

Abstract
More than 60% of low-grade non-invasive papillary urothelial cell carcinomas contain activating point mutations of FGFR3. The phenotypic consequences of constitutive activation of FGFR3 in bladder cancer have not been elucidated and further studies are required to confirm the consequences of inhibiting receptor activity in urothelial cells. We measured FGFR3 transcript levels and demonstrated that transcript levels were significantly more abundant in low-stage and grade tumours. We identified a tumour cell line, 97-7, expressing the most common FGFR3 mutation (S249C) at similar FGFR3 transcript levels to low-stage and grade tumours. In these cells S249C FGFR3 protein formed stable homodimers and was constitutively phosphorylated. We used retrovirus-mediated delivery of shRNA to knockdown S249C FGFR3. This induced cell flattening, decreased cell proliferation, and reduced clonogenicity on plastic and in soft agar. However, no effects of knockdown of wildtype FGFR3 were observed in telomerase immortalised normal human urothelial cells, indicating possible dependence of the tumour cell line on mutant FGFR3. Re-expression of S249C FGFR3 in shRNA-expressing 97-7 cells resulted in a reversal of phenotypic changes, confirming the specificity of the shRNA. These results indicate that targeted inhibition of S249C FGFR3 may represent a useful therapeutic approach in superficial bladder cancer.
Keywords: FGFR3, urothelial cell carcinoma, shRNA, therapeutic target
Introduction
Urothelial cell carcinoma (UCC) of the urinary bladder is the fifth most common cancer in industrialised countries (Parkin et al., 2005). Molecular and pathological studies suggest that low-grade non-invasive, and high-grade invasive UCC, arise via distinct pathways (Knowles, 2006; Wu, 2005). Low-grade non-invasive UCC represent the majority of tumours at presentation. A high proportion of patients with low-grade UCC develop recurrences but usually with no progression to invasive disease (Holmang et al., 1995; Kurth et al., 1995). The high rate of recurrence requires intensive monitoring by cystoscopy, which is an uncomfortable, invasive and expensive procedure, placing a heavy burden on both patients and health care providers. For these patients the development of non-invasive tests for recurrence and targeted therapies to prevent recurrence are needed. Invasive tumours (pT2 or greater) appear to arise from high-grade carcinoma in situ. Patients that present with invasive disease have a much worse prognosis, with a 50% 5-year survival, and therefore the development of effective systemic therapies is highly desirable.
These two phenotypic pathways to tumour development are also distinguished by specific genetic alterations. High-grade invasive UCC tend to have multiple and complex genomic changes and show functional defects in the p53 and Rb pathways. In contrast, few genetic changes have been identified in low-grade superficial UCC. The most common are loss of heterozygosity on chromosome 9 and mutations in fibroblast growth factor receptor 3 (FGFR3). Mutations of FGFR3 are significantly associated with low tumour stage and grade with a frequency >60% in Ta tumours (Billerey et al., 2001; van Rhijn et al., 2001). The same mutations, which cause constitutive activation of FGFR3, are found in autosomal dominant heritable disorders of skeletal development (Webster & Donoghue, 1997).
FGFs and their receptors play important roles in many biological processes including embryonic development, wound healing, hematopoiesis, and angiogenesis. The FGFR family comprises four main members of high-affinity receptors (FGFRs1-4). These consist of a core structure containing an extracellular domain, a hydrophobic transmembrane domain and an intracellular kinase domain. FGF ligands bind to the extracellular domain resulting in receptor dimerization and activation. The receptors and their isoforms are expressed in a cell- and tissue-specific manner, which reflects their differential roles in different tissues and cell lineages. In normal bladder cells, two major isoforms of FGFR3 have been identified; FGFR3b and FGFR3 Δ8-10. FGFR3b is the full-length receptor and FGFR3 Δ8-10 is a soluble isoform that can act as a dominant negative regulator of FGF1-induced proliferation (Tomlinson et al., 2005). A second full-length FGFR3 isoform, FGFR3c expressed by mesenchymal cells (Scotet & Houssaint, 1995), is produced by alternative splicing of exons 8 and 9 and binds a wider range of FGF ligands than FGFR3b (Ornitz et al., 1996). Although this isoform is not detected in normal urothelial cells, altered expression of FGFR3 isoforms has been observed in bladder cancer cell lines, many of which show a decrease in FGFR3 Δ8-10 expression and an isoform switch from FGFR3b to FGFR3c (Tomlinson et al., 2005).
