

Abstract
Background
The ability of an intracellular pathogen to establish infection depends on the capacity of the organism to survive and replicate inside the host. Mycobacterium fortuitum is a bacteria that contains genes involved in the detoxification of the oxygen reactive species such as those produced by the host during the infection. In this work, we investigate the effects of hydrogen peroxide on the transcription and expression of these genes by developing a real time quantitative PCR technique (qRT-PCR) using the ribosomal promoter region (rrnA-P1) as reference product for quantification of the mRNA levels.
Results
M. fortuitum cultures were treated with different hydrogen peroxide concentrations (0.02 to 20 mM) during several periods of time (30 to 120 minutes). The activity of the enzymes KatGII and SodA, and the transcription of corresponding genes were evaluated. The transcriptional regulator furAII gene was also studied.
The ribosomal promoter region rrnA-P1 was validated as referential product under the stress conditions checked by qRT-PCR.
Minor changes were observed under the conditions tested except when bacteria were incubated in the presence of 20 mM hydrogen peroxide. Under those conditions, the levels of transcription of the three genes under study increased at 30 minutes of treatment. The viability of the bacteria was not influenced under the conditions tested.
Conclusion
In this work, we have quantified transcriptional responses to stress suggesting that, the opportunistic pathogen M. fortuitum is more resistant and differs in behaviour in the presence of hydrogen peroxide, when compared to the major pathogen Mycobacterium tuberculosis and the saprophyte Mycobacterium smegmatis. Besides, we demonstrate the mycobacterial non-coding region rrnA-P1 to be a suitable reference product in the analysis of qRT-PCR transcriptional data of M. fortuitum.
Background
The genus Mycobacterium includes species found in a wide range of ecological niches. M. tuberculosis, M. leprae, and M. avium subsp. paratuberculosis, are pathogenic species causing clinical disease in humans or animals. Other species as M. smegmatis and M. phlei are saprophytes. In addition, an intermediate position is occupied by species opportunistic pathogens such as M. fortuitum, a rapidly growing mycobacteria that is ubiquitous in soil and water. This mycobacteria is known to cause cutaneous infection, typically in association with trauma or clinical procedures [1,2]. Due to its capacity to growth and to survive intracellularly, M. fortuitum has been previously used as a model for studying the intracellular killing of mycobacteria [3,4].
Most bacteria, when exposed to toxic oxygen metabolites, exhibit an adaptive response and express several genes involved in detoxification of oxygen reactive species, such as superoxide dismutases (SOD), catalases and peroxidases [5,6]. This type of adaptative response has been implicated in the intracellular survival of pathogenic mycobacteria, promoting their maintenance in the host [7-9].
The major pathogen, M. tuberculosis, produces an iron co-factored SOD encoded by the gene sodA [10] and a copper and zinc SOD encoded by the gene sodC [11] whose involvement in protection against oxidative stress of M. tuberculosis within macrophages is currently controversial [12,13]. SOD encoded by sodA is a major secreted protein [14] that inhibits host responses to M. tuberculosis [15]. The expression of genes sodA and sodC is considered constitutive and inducible respectively [15,16]. Homologues of these genes have been detected in several mycobacterial species, including M. fortuitum [13,17].
There are two classes of catalases identified in mycobacteria: the heat-labile catalase or T-catalase (coded by katG), which has also a peroxidase-like function, and the heat-stable catalase or M-catalase (coded by katE). The catalase codified by katG is synthesized preferentially in response to oxidative stress whereas the catalase codified by katE is produced in response to nutrient depletion as occurs in the stationary phase of growth [18]. M. fortuitum produces both catalases however M. tuberculosis produces only T-catalase whilst M. terrae only produces M-catalase [19]. The katG gene has been widely studied in M. tuberculosis [20,21]. The KatG protein contributes to the ability of M. tuberculosis to grow and survive within the infected host tissues [22] and is involved in the resistance of M. tuberculosis to the first-line antimicrobial drug isoniazid [23,24]. Due to their involvement in disease pathogenesis, both sodA and katG genes are considered virulence factors of M. tuberculosis [25].
The oxidative stress response in bacteria is linked to the iron metabolic pathway [26]. One of the proteins involved in that link is the ferric uptake regulator protein (FurA) an homologue of the Fur of E. coli, encoded by the furA gene [27]. The furA gene is located immediately upstream of the katG gene in M tuberculosis, an arrangement that appears to be common amongst mycobacterial genomes [28,29].
Only few studies have investigated the regulation of expression of detoxifying genes in mycobacteria. Two promoters have been detected that control katG expression in M. tuberculosis, one is located upstream of the furA gene and is co-transcribed with katG [30,31]. Moreover, FurA represses katG gene expression in M. tuberculosis and M. smegmatis [29] and autoregulates its own expression by binding to the Fur-A box, a 23 bp AT-rich sequence located upstream of furA gene, which is overlapping the -35 and -10 region of the corresponding promoter [32,33]. No studies have yet been published on the regulation of mycobacterial sod genes.
The genes sodA and katG have been previously studied by our group in M. fortuitum [17,34]. We have shown that protein SodA of M. fortuitum requires manganese as cofactor [17]. This mycobacteria has two genes encoding for two different enzymes with catalase-peroxidase activities, called katGI and katGII (KatGI and KatGII). KatGII is more related to the single M. tuberculosis KatG than KatGI [34]. Upstream of both genes we have also found copies of the furAI and furAII genes respectively [EMBL: Y17061 and Y17062]. The furAII-katGII spacer region spans only 33 nucleotides in the genome of M. fortuitum, thus suggesting that the two genes might be co-transcribed.
In this work we have further analyzed the transcriptional activity of katGII, furAII and sodA genes in M. fortuitum under oxidative stress. Putative promoters have been identified, and real-time quantitative PCR (qRT-PCR) applied to the transcriptional analysis of cultures, which have been exposed to hydrogen peroxide.
qRT-PCR technology has been widely used in the analysis of gene transcriptional activity due to its high efficiency, level of sensitivity and specificity [35-37]. Quantitative gene-expression assays using this technique are typically referenced to an internal control to account for differences in the RNA load. The conditions of the experimental design should not influence the detection level of the selected internal control. Thus the validation of reference controls is recommended in any such experimentation [38].
Two different products sigA [39-41] and rrs (16S rRNA) [42,43], have most often been used as reference standards in qRT-PCR assays involving mycobacteria. However both of these have disadvantages. sigA expression has been found to decrease threefold during entry of bacterial growth into the stationary phase [44]. Levels of rrs usually do not change during most phases of bacterial growth, however it is present in a much higher amounts than any mRNA in the cell [M. C. Nuñez & M. J. Garcia, unpublished results] probably because of its higher stability as part of the ribosomes, compared to the stability of other RNA molecules in the cytoplasm, which can cause problems with sensitivity.
