetic maps of the three hybrid species of Helianthus contain a record of recombination between the parental genomes involved in the initial hybridization and origin of these species. Our modeling suggests that recombination continued to shape the genomic composition of the hybrid species for hundreds of generations before stabilization. Relative to other estimates of the rate of speciation, even at the scale of hundreds of generations, diploid hybrid speciation would be considered one of the fastest forms of speciation (Coyne and Orr 2004). At the same time, the results indicate that the stabilization of hybrid species is somewhat slower than has been suggested previously (fewer than 60 generations) on the basis of modeling (Ungerer et al. 1998) and experimental results (Rieseberg et al. 1996; Rieseberg 2000).
The modeling approach taken in an early analysis of chromosomal blocks in H. anomalus was very similar to our modeling, with at least two important differences. In Ungerer et al. (1998), the simulations continued for some specified number of generations, after which the resulting block size distributions were compared to those in the map of H. anomalus. Simulations of 60 or fewer generations exhibited the closest match to the observed map data. However, the amount of variation segregating in the population at the end of the simulation was not tracked, and therefore it is not clear that the time to stabilization of the genome was estimated. Additionally, the earlier analysis considered the maximum sizes of chromosomal blocks, rather than junction density. This creates unnecessary ambiguity because the precise length of chromosomal blocks cannot be determined with the present dataset. In contrast, the number of junctions detected can be divided by the map size to get an unambiguous metric that is the rate parameter of the exponential distribution of block sizes. These two differences are likely to account for the differences in our rate estimates and those of Ungerer et al. (1998). In addition to the differences in modeling approach, the H. anomalus map employed in the present study has significantly greater resolution due to the addition of 85 new species-specific markers, which allows for more accurate estimates of junction density (or block size).
Experiments with hybrids between Helianthus are consistent with the potential for rapid hybrid speciation and indicate that aspects of the process are repeatable, even in an experimental setting. In one study, five generations of hybridization between H. annuus and H. petiolaris in three independent mating designs resulted in hybrid populations with patterns of introgression that were significantly similar to the composition of H. anomalus (Rieseberg et al. 1996). Because five generations of crossing were sufficient to detect this association between the genomic composition of the synthetic lineages and that of H. anomalus, the result was interpreted as evidence for the potential rapidity of hybrid speciation. Each of the three synthetic lineages exhibited substantial postzygotic reproductive isolation from each of the parental species, in the form of reduced seed set and pollen viability (Rieseberg 2000). A significant fraction of the intrinsic postzygotic isolation found between H. anomalus and the parental species was evident in the synthetic hybrid lineages (76.9–99.7% for seed set and 55.2–93.1% for pollen viability), which again supports the rapidity of hybrid speciation. More recent comparisons with H. deserticola and H. anomalus revealed similar results. Both hybrid species were more similar in genomic composition to the synthetic hybrids than expected by chance (Karrenberg et al. 2007) and showed reduced seed set and pollen viability when crossed with the parental species (Lai et al. 2005).
In terms of genomic composition and reproductive isolation, the synthetic hybrid lineages demonstrated that significant steps toward speciation could be taken in five generations. However, the genomes were far from stabilized, as there was still substantial variation segregating in the synthetic hybrid lineages and no single individual approached the genetic composition of the hybrid species (each of the 56–58 individuals in the fifth generation of the lineages possessed a unique genotype and most chromosomal segments that had gone to fixation in the hybrid lineages were those of H. annuus, the recurrent parent in backcrosses). Similarly, the synthetic lineages had developed significant postzygotic isolation in crosses with the parental species, but the expression of isolation in crosses with H. annuus was appreciably less than between H. anomalus and H. annuus (average of 85.8% and 66.6% of interspecific isolation due to loss of seed set and pollen viability; Rieseberg 2000).
Our modeling and the results of previous experimental studies of hybridization in sunflowers (Rieseberg et al. 1996; Rieseberg 2000; Lai et al. 2005; Karrenberg et al. 2007) make it clear that the evolution of reproductive isolation in a hybrid lineage does not require that the recombined genome within that lineage be fully stabilized. Because the precise relationship of genome stabilization to hybrid speciation has not been clearly delineated previously, we discuss the relationship in the following paragraphs.