Mutated FGFR3c isoforms show transforming ability in NIH-3T3 cells (Chesi et al., 2001; Hart et al., 2001; Hart et al., 2000; Ronchetti et al., 2001) and a recent study has demonstrated that the most common mutant form of FGFR3b found in bladder cancer (S249C) is also able to transform NIH-3T3 cells, inducing anchorage-independent growth and tumour formation in nude mice (Bernard-Pierrot et al., 2006). Although transforming ability of this mutant form has not yet been demonstrated in urothelial cells, the combined evidence of high frequency of somatic mutation in bladder cancer and transforming ability in rodent fibroblasts, indicates that mutation of FGFR3b is a key event in the pathway leading to low-grade superficial bladder cancer and is potentially a good therapeutic target.
In addition to bladder carcinoma, mutations of FGFR3 identical to those found in skeletal dysplasia syndromes have been identified in multiple myeloma (Chesi et al., 2002), cervical carcinoma (Cappellen et al., 1999) and benign skin tumours (Logie et al., 2005). Increased expression of wildtype FGFR3 is also found in multiple myeloma. Around 10-20% of myeloma patients show a t(4;14)(p16;q32) that translocates FGFR3 on chromosome 4 into the IgH locus on chromosome 14, resulting in some cases, in overexpression of FGFR3 (Chesi et al., 1997; Chesi et al., 1998; Richelda et al., 1997). Inhibition of FGFR3 in t(4;14) multiple myeloma cell lines, by receptor tyrosine kinase (RTK) inhibitors, antibodies and RNA interference induces cytostatic and cytotoxic responses (Chen et al., 2005; Grand et al., 2004; Paterson et al., 2004; Trudel et al., 2004; Trudel et al., 2005; Trudel et al., 2006; Zhu et al., 2005). Thus targeting either mutant or over-expressed wildtype FGFR3 appears to be a valid therapeutic approach in multiple myeloma.
Recent studies have demonstrated that not only is FGFR3 mutated but FGFR3 protein is also overexpressed in UCC (Gomez-Roman et al., 2005). Both increased expression of wildtype isoforms and altered ligand-binding affinity via isoform switching, could lead to unregulated signaling with similar effects to mutational activation. Indeed, initial studies suggest that targeting FGFR3 that is highly expressed or mutated, may be a valid therapeutic approach for bladder cancer (Bernard-Pierrot et al., 2006; Martinez-Torrecuadrada et al., 2005).
To date no study has examined the effect of targeted inhibition or downregulation of the most common mutant form of FGFR3 (S249C) found in bladder cancer. Here, we have assessed the phenotypic consequences of specific inhibition of FGFR3 in normal urothelial cells and in bladder tumour cells with the S249C FGFR3 mutation. We examined the relationship of FGFR3 expression levels to tumour stage and identified a cell line with FGFR3 mutation that expresses FGFR3 at similar transcript levels to low-stage UCC. We used an shRNA retrovirus to stably knockdown FGFR3 and examined the effects on the phenotype of these mutant cells and of normal urothelial cells. Our results are consistent with an oncogenic role for mutant FGFR3b and demonstrate the potential of targeting FGFR3 as a therapy to prevent recurrence in low-stage superficial bladder cancer and to treat tumours of all grades and stages that show over-expression and/or mutation of FGFR3.