Recently, our laboratory described [45] a putative new reference product (rrnA-P1) suitable for qRT-PCR. Here we demonstrate the usefulness of this promoter as reference in the qRT-PCR analysis of cultures of M. fortuitum during oxidative stress. This methodology may be applied to other quantitative gene-expression analysis of mycobacteria and other bacterial genera in order to evaluate the influence of any experimental treatment on the transcriptional level of gene expression.
Results
Enzymatic activity upon oxidative stress of KatG and SodA proteins
The enzymatic activities of catalase-peroxidase and superoxide dismutase proteins were examined in the extracts collected from M. fortuitum cultures (see Methods). The results (Figure 1a) indicated a low level of activity of the KatGI under each of the test conditions. Further analysis of the mRNA levels for katGI (gene coding for KatGI) was unsuccessful, probably due to the scarce expression level of that gene under the experimental conditions applied. The activity of KatGII remains similar from 30 to 120 minutes under 0.02 and 2 mM of H2O2 (Figure 1a). KatGII showed its lowest activity relative to the control at 30 min of treatment with 20 mM hydrogen peroxide. At this highest concentration, the catalase-peroxidase activity of KatGII increased from cultures treated during 30 min to cultures treated during 120 min (Figure 1a). See Additional file 1 for densitometry values.
Figure 1.
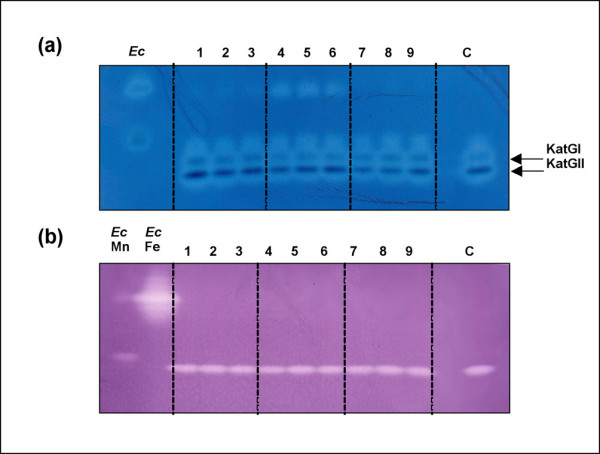
Protein activity of KatG and SodA using ND-PAGE gels. (a) Catalase and peroxidase activities; (b) Superoxide dismutase activity. M. fortuitum cultures treated with H2O2 at the following concentrations: Lanes 1 to 3, 0.02 mM; lanes 4 to 6, 2 mM; lanes 7 to 9, 20 mM and lane C, control. Samples of each concentration were treated using increasing length of periods as follows: 30 min (lanes 1, 4, and 7); 60 min (lanes 2, 5, and 8); and 120 min (lanes 3, 6, and 9). The bands corresponding to KatGI and KatGII are indicted with arrows. E. coli extracts are incorporated in the catalase-peroxidase activity gel and commercial Fe- and Mn-SOD in the superoxide dismutases activity gel. Abbreviations: Ec: E. coli, Mn: Manganese-SOD, Fe: Iron-SOD.
The SodA enzymatic activity did not show detectable variation with the control comparing different extracts under all the conditions tested (Figure 1b). This result is in agreement with the considered constitutive expression of the sodA gene [15].
Identification of the transcriptional start points (tsp) of the genes under study
We detected two putative transcriptional start points (tsp P1 and tsp P2) in each the three genes examined by RNA protection assay (RPA) (Table 1a). Putative -35 and -10 boxes were identified according the consensus mycobacterial promoters previously described in the literature [46,47]. The presence of furAII-katGII co-transcripts was tested by standard RT-PCR. Co-transcripts furAII-katGII were detected in all the stress conditions tested (data not shown). These results are in agreement to those described previously in M. tuberculosis [30,33].
Table 1.
Promoter regions of the sodA, katGII and furAII genes of M. fortuitum
| (a) | ||||||||
| Gene | P | -35 box | spacing | -10 box | spacing | tsp (+1) | spacing | start codon |
| sodA | P1 | TTGTTC | 19 | TATACG | 4 | G | 95 | GTG |
| P2 | TTGCGG | 18 | AGTGTT | 6 | G | 44 | GTG | |
| katGII | P1 | TGCCTG | 17 | CGGCTT | 13 | G | 90 | ATG |
| P2 | CACGGC | 16 | CCGAGG | 7 | T | 76 | ATG | |
| furAII | P1 | TCGTCT | 20 | CGGAAT | 12 | T | 40 | GTG |
| P2 | TTCTCG | 12 | TATTCT | 5 | A | 28 | GTG | |
| (b) | ||||||||
| Mycobacterium species | AT-rich region | spacing | start codon | |||||
| M. tuberculosis | AGT CTT GAC TAA TTC CAG AAA AG | 9 | TTG | |||||
| M. smegmatis | ATT CTT GAC TAA TTC CAG AAA AG | 16 | GTG | |||||
| M. fortuitum (furAII) | ATTCTT GAC TAA TTC CAG AAA AC | 16 | GTG | |||||
(a) Location of the promoters of the sodA, katGII and furAII genes of M. fortuitum. Abbreviations: P: promoter; -10 box and -35 box: putative -10 and -35 boxes; tsp (+1): transcription start point.
(b) Alignment of the AT-rich sequences upstream of the furA genes in mycobacteria. The -35 boxes of M. tuberculosis and M. smegmatis [33] and the -10 box of M. fortuitum are underlined. The tsp+1 nucleotide of the P2 promoter of the furAII gene of M. fortuitum is bolded.
Validation of rrnA-P1 as reference product in qRT-PCR under stress conditions
The rrnA-P1 promoter-usage of M. fortuitum cultures treated with hydrogen peroxide was analyzed by primer extension (PE) [48,49]. The relative activities of the five rrn promoters of M. fortuitum, corresponding to different PE products, were determined by quantification of the radioactive levels for each of the conditions tested (Figure 2a).
Figure 2.

rrn promoter-usage under hydrogen-peroxide treatment. (a) Percentage of usage corresponding to representative experiment of the M. fortuitum rrn promoters. rrnA-P1 to PCL1 and rrnB-P1 indicate the five promoter of rrn operons in M. fortuitum [48]. (b) Validation of the rrnA-P1 promoter as qRT-PCR reference product; by definition, the value of the control is one (see Methods), the statistical significance of data was determined by applying one-way analysis of variance. M. fortuitum cultures treated with H2O2 at the following concentrations: lanes 1 to 3, 0.02 mM; lanes 4 to 6, 0.2 mM; lanes 7 to 9, 2 mM; lanes 10 to 12, 20 mM and lane C, control. Samples of each concentration were treated using increasing length of periods as follows: 30 min (lanes 1, 4, 7 and 10); 60 min (lanes 2, 5, 8 and 11); and 120 min (lanes 3, 6, 9 and 12). Abbreviations: Ct: Cycle threshold.