In hybrid speciation, a novel lineage originates through hybridization and becomes substantially isolated as segregating genic or chromosomal factors that contribute directly to initial isolation approach fixation (i.e., become stabilized). The hybrid lineage then increases in frequency within a small number of generations. Evidence for the rapidity of this process not only comes from the studies of synthetic hybrid lineages (described above), but also from simulation models of hybrid speciation (McCarthy et al. 1995; Buerkle et al. 2000), in which a novel hybrid lineage became established within tens of generations. These models demonstrated that the successful origin of an independent hybrid lineage requires the rapid development of substantial isolation; gene flow from parental taxa will lead to nonindependence, and a hybrid zone rather than an independent hybrid species (Buerkle et al. 2000, 2003).
The genomic regions that are subject to selection in the establishment of a hybrid species would initially be in a genetic background of segregating variation that would be stabilized over a longer period by recombination and drift in the isolated population. Thus, although reproductive isolation is the sine qua non of hybrid speciation, genome-wide stabilization is not required for speciation. Rather, its time course provides an upper bound of the interval over which reproductive isolation was established. This represents a modification of previous treatments that implicitly or explicitly included both the establishment of reproductive isolation and genome stabilization in speciation (e.g., Grant 1971; Ungerer et al. 1998; Rieseberg 2006; Mallet 2007).
The inclusion of genome stabilization in the concept of hybrid speciation probably traces back to the early recognition that the assortment of chromosomal rearrangements in hybrids may contribute to reproductive isolation (Stebbins 1957; Grant 1958). The establishment of a lineage that is homozygous for a novel combination of chromosomal rearrangements or genes is the basis for homoploid hybrid speciation (also referred to as recombinational speciation, Grant 1971). Grant (1971) did not explicitly extend the establishment of homozygosity beyond rearranged chromosomes to the whole genome, but this idea was suggested by the results of experimental hybridizations in sunflowers (Rieseberg et al. 1996; Karrenberg et al. 2007). These studies showed that fertility and viability selection rapidly pushed the populations in artificial hybrid lineages to have a significant overrepresentation of those parental chromosomal blocks found in the hybrid species of Helianthus. However, the modeling and analysis in this article make clear that selection for restored fertility and ecological adaptation did not lead to the rapid fixation of very large chromosomal regions and stabilization of the majority of the genome. Instead, the size of chromosomal blocks is consistent with a history of recombination that extends over hundreds of generations.
The genetic maps for the hybrid species of sunflowers are for particular parents used in the mapping studies (Rieseberg et al. 2003) and are informative about the history of junction formation in their ancestry. Given phylogeographic evidence that is consistent with multiple origins of two of the three hybrid species (H. anomalus and H. deserticola; Schwarzbach and Rieseberg 2002; Gross et al. 2003), it is possible that maps based on recombination between other sets of parents would contain a different distribution of junctions and would imply a different time course for genome stabilization. However, any intraspecific variation in genome stabilization is likely to fall within the range of relatively consistent time estimates for the three hybrid species. It is noteworthy that the different ecological and habitat affinities and associated histories of selection for H. anomalus, deserticola and paradoxus are not reflected in clear differences in the estimated times to overall genome stabilization. This lack of sensitivity in overall genome stabilization is perhaps unsurprising and instead variation in the adaptive responses of each hybrid lineage might be recorded in the genes and chromosomal regions that contributed directly to the response to fertility and ecological selection. As our knowledge of the genetic basis of reproductive isolation and adaptation improves for these taxa, it will be interesting to compare the genomic composition and history of recombination in these regions among species, and among multiple phylogeographic lineages within each species.
In the future, a genome sequence will be assembled for Helianthus and a truly large number of species-specific nucleotide polymorphisms will be identified. The polymorphisms will give rise to a precise map of junctions in the hybrid species. Based on these genomic data, we anticipate identifying portions of linkage groups that went rapidly to fixation and led the way in establishing reproductive isolation in the first phase of hybrid speciation. These should be regions of physically extensive interspecific linkage disequilibrium and low junction density. In contrast, regions of the genome that did not contribute directly to isolation are expected to harbor a majority of the junctions on the map.