Results
Association of FGFR3 expression levels with tumour stage and grade
To examine the effects of downregulation of mutant FGFR3b it was important to use a bladder cancer cell line that was representative of the type of bladder tumours that have FGFR3 mutation. Numerous reports have demonstrated that FGFR3 mutation is most frequent in low-stage and grade bladder cancers (Bakkar et al., 2003; Kimura et al., 2001; van Rhijn et al., 2001) and these tumours express increased levels of FGFR3 (Gomez-Roman et al., 2005). To identify a bladder cancer cell line that expressed similar levels of FGFR3 to low-stage and grade bladder tumours, we measured FGFR3 transcript levels in bladder tumours and bladder cancer cell lines by real-time RT-PCR (Figure 1). We have previously demonstrated that transcript levels measured by real-time RT-PCR represent protein levels in bladder cell lines (Tomlinson et al., 2005). We examined FGFR3 transcript levels in 34 pTa, 19 pT1 and 12 pT2 tumours. Levels of FGFR3 in pTa, pT1 and pT2 tumours were increased by 5.8, 4.8 and 1.3 fold, respectively, compared to normal human urothelial cells (NHUC) (Figure 1A). Superficial bladder tumours (pTa and pT1) expressed significantly higher levels of FGFR3 than invasive carcinomas (Student's t test, p < 0.005). FGFR3 levels were also higher in grade 2 compared with grade 3 tumours (p < 0.05) (Figure 1B). Eighty five percent (23 of 27) of bladder cancer cell lines expressed lower levels of FGFR3 than NHUC (Figure 1C). Three of the cell lines have been found to contain FGFR3 mutations (Jebar et al., 2005; Sibley et al., 2000). These are 97-7 (S249C), 97-29 (S249C), and J82 (K652E). The cell line 97-7 was used in subsequent experiments as it contained the most frequently identified mutation in bladder cancer (S249C) and expressed similar levels of FGFR3 to low-stage and grade tumours. This cell line was established from a T1 grade II/III primary tumour and was shown to contain a mutation in TP53 (Sarkar et al., 2000). We have shown previously that this line does not contain a Ras gene mutation (Jebar et al., 2005).
Figure 1.
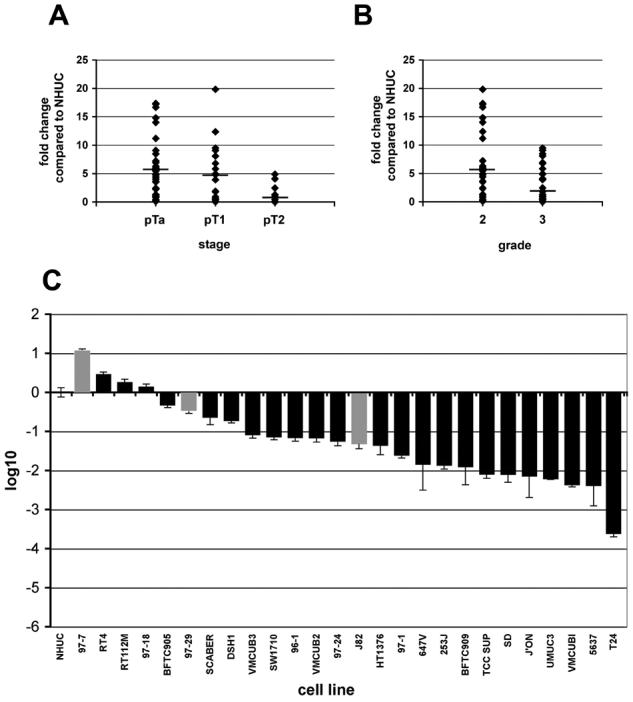
Expression levels of FGFR3 in bladder tumours and bladder cancer cell lines. FGFR3 transcript levels were measured by real-time RT-PCR and normalised to SDHA. Values represent fold difference compared to FGFR3 transcript levels in NHUC. A, FGFR3 transcript levels were measured in 34 pTa, 19 pT1 and 12 pT2 bladder tumours. B, the tumours were separated according to grade; 31 grade 2 and 33 grade 3. Only one grade 1 tumour was harvested and therefore not included in the graph. C, FGFR3 transcript levels were measured in 26 bladder cancer cell lines and the fold change was represented using log10. The bars for bladder cancer cell lines containing FGFR3 mutations are shown in grey.