All the promoters were differentially expressed under each of the conditions tested with the exception of the rrnA-P1 promoter whose contribution to the pre-rRNA synthesis was less than 1%, and evenly maintained in each of the stress conditions tested (Figure 2a). These results suggested the suitability of the rrnA-P1 promoter as reference standard in qRT-PCR analysis of M. fortuitum under oxidative stress.
Experiments were conducted to validate the usefulness of rrnA-P1 product as a reference in qRT-PCR, when M. fortuitum cultures were grown under oxidative stress. Validation of the rrnA-P1 promoter as a constitutive product was investigated using the derivation of the 2-ΔΔCt equation as indicated in Methods: 2-ΔCt (ΔCt = Ct sample - Ct control). In this equation the control corresponded to the untreated culture, and the sample corresponded to each stress culture condition. The results are showed in the Figure 2b. No significant differences were found comparing data from all the culture conditions tested with the control (p = 0.445). This result validates the product corresponding to rrnA-P1 promoter as a reference in the analysis undertaken.
Analysis of the M. fortuitum gene expression upon oxidative stress
The transcription of katGII and sodA in M. fortuitum during the oxidative stress response was initially analyzed using Northern Blot assay (Figure 3). The transcripts were detected by hybridization with internal probes corresponding to each of the genes. An internal probe of the rrs gene was also used to test the relative amount of RNA fixed on the filter (Figure 3). A single band was detected per each probe at the expected sizes: 1.5 Kb, 2.2 Kb, and 620 bp for rrs, katGII, and sodA respectively (Figure 3).
Figure 3.

Northern blot analysis of the sodA, katGII and rrs under hydrogen-peroxide treatment. Total RNA from M. fortuitum cultures treated with H2O2: lanes 1 to 3, 0.02 mM; lanes 4 to 6, 0.2 mM; lanes 7 to 9, 2 mM; lanes 10 to 12, 20 mM and lane C, control. Samples of each concentration were treated using increasing length of periods as follows: 30 min (lanes 1, 4, 7 and 10); 60 min (lanes 2, 5, 8, and 11); 120 min (lanes 3, 6, 9 and 12). The same filter was stripped out and re-hybridized sequentially with the several radiolabelled probes: rrs, katGII and sodA. The sizes of the corresponding mRNAs are indicated.
The amounts of radioactive signals corresponding to katGII, and sodA genes in each lane were also quantified and normalized with the amount of radioactivity obtained using the rrs as a probe (see Additional file 1 for the complete set of the cpm values). No significant changes were obtained in the level of mRNA corresponding to either of the genes under stress conditions, with the exception of mRNA of the katGII gene which increased 10 fold after 30 min of treatment at 20 mM H2O2; and the mRNA of the sodA gene which increased two fold after 30 and 120 min of treatment at the same H2O2 concentration (Figure 3).
In order to determine more accurately the amount of the transcripts of M. fortuitum in response to oxidative stress, qRT-PCR technique was applied to quantify the mRNAs. The transcription of sodA, katGII and its putative regulator furAII were analyzed (Figure 4). The genes under study did not show main variations in the quantity of products in relation to the different H2O2 concentrations applied, as well as in relation to the duration of the treatment. Again, the only significant change was detected at the highest concentration tested (20 mM H2O2). At this concentration, the highest level of katGII, furAII and sodA expressions corresponded to the culture treated with H2O2 during 30 min. Compared to the control culture, katGII and furAII mRNA levels product increased in this point more than twenty and fifteen fold respectively. The products corresponding to sodA suffered a three-fold increase in this same point of analysis (Figure 4).
Figure 4.

qRT-PCR analysis of katGII, furAII and sodA under hydrogen-peroxide treatment. (a) katGII gene expression; (b) furAII gene expression; (c) sodA gene expression. The 2-ΔΔCt method was applied to calculate the relative amount of the cDNAs of interest, comparing treated and untreated cultures (see Methods). M. fortuitum cultures were treated with hydrogen-peroxide at the following concentrations: lanes 1 to 3, 0.02 mM; lanes 4 to 6, 0.2 mM; lanes 7 to 9, 2 mM; lanes 10 to 12, 20 mM and lane C, control. Samples of each concentration were treated using increasing length of periods as follows: 30 min (lanes 1, 4, 7 and 10); 60 min (lanes 2, 5, 8 and 11); and 120 min (lanes 3, 6, 9 and 12).
Under other H2O2 concentrations tested (0.02 mM, 0.2 mM and 2 mM) the mRNA levels of katGII, furAII and sodA either increased to a lesser extent or remain unchanged with the duration of the treatment (Figure 4).
Viability of M. fortuitum under different H2O2 stress treatment
To analyse the influence of the stress on the viability of the M. fortuitum, we determined the colony forming units (CFUs) of each of the cultures (Figure 5). No main differences were found when controls and test cultures were compared, indicating that changes detected at the mRNA and proteins levels were not related to changes in the bacterial viability.
Figure 5.
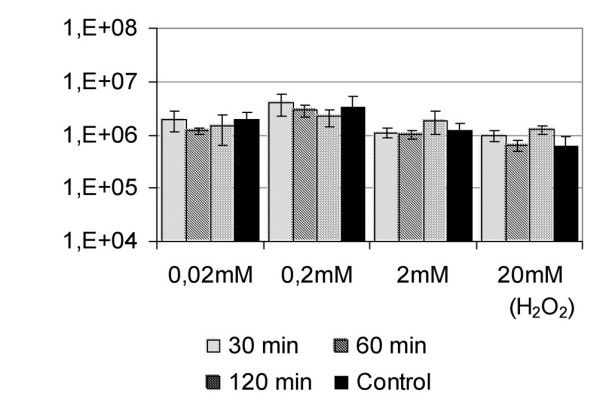
Viability of the M. fortuitum cultures treated with H2O2. The colony forming units (CFUs) were determined for the untreated control and for each culture treated with 0.02, 0.2, 2 and 20 mM of H2O2 after 30, 60 and 120 minutes. One control was used for each H2O2 concentration (showed as black columns in the figure). The error bars corresponding to data from three different cultures are showed.
Discussion
Mycobacteria are ubiquitous bacteria and are therefore exposed to a wide range of environmental conditions, including animals and humans. One of these conditions is the host stress response which includes induction of reactive oxygen intermediates (ROI) against which mycobacteria synthesize enzymes such as superoxide dismutases, catalases and peroxidases. This work studies these enzymatic responses in the opportunistic pathogen M. fortuitum growing in the presence of hydrogen peroxide measured using real-time PCR to quantify mRNA specific products corresponding to genes sodA, katGII and furAII involved in the oxidative stress response.
The development of a reliable qRT-PCR method requires corrections for experimental sample variations, such as reverse transcription efficiency. Those variations are best controlled by the inclusion of an internal reference standard. Ideally this reference standard should have a constant expression under each of the experimental conditions [50]. Moreover, validation of the internal control is recommended when designing quantitative gene expression studies [50].