Our modeling establishes that the time course for the stabilization of a hybrid species’ genome is on the order of hundreds of generations. As with other forms of speciation, there is no way to know at what time the last significant or critical component of isolation became fixed and speciation was complete. Even with this uncertainty, our results show that diploid hybrid speciation is remarkably rapid relative to most other forms of speciation.
Acknowledgments
We thank E. Baack, H. Marriott, three anonymous reviewers, and H. Hollocher for their careful reading of this manuscript and their constructive suggestions for improvement. The research was supported by a National Institutes of Health grant (GM059065) to L. Rieseberg.
LITERATURE CITED
- Baird SJE. A simulation study of multilocus clines. Evolution. 1995;49:1038–1045. doi: 10.1111/j.1558-5646.1995.tb04431.x. [DOI] [PubMed] [Google Scholar]
- Barton NH. Multilocus clines. Evolution. 1983;37:454–471. doi: 10.1111/j.1558-5646.1983.tb05563.x. [DOI] [PubMed] [Google Scholar]
- Buerkle CA, Morris RJ, Asmussen MA, Rieseberg LH. The likelihood of homoploid hybrid speciation. Heredity. 2000;84:441–451. doi: 10.1046/j.1365-2540.2000.00680.x. [DOI] [PubMed] [Google Scholar]
- Buerkle CA, Wolf DE, Rieseberg LH. The origin and extinction of species through hybridization. In: Brigham CA, Schwartz MW, editors. Population viability in plants: conservation, management, and modeling of rare plants. Springer Verlag; New York: 2003. pp. 117–141. [Google Scholar]
- Chapman NH, Thompson EA. The effect of population history on the lengths of ancestral chromosome segments. Genetics. 2002;162:449–458. doi: 10.1093/genetics/162.1.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NH, Thompson EA. A model for the length of tracts of identity by descent in finite random mating populations. Theor Popul Biol. 2003;64:141–150. doi: 10.1016/s0040-5809(03)00071-6. [DOI] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. Speciation. Sinauer Associates; Sunderland, MA: 2004. [Google Scholar]
- Crow J, Kimura M. An introduction to population genetics. Harper and Row; New York: 1970. [Google Scholar]
- Fisher RA. A fuller theory of “junctions” in inbreeding. Heredity. 1954;8:187–197. [Google Scholar]
- Gompert Z, Fordyce JA, Forister ML, Shapiro AM, Nice CC. Homoploid hybrid speciation in an extreme habitat. Science. 2006;314:1923–1925. doi: 10.1126/science.1135875. [DOI] [PubMed] [Google Scholar]
- Grant V. Exchange of genetic material: mechanisms and consequences. Vol. 23. Cold Spring Harbor Symposium on Quantitative Biology; Cold Spring Harbor, New York: 1958. The regulation of recombination in plants; pp. 337–363. [DOI] [PubMed] [Google Scholar]
- Grant V. Plant speciation. Columbia Univ. Press; New York: 1971. [Google Scholar]
- Gross BL, Rieseberg LH. The ecological genetics of homoploid hybrid speciation. J Heredity. 2005;96:241–252. doi: 10.1093/jhered/esi026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross BL, Schwarzbach AE, Rieseberg LH. Origin(s) of the diploid hybrid species Helianthus deserticola (Asteraceae) Am J Bot. 2003;90:1708–1719. doi: 10.3732/ajb.90.12.1708. [DOI] [PubMed] [Google Scholar]
- Howarth DG, Baum DA. Genealogical evidence of homoploid hybrid speciation in an adaptive radiation of Scaevola (Goodeniaceae) in the Hawaiian islands. Evolution. 2005;59:948–961. [PubMed] [Google Scholar]