Establishment of stable FGFR3 knockdown by retrovirally-delivered shRNA
Knockdown of transcripts using small hairpin RNA (shRNA) is a powerful tool for studying gene function. We sub-cloned a human U6 promoter (Matsukura et al., 2003) into the retroviral vector pRS-puro. Eight shRNAs targeting FGFR3 were designed and tested in 97-7 cells. A non-specific shRNA was constructed and used as a control for shRNA transduction. Only one shRNA was effective and this knocked down FGFR3 transcript levels by 91% (Figure 2A). FGFR3 protein levels were reduced in cells expressing the shRNA at week 3 and week 7, demonstrating efficient and stable knockdown (Figure 2B).
Figure 2.

Retrovirus-mediated delivery of shRNA in bladder cancer cells. A, FGFR3 transcript levels in 97-7 cells transduced with vector, non-specific shRNA or FGFR3 shRNA retroviruses were measured by real-time RT-PCR. B, lysates from 97-7 cells harvested three and seven weeks post-transduction and FGFR3 protein levels and examined by western blot.
S249C mutation stabilizes receptor dimerization and leads to constitutive phosphorylation
The 97-7 cell line contains a heterozygous S249C FGFR3 mutation (Jebar et al., 2005). To confirm that the FGFR3 transcripts in these cells contain the mutation, we performed RT-PCR across exon 7 and sequenced the product. FGFR3 transcripts were homozygous for the S249C mutation (data not shown). This suggests that only the allele containing the mutation is expressed. The cysteine residue in the S249C mutant receptors may allow disulphide bonds to form between the extracellular domain of mutant monomers, thus inducing activation. The presence of mutant cysteine residues in the Ig2-Ig3 linker domain or the transmembrane region of FGFR3c results in abnormal dimerization and ligand-independent activation (Adar et al., 2002; d'Avis et al., 1998). We demonstrated that the mutant FGFR3b protein in 97-7 formed stable dimers unlike the wildtype receptor in TERT-NHUC (Figure 3A). Dimerization of FGFR3 is predicted to result in receptor phosphorylation. We examined the phosphorylation status of wild-type FGFR3b expressed in TERT-NHUC and S249C FGFR3b expressed in 97-7 cells by immunoprecipitation and western blotting (Figure 3B). S249C FGFR3b had a higher phosphorylation level than wild-type FGFR3b.
Figure 3.

S249C FGFR3b is dimerized and phosphorylated and its knockdown induces cell flattening. A, protein lysates from TERT-NHUC over-expressing FGFR3b and 97-7 were western blotted with (+) and without (−) denaturation. (−) samples contained no ß-mercaptoethanol and were not heat denatured. S249C mutation caused stabilization of dimers compared to wildtype protein (arrows). B, protein lysates were immunoprecipitated with FGFR3 E antibody and blotted with anti-phosphotyrosine 4G10 (pTYR) antibody. Blots were stripped and re-probed with the FGFR3 B9 antibody as a loading control. C, phase contrast micrograph of 97-7 cells ten days post-transduction. (bar=100μm). D, immunofluorescence images of alpha-tubulin demonstrating changes in the cytoskeleton of S249C FGFR3b knockdown cells compared to non-specific shRNA (bar=30μm).