We have previously reported that the first promoter of the rrnA operon (rrnA-P1) gave better results in M. fortuitum [45] than sigA and 16S rRNA, the standards more frequently used in M. tuberculosis [40,43]. This study has tested this product for suitability as reference standard in the study of the oxidative stress response of M. fortuitum.
The primer extension results showed that the product of the rrnA-P1 promoter was synthesized at a low and constant level when M. fortuitum cultures were placed under oxidative stress (Figure 2a). Experiments conducted to validate rrnA-P1 as reference standard in qRT-PCR (Figure 2b) found no statistically significant changes (p = 0.445), in expression levels during several of the experimental conditions. It was concluded therefore that rrnA-P1 is a suitable reference standard for qRT-PCR studies of M. fortuitum growing under oxidative stress.
In contrast to the single promoter previously detected upstream of furA and katG genes of M. tuberculosis [30,31], two promoters were identified in the M. fortuitum genome upstream of furAII, katGII and sodA (Table 1a). furAII and katGII genes were co-transcribed in M. fortuitum in a similar manner to M. tuberculosis [30,33], showing the previously described conserved organization of those genes among mycobacteria [29]. Recently, it has been demonstrated in M. smegmatis that the single transcript containing furA and katG is processed to originate two different transcripts, one covering furA and the second covering katG [51], that result explain why furA-katG co-transcripts were not detected in this species previously [32]. Nevertheless the occurrence of a similar phenomenon in M. fortuitum cannot be excluded, differently to M. smegmatis, furAII-katGII co-transcripts were detected in M. fortuitum supporting that a different transcriptional organization could occurs in this last mycobacteria.
Several genomes of mycobacterial species have been sequenced or are in progress [52], including that of the rapidly growing mycobacteria M. smegmatis (strain mc2155). This strain is used frequently in the study of mycobacteria, because of its rapid growth, and low biosafety requirements. A search of the M. smegmatis genomeidentified three putative katG genes. Two of these are associated with a corresponding furA gene immediately upstream, including a Fur-A box in one of them. Also of note is that the third katG gene has a long deletion in its 5' end (190 nucleotides), which could explain the lack of the sequence corresponding to a paired furA gene upstream.
Studies in M. tuberculosis have clearly established that Fur-A is a negative regulator of the katG expression in mycobacteria [29,33]; this regulation is mediated by the FurA-box identified upstream of the furA gene in the region corresponding to the -35 promoter sequence, also known as the AT-rich sequence. The linkage of FurA to the FurA-box is inhibited in the presence of hydrogen peroxide thus allowing the transcription of the downstream genes. Sala et al. [33] have analyzed the FurA box of M. smegmatis, which presumably included the single furA-katG copy associated with this regulatory sequence. In this work we identify that AT-rich sequence 16 nucleotides upstream the furAII start codon of M. fortuitum (Table 1b). This sequence includes the -10 box corresponding to the P2 promoter identified (Table 1). Similarly to M. smegmatis, only one of the two furA-katG tandem genes of M. fortuitum has a sequence equivalent to the FurA box upstream.
By using qRT-PCR, we observed that the influence of hydrogen peroxide in the level of expression of katGII and furAII of M. fortuitum differs from other mycobacteria [29,31,33,53] by not showing variation in the protein and mRNA levels of these genes when M. fortuitum was treated with up to 2 mM hydrogen peroxide (Figures 1, 3 and 4). These results could indicate that M. fortuitum appears to be more resistant to the hydrogen peroxide stress than other mycobacterial species.
We did find significant changes however in the stress response in cultures of M. fortuitum when treated with 20 mM H2O2. Under such conditions, the mRNA levels of the two genes (katGII and furAII) increased significantly, and the level of sodA mRNA rose to its higher level after 30 minutes of treatment (Figure 4). Considering also that only a low level of mRNA could be detected when cultures were treated with 20 mM of H2O2 during 60 and 120 minutes (Figure 4), we hypothesized that 20 mM H2O2 may be deleterious for the bacteria, however M. fortuitum cultures studied under stress found no decrease in CFU under each of the conditions tested (Figure 5). We conclude therefore that the changes detected in genes related to the stress response (Figure 4) are not associated to changes in the viability of the bacteria, however they could indicate a compensatory reaction, at the transcriptional level, to the decrease detected at the protein level (Figure 1) indicating some adaptative response of the transcriptional machinery to that extremely adverse environmental conditions. Further studies are required to determine if the response to 20 mM H2O2 might indicate a global alteration in the transcriptional activity or possibly post-transcriptional regulation of M. fortuitum.
Conclusion
Our results indicate that mycobacteria differ in their response against oxidative stress. The opportunistic pathogen M. fortuitum is more resistant to hydrogen peroxide than the main pathogen M. tuberculosis and the saprophyte M. smegmatis. Even though changes in the mRNA and proteins related to oxidative stress were detected, the M. fortuitum viability was not affected at a H2O2 treatment as strong as 20 mM during 120 minutes.
This work also demonstrated the suitability of the non-coding region (pre-rrnA-P1) as reference standard in the quantification of the transcriptional activity of genes associated to oxidative stress in M. fortuitum. The reference product applied (pre-rrnA-P1) is related to the ribosomal RNA, a bacterial component frequently used as reference in the analysis of the bacterial transcriptional activity.
Methods
Bacterial strain and growth conditions
M. fortuitum ATCC 6841T was maintained on Lowenstein Jensen at 4°C for short-term storage and 40% glycerol at -70°C for long-term storage.
Three different cultures of M. fortuitum were grown at 37°C until exponential phase (OD = 0.6–0.8 U) in 100 ml of Sauton broth [54]. Hydrogen peroxide (H2O2) was then added to obtain 0.02 to 20 mM final concentrations. Cells were further incubated at 37°C with shaking for 1/2 h, 1 h and 2 h, per each H2O2 concentration. Total RNA and proteins were purified from the cultures (75 ml and 25 ml respectively) following to the treatment with hydrogen peroxide. Untreated cultures were also tested as reference.
Viability of bacteria recovered from the cultures was checked, by determining the colony forming units (CFUs) on Middlebrook 7H10 petri dishes. To reduce possible variation introduced by individual inocula over the several sets of cultures, a separate untreated control culture was included for each H2O2 concentration tested.
Protein isolation and analysis of the protein activity
The protein extracts were prepared by vigorous mixing of bacterial cells in the presence of 0.15–0.212 mm of diameter glass beads (SIGMA) during three sequential pulses of 45 s at 4 m/s in a Fast-Prep (Savant-Instruments). Bacterial extracts were centrifuged and the supernatants were recovered and stored at -70°C. The analysis of the extracts was made by Polyacrilamide Gel Electrophoresis (PAGE). Activity gels were performed in non-denaturing polyacrylamide gel electrophoresis (ND-PAGE) [55], 10% and 15% [wt/vol] ND-PAGE were applied for catalase and peroxidase and superoxide dismutase analysis respectively. Catalase and peroxidase activities of the mycobacterial extracts were examined by the double-staining method as described previously [56]. Superoxide dismutase activity was examined as described previously [57,58]. Densitometry of the activity bands was determined directly on the gels using ID-Manager (TDI) for comparative purposes.