- Karrenberg S, Lexer C, Rieseberg LH. Reconstructing the history of selection during homoploid hybrid speciation. Am Nat. 2007;169:725–737. doi: 10.1086/516758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z, Nakazato T, Salmaso M, Burke JM, Tang S, Knapp SJ, Rieseberg LH. Extensive chromosomal repatterning and the evolution of sterility barriers in hybrid sunflower species. Genetics. 2005;171:291–303. doi: 10.1534/genetics.105.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod AK, Haley CS, Woolliams JA, Stam P. Marker densities and the mapping of ancestral junctions. Genet Res. 2005;85:69–79. doi: 10.1017/s0016672305007329. [DOI] [PubMed] [Google Scholar]
- Mallet J. Hybrid speciation. Nature. 2007;445:279–283. doi: 10.1038/nature05706. [DOI] [PubMed] [Google Scholar]
- Mavárez J, Salazar CA, Bermingham E, Salcedo C, Jiggins CD, Linares M. Speciation by hybridization in Heliconius butterflies. Nature. 2006;441:868–871. doi: 10.1038/nature04738. [DOI] [PubMed] [Google Scholar]
- McCarthy EM, Asmussen MA, Anderson WW. A theoretical assessment of recombinational speciation. Heredity. 1995;74:502–509. [Google Scholar]
- Meyer A, Salzburger W, Schartl M. Hybrid origin of a swordtail species (Teleostei: Xiphophorus clemenciae) driven by sexual selection. Mol Ecol. 2006;15:721–730. doi: 10.1111/j.1365-294X.2006.02810.x. [DOI] [PubMed] [Google Scholar]
- Mir C, Toumi L, Jarne P, Sarda V, Di Giusto F, Lumaret R. Endemic North African Quercus afares Pomel originates from hybridisation between two genetically very distant oak species (Q. suber L. and Q. canariensis Willd.): evidence from nuclear and cytoplasmic markers. Heredity. 2006;96:175–184. doi: 10.1038/sj.hdy.6800782. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH. Homoploid reticulate evolution in Helianthus (Asteraceae): evidence from ribosomal genes. Am J Bot. 1991;78:1218–1237. [Google Scholar]
- Rieseberg LH. Hybrid origins of plant species. Ann Revi Ecol Syst. 1997;28:359–389. [Google Scholar]
- Rieseberg LH. Crossing relationships among ancient and experimental sunflower hybrid lineages. Evolution. 2000;54:859–865. doi: 10.1111/j.0014-3820.2000.tb00086.x. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH. Hybrid speciation in wild sunflowers. Ann Mo Bot Gard. 2006;93:34–48. [Google Scholar]
- Rieseberg LH, Sinervo B, Linder CR, Ungerer M, Arias DM. Role of gene interactions in hybrid speciation: evidence from ancient and experimental hybrids. Science. 1996;272:741–745. doi: 10.1126/science.272.5262.741. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Raymond O, Rosenthal DM, Lai Z, Livingstone K, Nakazato T, Durphy JL, Schwarzbach AE, Donovan LA, Lexer C. Major ecological transitions in wild sunflowers facilitated by hybridization. Science. 2003;301:1211–1216. doi: 10.1126/science.1086949. [DOI] [PubMed] [Google Scholar]
- Schwarz D, Matta BM, Shakir-Botteri NL, McPheron BA. Host shift to an invasive plant triggers rapid animal hybrid speciation. Nature. 2005;436:546–549. doi: 10.1038/nature03800. [DOI] [PubMed] [Google Scholar]
- Schwarzbach AE, Rieseberg LH. Likely multiple origins of a diploid hybrid sunflower species. Mol Ecol. 2002;11:1703–1715. doi: 10.1046/j.1365-294x.2002.01557.x. [DOI] [PubMed] [Google Scholar]
- Stam P. The distribution of the fraction of the genome identical by descent in finite random mating populations. Genet Res. 1980;35:131–155. [Google Scholar]
- Stebbins GL. The hybrid origin of microspecies in the Elymus glaucus complex. Cytologia Supplement. 1957;36:336–340. [Google Scholar]
- Ungerer MC, Baird SJE, Pan J, Rieseberg LH. Rapid hybrid speciation in wild sunflowers. Proc Natl Acad of Sci USA. 1998;95:11757–11762. doi: 10.1073/pnas.95.20.11757. [DOI] [PMC free article] [PubMed] [Google Scholar]