Knockdown of S249C FGFR3b alters cell morphology, inhibits proliferation and decreases colony-forming ability
97-7 cells expressing the shRNA developed a flattened appearance (Figure 3C). Cell flattening was not observed in control cells transduced with the empty vector (data not shown) or the non-specific shRNA control (Figure 3 C). We performed immunofluorescence for alpha-tubulin to compare the cytoskeleton of 97-7 knockdown cells (Figure 3D). The alpha-tubulin distribution highlights the changes in cell morphology of shRNA treated 97-7 cells. Cells expressing the FGFR3 shRNA increased in number at a slower rate compared to vector and non-specific shRNA controls (Figure 4A). The difference in cell number was due to changes in proliferation and not apoptosis as no difference in acridine orange staining (apoptosis marker) was observed between cells expressing FGFR3 shRNA and vector or non-specific controls (data not shown). To determine if the change in proliferation rate was a specific effect of knocking down mutant FGFR3b, we knocked down wildtype FGFR3b in TERT-NHUC. More than 80% knockdown was confirmed by real-time RT-PCR (data not shown). No change in morphology or proliferation rate was observed in shRNA treated TERT-NHUC compared to vector or non-specific shRNA controls (Figure 4A).
Figure 4.

Differential effects of knockdown of S249C FGFR3b in tumour cells and of wild-type FGFR3b in normal cells. 97-7 cells (A) or TERT-NHUC (B) transduced with vector, non-specific shRNA or FGFR3 shRNA retroviruses were plated at 3×104 cells per well in triplicate in full medium and counted at three time points up to day six. Cells expressing vector are represented by square points and a solid line, cells expressing non-specific shRNA are represented by triangle points and a dashed line, cells expressing FGFR3 shRNA are represented by round points and a solid line.
The ability of 97-7 cells to form colonies at low density was assessed by seeding 500 or 3000 cells in a 10 cm plate. Cells expressing FGFR3 shRNA showed reduced colony-forming ability (Figure 5A). In addition, these cells showed a decreased ability to grow in agar (Figure 5B). Colonies were smaller and fewer compared to vector and non-specific controls (n=3).
Figure 5.

FGFR3 knockdown reduces clonogenicity of 97-7 cells. A, 12 day colonies from cells transduced with vector, non-specific or shRNA retroviruses (3000 cells plated). B, viable colonies in agarose were stained with p-iodonitrotretrazolium violet. The grey and black bars represent the number of viable colonies < 1mm2 or > 1mm2, respectively.
Reversal of phenotypic changes by re-expression of S249C FGFR3b
Specificity of shRNA remains one of the biggest obstacles for studying gene function using RNA interference. As shRNA knockdown was only achieved by one construct, we sought to validate the specificity of the FGFR3 shRNA in our experiments. The shRNA was targeted to the 3' UTR of FGFR3 and this allowed us to re-express the S249C FGFR3b from a cDNA clone that did not contain the 3' UTR. Re-expression of S249C FGFR3b reversed cell flattening (Figure 6A). To confirm re-expression of FGFR3b we performed real-time RT-PCR (data not shown) and western blots (Figure 6B). These confirmed re-expression of mutant FGFR3b and demonstrated an increase in expression by approximately 20-fold compared with the parental cell line. Re-expression of S249C increased cell proliferation (Figure 6C) and colony forming ability (Figure 6D). Interestingly, over-expression of the mutant receptor caused a ten-fold increase in the number of viable colonies in soft agar. To further control for off-target effects of shRNA we transduced two bladder cancer cell lines (UMUC3 and 5637) with shRNA. These cell lines express relatively low levels of FGFR3 (Figure 1C) and therefore any changes in phenotype would be due to off-target knockdown. No change in morphology, cell proliferation or ability to form colonies was observed in either cell line (data not shown). This confirms the effects of FGFR3 shRNA observed in 97-7 cells were specifically due to the knockdown of S249C FGFR3.
Figure 6.