RNA isolation
Mycobacterial cultures were collected, and total RNA isolated as described previously [59]. Quality of the RNA isolation was checked by gel electrophoresis followed by quantification in spectrophotometer [55].
Analysis of precursors rRNA (pre-rrn) by primer extension
The ribosomal promoter usage of M. fortuitum in Sauton cultures, treated and untreated with H2O2, was determined using the primer extension (PE) procedure. The primer JY15 (Table 2) was end labelled with [γ-32P]ATP by T4 polynucleotide kinase, and primer extension was carried out with Avian Myeloblastosis Virus (AMV) reverse transcriptase (Promega) as described previously [59]. PE experiments were performed with 25 μg of total RNA per sample. Transcriptional products, corresponding to each rrn promoter, were quantified in the gels by using an Instant Imager system (Packard-Izasa).
Table 2.
Oligonucleotides used in this study
| DNA target | Forward Primer [Sequence 5'→3'] | Reverse primer [Sequence 5'→3'] | Annealing T (°C) | Size amplicon (bp) |
| Northen Blot (probes) | ||||
| rrs | kk4 [TTGGAGAGTTTGATCCTGGCTCAG] | Rac6 [GTGGACTACCAGGGTATCTAATCCT] | 58 | 791 |
| sodA | Sod3.1 [ACATCTCGGGTCAGATCAACGAG C] | Sod4 [GACGTTCTTGTACTGCAGGTAG] | 58 | 465 |
| katGII | Cat39 [AAACCCATCCTACTATCG] | Cat40 [GGTTCACGTAGATCAGG] | 50 | 696 |
| qRT-PCR | ||||
| rrnA-P1 | FoP1 [CTGCTCGTCAGCCTCGAAATCG] | P1R-LC [ACTTCAAAAGATTAGCGCGG] | 55 | 75 |
| sodA | Sod12 [CCTTCCAGCTCTACGACCAG] | Sod13 [CCTTCCAGCTCTACGACCAG] | 55 | 114 |
| katGII | katLC1 [GTGGTCCGATCTCATCGTCT] | katLC2 [CAGTAGATGTCCTCCTCGGG] | 55 | 117 |
| furAII | furA-Fw [ACGGCGGACTTCCAGCAG] | furA-Rev [ACGGATGATCGTTTCGGTG] | 62 | 120 |
| RPA | ||||
| sodA | Sod F8 [ACCTGCACACCCGCAACC] | Sod 3T [PT7-GCTCGTTGATCTGCCCCGA] | 60 | 324 |
| katGII | Cat35 [GACAACCACCACCACGT] | P1.1 T [PT7-TCTCGGCGGGTTCAGTCT] | 50 | 292 |
| furAII | FUR-II-10 [TCGTCTAGCGCCCTCACC] | FUR-II-1T [PT7-ACTCGGGGTCGGGTCACA] | 60 | 177 |
| Other primers | ||||
| furAII-katGII | furAII-Fo [GACCTACCCGAGGTCTCTCA] | P1.1 [TCTCGGCGGGTTCAGTCT] | 54 | 388 |
| pre-rrnA | FoPCL1 [CAAAACAGGGCGGCAAAGC] | cKK4 [GAGCCAGGATCAAACTCTCC] | 58 | 225 |
| 16S-rrnA | JY15 [CACACTATTGAGTTCTC] | - | - | |
Oligonucleotides used for Norther Blot, RNAse protection assay, real-time qRT-PCR and conventional PCR.
Abbreviations: PT7: RNA polymerase promoter sequence for the phage T7 (AATTCTAATACGACTCACTATAGGG).
Identification of transcription starts points using RNAse Protection Assay
RNase protection assay (RPA) was carried out as described previously [60]. The RPA method requires previous preparation of a radiolabeled RNA probe complementary to the RNA transcript by transcription of a minigene corresponding to the region under analysis [61]. Minigenes were synthesized by PCR amplification of the M. fortuitum corresponding genomic regions by using oligonucleotides described in Table 2.
Radiolabelled transcription products were obtained by using the Riboprobe kit (Promega) as specified by the manufacturer; this yielded probes of 324, 292 and 177 nucleotides for sodA, katGII and furAII genes respectively. The products were purified on a polyacrylamide gel electrophoresis (6% [wt/vol] polyacrylamide) sequencing gel. The appropriate bands were excised and eluted as described previously [60].
Hybridization was carried out in PES buffer [60], with radiolabeled samples (2.5 × 105 cpm) and 20 μg of DNA-free RNA; the reaction products were digested with different amounts of an RNase cocktail (Ambion). Stock RNase cocktail comprises both RNase T1 (20 U/ul) and RNase A (1 μg/μl), and an optimum concentration (up to a 10-fold dilution) was established for use in the RNA protection assays.
Products of both primer extension and RNase protection assays were analyzed by electrophoresis through 6% (wt/vol) polyacrylamide-8 M urea sequencing gels. Radioactive products were located by autoradiography either at 20°C or at -70°C using an intensifying screen. The gels were calibrated with the products of appropriate DNA-sequencing reactions. DNA sequences were determined by the dideoxy chain termination procedure with [α-35S]dATP [55]. The putative promoter regions (-10 and -35 boxes) were determined (Table 1).
Analysis of furAII-katGII region
The cDNA from M. fortuitum under the different stresses was analysed by PCR using the primers furAII-Fo and P1.1 (Table 2) to test the presence of furAII and katGII cotranscripts. PCR cycling was performed as follows: a denaturizing step at 95°C for 5 minutes; 36 cycles of 95°C for 45 seconds, 54°C for 45 seconds and 72°C for 1 minute; and a final extension at 72°C for 10 minutes.
Analysis of mRNA by Northern blot
Northern blot analysis was performed by fractionation of 20 to 30 μg of total RNA samples on a 1.5% agarose gel containing 6.6% formaldehyde. RNA ladder (0.24 to 9.5 kb; Gibco BRL) was used as the molecular size standard. The gels were transferred to a nylon membrane (Hybond-N; Amersham) by capillary blotting in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), and further cross-linked by UV irradiation [55].
The membranes were hybridized with three probes: katGII, sodA and the 16S rRNA (rrs) genes sequentially. The probes were prepared by standard PCR. Amplifications were performed on M. fortuitum chromosomal DNA (100 ng) in a final volume of 50 μl containing 1 μM of each primer and 1 U of Taq polymerase (Amplitaq Gold, Roche). All amplifications were carried out as follows: 36 cycles of 1 min at 95°C; 45 s at the annealing temperature (Table 2); and 1 min at 72°C, with subsequent 10 min final extension at 72°C. The primers used amplify the coding regions of the corresponding gene. Primers characteristics, annealing temperatures, and sizes of each amplicon are indicated in the Table 2. Hibridizations were made with 90 ng of each PCR product labeled with 32P α-dCTP using Megaprime DNA labeling system (Amersham) at 65°C overnight. Standard procedures were used for both hybridization and washes [55]. Filters were spread out and rehybridized with the three probes sequentially. The radioactivity hybridized for each gene was quantified on the filter using an Instant Imager system (Packard-Izasa) and also detected by exposition of the membranes to X-ray films. Data derived from hybridization with the rrs gene were used as standard to normalize the amounts of RNA fixed on the filter.
Analysis of mRNA by qRT-PCR
One hundred ng of the mycobacterial RNA isolated was reverse transcribed by using 30 U AMV reverse transcriptase and random primer hexamers (Promega). The absence of DNA following DNase treatment was checked by performing conventional PCR using FoPCL1 and cKK4 (Table 2). Conventional PCR cycling was performed as follows: a denaturizing step at 95°C for 5 minutes; 36 cycles of 95°C for 1 minute, 58°C for 45 seconds and 72°C for 2 minutes; and a final extension at 72°C for 10 minutes. The RNA product was considered suitable for reverse transcription when the control-DNA PCR was negative.
The qRT-PCR method was used to determine the level of each transcriptional-product derived from M. fortuitum under the different stresses tested. The qRT-PCR was performed with cDNAs prepared from 2 separate cultures per treatment. A total of 16 data were obtained per point derived from two cDNA separate preparations from each culture. Each of the four cDNA samples obtained was amplified twice in duplicated experiments.
Real-time PCR was carried out using a capillary PCR instrument (Light Cycler; Roche). The amplification of the target sequence was detected using SYBR green Conditions were used as follows: LightCycler Fast Start DNA master SYBR Green I reagent (1 μl) was supplemented with 3.5 mM (final concentration) MgCl2 and 0.5 μM of each primer in 7 μl of volume. Sample cDNA (3 μl) was added to the mix. The PCR cycling programme was as follows: denaturizing, 1 cycle of 95°C for 10 min with a transition rate of 20°C/s; amplification, 45 cycles at 95°C for 0 s, the corresponding annealing temperature for each product (Table 2) for 5 s and an extension at 72°C for 10 s with a single fluorescence acquisition, in all the cases the transition rate was 20°C/s. Specificity of the reaction was checked by analysis of the melting curve of the final amplified product. The oligonucleotides used in qRT-PCR are indicated in the Table 2. In all cases, the negative control was undetectable, because the PCR experiments were stopped before its fluorescence increased.
qRT-PCR data analysis
The qRT-PCR data were plotted as the fluorescence signal versus the cycle number. An arbitrary threshold was set at the midpoint of the log of fluorescent level versus cycle number plot. The Ct value is defined as the cycle number at which the fluorescent level crosses this threshold. In the present study, the fold change in the cDNA amounts was calculated from the formula 2-ΔΔCt (Applied Biosystem User Bulletin). Using the 2-ΔΔCt method, the data are presented as the fold change in gene expression normalized to an endogenous reference gene and relative to the untreated control. This calculation transforms the logarithmic Ct data to a linear value. Error of the data was calculated by converting the data from logarithmic to linear using the expression 2-Ct. Raw Ct values falsely represents the error and should be avoided [50]. For the untreated control sample, ΔΔCt equals zero, therefore by definition, the fold change in gene expression relative to the untreated control is one. For the treated samples, 2-ΔΔCt indicates the fold change in gene expression relative to the control [38]. The fold change in cDNA (per each target gene) relative to the rrnA-P1 control [45] was determined as follows: fold change = 2-ΔΔCt; where ΔΔCt = (CtTarget - CtrrnA-P1)Sample - (CtTarget - CtrrnA-P1)Control. The sample corresponds to each H2O2 treated cultures, and controls are the untreated cultures in the formula. According to Winer [62] the relative quantification of gene expression by using the 2-ΔΔCt method correlates with the absolute gene quantification obtained using standard curves. The statistical significance of data was determined by applying one-way analysis of variance.
Authors' contributions
MdCN carried out the molecular biology and protein studies, and participated in the drafted the manuscript. MdCM participated and supervised the design of the study and helped to the elaboration of the manuscript. MJR carried out the identification of tsp of the genes in this study. MJG conceived the study, and participated in its design and follow up and coordinate the manuscript. All authors read and approved the final version of the manuscript.
Supplementary Material
Quantitative data corresponding to Figures 1 and 3. Data corresponding to Figure 1 show the densitometry of a representative activity gel of KatG and SOD in protein extracts of Mycobacterium fortuitum. Data corresponding to Figure 3 show the cpm values of a representative experiment of hybridization of a Northern Blot with KatGII, sodA and rrs (16SrRNA) before and after normalization.
Acknowledgments
Acknowledgements
This work was supported by grants from the Spanish Ministry of Education (SAF2005-05733) and EU INCO-DEV program (ICA4-CT-2002-10063). We would like to acknowledge to TJ Bull for his revision of the manuscript and helpful advice.
Contributor Information
María Carmen Núñez, Email: cnserrano@inia.es.
María Carmen Menéndez, Email: carmen.menendez@uam.es.
María José Rebollo, Email: maria-jose@gsk.com.
María J García, Email: mariaj.garcia@uam.es.
References
- Winthrop KL, Albridge K, South D, Albrecht P, Abrams M, Samuel MC, Leonard W, Wagner J, Vugia DJ. The Clinical Management and Outcome of Nail Salon-Acquired Mycobacterium fortuitum Skin Infection. Clin Infect Dis. 2004;38:38–44. doi: 10.1086/380459. [DOI] [PubMed] [Google Scholar]
- Angeli K, Lacour JP, Mantoux F, Roujeau JC, Andre P, Truffot-Pernot C, Ortonne JP. Mycobacterium fortuitum skin infection occurring after a facelift. Ann Dermatol Venereol. 2004;131:198–200. doi: 10.1016/s0151-9638(04)93571-5. [DOI] [PubMed] [Google Scholar]
- Geertsma MF, Nibbering PH, Pos O, Van Furth R. Interferon-gamma-activated human granulocytes kill ingested Mycobacterium fortuitum more efficiently than normal granulocytes. Eur J Immunol. 1990;20:869–873. doi: 10.1002/eji.1830200423. [DOI] [PubMed] [Google Scholar]
- Nibbering PH, Pos O, Stevenhagen A, Van Furth R. Interleukin-8 enhances nonoxidative intracellular killing of Mycobacterium fortuitum by human granulocytes. Infect Immun. 1993;61:3111–3116. doi: 10.1128/iai.61.8.3111-3116.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman MF, Morgan RW, Jacobson FS, Ames BN. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell. 1985;41:753–762. doi: 10.1016/s0092-8674(85)80056-8. [DOI] [PubMed] [Google Scholar]
- Demple B, Halbrook J. Inducible repair of oxidative DNA damage in Escherichia coli. Nature. 1983;304:466–468. doi: 10.1038/304466a0. [DOI] [PubMed] [Google Scholar]
- Gordon AH, Hart PD. Stimulation or inhibition of the respiratory burst in cultured macrophages in a mycobacterium model: initial stimulation is followed by inhibition after phagocytosis. Infect Immun. 1994;62:4650–4651. doi: 10.1128/iai.62.10.4650-4651.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe TR, Hibler NS, Deretic V. Response of Mycobacterium tuberculosis to reactive oxygen and nitrogen intermediates. Mol Med. 1996;2:134–142. [PMC free article] [PubMed] [Google Scholar]
- Laochumroonvorapong P, Paul S, Manca C, Freedman VH, Kaplan G. Mycobacterial growth and sensitivity to H2O2 killing in human monocytes in vitro. Infect Immun. 1997;65:4850–4857. doi: 10.1128/iai.65.11.4850-4857.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lathigra R, Garbe T, Catty D, Young D. Genetic analysis of superoxide dismutase, the 23 kilodalton antigen of Mycobacterium tuberculosis. Mol Microbiol. 1991;5:381–391. doi: 10.1111/j.1365-2958.1991.tb02120.x. [DOI] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream M-A, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Dussurget O, Stewart G, Neyrolles O, Pescher P, Young D, Marchal G. Role of Mycobacterium tuberculosis copper-zinc superoxide dismutase. Infect Immun. 2001;69:529–533. doi: 10.1128/IAI.69.1.529-533.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piddington DL, Fang FC, Laessig T, Cooper AM, Orme IM, Buchmeier NA. Cu, Zn Superoxide Dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect Immun. 2001;69:4980–4987. doi: 10.1128/IAI.69.8.4980-4987.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harth G, Horwitz MA. Export of recombinant Mycobacterium tuberculosis superoxide dismutase is dependent upon both information in the protein and mycobacterial export machinery. J Biol Chem. 1999;274:4281–4292. doi: 10.1074/jbc.274.7.4281. [DOI] [PubMed] [Google Scholar]
- Edwards KM, Cynamon MH, Voladri RK, Hager CC, DeStefano MS, Tham KT, Lakey DL, Bochan MR, Kernodle DS. Iron-cofactored superoxide dismutase inhibits host responses to Mycobacterium tuberculosis. Am J Respir Crit Care Med. 2001;164:2213–2219. doi: 10.1164/ajrccm.164.12.2106093. [DOI] [PubMed] [Google Scholar]
- Volpe E, Cappelli G, Grassi M, Martino A, Serafino A, Colizzi V, Sanarico N, Mariani F. Gene expression profiling on human macrophages at late time of infection with Mycobacterium tuberculosis. Immunology. 2006;118:449–460. doi: 10.1111/j.1365-2567.2006.02378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez MC, Domenech P, Prieto J, Garcia MJ. Cloning and expression of the Mycobacterium fortuitum superoxide dismutase gene. FEMS Microbiol Lett. 1995;134:273–278. doi: 10.1111/j.1574-6968.1995.tb07950.x. [DOI] [PubMed] [Google Scholar]
- Miller RA, Britigan BE. Role of oxidants in microbial pathophysiology. Clin Microbiol Rev. 1997;10:1–18. doi: 10.1128/cmr.10.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne LG, Diaz GA. Serological, taxonomic and kinetic studies of the T and M classes of mycobacterial catalase. Intern J System Bacteriol. 1982;32:296–304. [Google Scholar]
- Zhang Y. Life without katG. Trends Microbiol. 1996;4:415–416. doi: 10.1016/0966-842x(96)30030-9. [DOI] [PubMed] [Google Scholar]
- Slayden RA, Barry CE., 3rd The genetics and biochemistry of isoniazid resistance in Mycobacterium tuberculosis. Microbes Infect. 2000;2:659–669. doi: 10.1016/s1286-4579(00)00359-2. [DOI] [PubMed] [Google Scholar]
- Li Z, Kelley C, Collins F, Rouse D, Morris S. Expression of katG in Mycobacterium tuberculosis is associated with its growth and persistence in mice and guinea pigs. J Infect Dis. 1998;177:1030–1035. doi: 10.1086/515254. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992;358:591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Garbe T, Young D. Transformation with katG restores isoniazid-sensitivity in Mycobacterium tuberculosis isolates resistant to a range of drug concentrations. Mol Microbiol. 1993;8:521–524. doi: 10.1111/j.1365-2958.1993.tb01596.x. [DOI] [PubMed] [Google Scholar]
- Smith I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin Microbiol Rev. 2003;16:463–496. doi: 10.1128/CMR.16.3.463-496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussurget O, Rodriguez M, Smith I. protective role of the Mycobacterium smegmatis IdeR against reactive oxygen species and isoniazid toxicity. Tuber Lung Dis. 1998;79:99–106. doi: 10.1054/tuld.1998.0011. [DOI] [PubMed] [Google Scholar]
- Zheng M, Doan B, Schnerider TD, Storz G. OxyR and SoxRS regulation of fur. J Bacteriol. 1999;181:4639–4643. doi: 10.1128/jb.181.15.4639-4643.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagan-Ramos E, Song J, McFalone M, Mudd MH, Deretic V. Oxidative stress response and characterization of the oxyR-ahpC and furA-katG loci in Mycobacterium marinum. J Bacteriol. 1998;180:4856–4864. doi: 10.1128/jb.180.18.4856-4864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahrt TC, Song J, Siple J, Deretic V. Mycobacterial FurA is a negative regulator of catalase-peroxidase gene katG. Mol Microbiol. 2001;39:1174–1185. doi: 10.1111/j.1365-2958.2001.02321.x. [DOI] [PubMed] [Google Scholar]
- Master S, Zahrt TC, Song J, Deretic V. Mapping of Mycobacterium tuberculosis katG promoters and their differential expression in infected macrophages. J Bacteriol. 2001;183:4033–4039. doi: 10.1128/JB.183.13.4033-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pym AS, Domenech P, Honore N, Song J, Deretic V, Cole ST. Regulation of catalase-peroxidase (KatG) expression, isoniazid sensitivity and virulence by furA of Mycobacterium tuberculosis. Mol Microbiol. 2001;40:879–889. doi: 10.1046/j.1365-2958.2001.02427.x. [DOI] [PubMed] [Google Scholar]
- Milano A, Forti F, Sala C, Riccardi G, Ghisotti D. Transcriptional regulation of furA and katG upon oxidative stress in Mycobacterium smegmatis. J Bacteriol. 2001;183:6801–6806. doi: 10.1128/JB.183.23.6801-6806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Forti F, Di Florio E, Canneva F, Milano A, Riccardi G, Ghisotti D. Mycobacterium tuberculosis FurA autoregulates its own expression. J Bacteriol. 2003;185:5357–5362. doi: 10.1128/JB.185.18.5357-5362.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez MC, Ainsa JA, Martin C, García MJ. katGI and katGII encode two different catalases-peroxidases in Mycobacterium fortuitum. J Bacteriol. 1997;179:6880–6886. doi: 10.1128/jb.179.22.6880-6886.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson UE, Heid CA, Williams PM. A novel method for real time quantitative RT-PCR. Genome Res. 1996;6:995–1001. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Johnson MR, Wang K, Smith JB, Heslin MJ, Diasio RB. Quantitation of dihydropyrimidine dehydrogenase expression by real-time reverse transcription polymerase chain reaction. Anal Biochem. 2000;278:175–184. doi: 10.1006/abio.1999.4461. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔCT) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Dubnau E, Fontan P, Manganelli R, Soares-Appel S, Smith I. Mycobacterium tuberculosis genes induced during infection of human macrophages. Infect Immun. 2002;70:2787–2795. doi: 10.1128/IAI.70.6.2787-2795.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud C, Papavinasasundaram KG, Speight RA, Springer B, Sander P, Bottger EC, Colston MJ, Draper P. The functions of OmpATb, a pore-forming protein of Mycobacterium tuberculosis. Mol Microbiol. 2002;46:191–201. doi: 10.1046/j.1365-2958.2002.03152.x. [DOI] [PubMed] [Google Scholar]
- Timm J, Post FA, Bekker LG, Walther GB, Wainwright HC, Manganelli R, Chan WT, Tsenova L, Gold B, Smith I, Kaplan G, McKinney Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc Natl Acad Sci USA. 2003;100:14321–14326. doi: 10.1073/pnas.2436197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordillo S, Guirado E, Gil O, Diaz J, Amat I, Molinos S, Vilaplana C, Ausina V, Cardona P-J. Usefulness of acr expression for monitoring latent Mycobacterium tuberculosis bacilli in in vitro and in vivo experimental models. Scand J Immunol. 2006;64:30–39. doi: 10.1111/j.1365-3083.2006.01765.x. [DOI] [PubMed] [Google Scholar]
- Dubnau E, Chan J, Mohan VP, Smith I. Responses of Mycobacterium tuberculosis to growth in mouse lung. Infect Immun. 2007;73:3754–3757. doi: 10.1128/IAI.73.6.3754-3757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli R, Dubnau J, Tyagi S, Kramer FR, Smith I. Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol Microbiol. 1999;31:715–724. doi: 10.1046/j.1365-2958.1999.01212.x. [DOI] [PubMed] [Google Scholar]
- Menendez MC, Rebollo MJ, Nuñez MC, Cox RA, Garcia MJ. Analysis of the precursor-rRNA fractions of rapidly growing mycobacteria: quantification by methods that include the use of a constitutive promoter (rrnA P1) as a novel standard. J Bacteriol. 2005;187:534–543. doi: 10.1128/JB.187.2.534-543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez M, Smith I. Determinants of Mycobacterial gene expression. In: Hatfull GF, Jacobs WR Jr, editor. Molecular genetics of Mycobacteria. ASM Press, Washington. DC; 2000. pp. 111–129. [Google Scholar]
- Smith I, Bishai WR, Nagaraja V. Control of the mycobacterial transcription. In: Cole ST et al, editor. Tuberculosis and the tubercle bacillus. ASM Press, Washington DC; 2005. pp. 219–234. [Google Scholar]
- Gonzalez-y-Merchand JA, Garcia MJ, Gonzalez-Rico S, Colston MJ, Cox RA. Strategies used by pathogenic and nonpathogenic mycobacteria to synthesize rRNA. J Bacteriol. 1997;179:6949–6958. doi: 10.1128/jb.179.22.6949-6958.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-y-Merchand JA, Colston MJ, Cox RA. Roles of multiple promoters in transcription of ribosomal DNA: effects of growth conditions on precursor rRNA synthesis in micobacteria. J Bacteriol. 1998;180:5756–5761. doi: 10.1128/jb.180.21.5756-5761.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Zakrajsek BA. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods. 2000;46:69–81. doi: 10.1016/s0165-022x(00)00129-9. [DOI] [PubMed] [Google Scholar]
- Sala C, Forti F, Magnoni F, Ghisoti D. The katG mRNA of Mycobacterium tuberculosis and Mycobacterium smegmatis is processed at its 5' end and is stabilized by both a polypurine sequence and translation initiation. BMC Mol Biol. 2008;9:33. doi: 10.1186/1471-2199-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes OnLine Database http://www.genomesonline.org
- Sherman DR, Sabo PJ, Hickey MJ, Arain TM, Mahairas GG, Yuan Y, Barry CE, III, Stover CK. Disparate responses to oxidative stress in saprophytic and pathogenic mycobacteria. Proc Natl Acad Sci USA. 1995;92:6625–6629. doi: 10.1073/pnas.92.14.6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder KM, Horwitz MA. Identification of iron-regulated proteins of Mycobacterium tuberculosis and cloning of tandem genes encoding a low iron-induced protein and a metal transporting ATPase with similarities to two-component metal transport systems. Microb Pathog. 1998;24:133–143. doi: 10.1006/mpat.1997.9999. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning: A laboratory manual 3rd. Cold Spring Harbor YN, Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Wayne LG, Diaz GA. A double staining method for differentiating between two classes of mycobacterial catalase in polyacrylamide electrophoresis gels. Anal Biochem. 1986;157:89–92. doi: 10.1016/0003-2697(86)90200-9. [DOI] [PubMed] [Google Scholar]
- Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- Wayne LG, Fridovich I. Assaying for Superoxide dismutase activity: some large consequences of minor changes in conditions. Anal Biochem. 1987;161:559–566. doi: 10.1016/0003-2697(87)90489-1. [DOI] [PubMed] [Google Scholar]
- Gonzalez-y-Merchand JA, Colston MJ, Cox RA. The rRNA operons of Mycobacterium smegmatis and Mycobacterium tuberculosis: comparison of promoter elements and of neighbouring upstream genes. Microbiology. 1996;142:667–674. doi: 10.1099/13500872-142-3-667. [DOI] [PubMed] [Google Scholar]
- Movahedzadeh F, Gonzalez-y-Merchand JA, Cox RA. Transcription start-site mapping. In: Parish T, Stoker NG, editor. Mycobacterium tuberculosis protocols. Humana Press. Totowa, NJ; 2001. pp. 105–124. [DOI] [PubMed] [Google Scholar]
- Mironov VN, Van Montagu M, Inzé D. High throughput RNase protection assay. Nucleic Acids Res. 1995;23:3359–3360. doi: 10.1093/nar/23.16.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270:41–49. doi: 10.1006/abio.1999.4085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Quantitative data corresponding to Figures 1 and 3. Data corresponding to Figure 1 show the densitometry of a representative activity gel of KatG and SOD in protein extracts of Mycobacterium fortuitum. Data corresponding to Figure 3 show the cpm values of a representative experiment of hybridization of a Northern Blot with KatGII, sodA and rrs (16SrRNA) before and after normalization.