Re-expression of S249C FGFR3b reverses phenotypic changes in FGFR3 knockdown 97-7 cells. A, phase contrast micrographs of 97-7 cells expressing shRNA and shRNA expressing cells transduced with retrovirus expressing S249C FGFR3b (bar=100μm). Pictures at ten days post transduction. B, western blot showing FGFR3b expression in 97-7 cells and in shRNA cells re-expressing S249C FGFR3b. C, fold change in cell number over four days. D, Number of colonies at low density and in soft agar. For low density, 500 cells were plated in 10 cm dishes and colonies counted on day 12. For soft agar, viable colonies were stained with p-iodonitrotretrazolium violet at 21 days. Graph represents the number of viable colonies per 10cm2.
Discussion
Mutation of FGFR3b is the most common genetic event described in bladder tumours to date. Importantly, mutation is highest in the large group of low-grade non-invasive tumours that form the bulk of urologic oncology practise. Currently, little is known about the molecular changes that lead to development of these tumours. Although it has been clear for many years that there is frequent deletion of chromosome 9 sequences, the identity and role of the genes involved has not been clear and no other potential molecular markers or therapeutic targets had been identified until recently. Mutant FGFR3 now provides not only an objective marker that may be used to detect disease recurrence (van Rhijn et al., 2003) but also a potential therapeutic target that is present in up to 80% of these tumours.
The high frequency of somatic mutation in bladder cancer implies an oncogenic role for mutant FGFR3b. However, despite the correlation of mutation and expression with disease, which suggests a role in disease causation, functional studies in urothelial cells have been lacking. Our objective in this study was to examine the effects of specific inhibition of the receptor in tumour cells bearing the common S249C mutation to determine if mutant FGFR3b is a valid therapeutic target.
We have demonstrated that levels of FGFR3 transcripts in bladder tumour samples follow the distribution of FGFR3 mutations, with significantly higher levels of expression in low-stage and grade bladder tumours than in invasive carcinomas. Two previous studies have reported on transcript levels in bladder tumour samples with broadly similar results. Levels of FGFR3 transcripts derived from expression array data for 22 tumours showed higher levels in pTa and pT1 compared to pT2 tumours and no detectable expression in normal urothelium (Gomez-Roman et al., 2005). In a second study, FGFR3 levels were examined by semi-quantitative RT-PCR, and comparable levels of FGFR3 were found in normal urothelium and tumours with no mutation (the majority of which were high grade and stage). Interestingly, high levels of FGFR3 expression correlated with the presence of mutated FGFR3 (Bernard-Pierrot et al., 2006). Lack of expression in normal urothelium in the first study may reflect the relative insensitivity of expression array detection compared with RT-PCR. FGFR3 protein levels have also been examined in bladder tumours by immunohistochemistry (Gomez-Roman et al., 2005; Matsumoto et al., 2004; Mhawech-Fauceglia et al., 2006). One of these studies found no association of expression with either tumour grade or stage (Matsumoto et al., 2004) but the other 2 studies both found an association of FGFR3 expression with low tumour grade and stage (Gomez-Roman et al., 2005; Mhawech-Fauceglia et al., 2006).
To identify a bladder cancer cell line that expressed similar levels of FGFR3 to low-stage superficial UCC, we measured FGFR3 transcript levels and compared them to known mutation status in thirty-one bladder cancer cell lines. Only one cell line, 97-7, expressed FGFR3 transcripts at similar levels to low-grade UCC. Similarly, a previous report only detected FGFR3 expression in 3 of 17 bladder cancer cell lines (Bernard-Pierrot et al., 2006). This low level of expression may be due to loss of expression of FGFR3 when tumour cells are cultured. We have previously observed that culture of normal bladder urothelium leads to a decrease in expression of FGFR3b and there are lower FGFR3b expression levels in proliferating cells compared to cells at confluence (Tomlinson et al., 2005). All cells used here were harvested at sub-confluence. Another contributing factor, not only to expression levels but also to the low frequency of FGFR3 mutation, is that the majority of bladder cancer cell lines have been derived from high-stage and grade cancers. Indeed, the frequency of FGFR3 mutation and levels of FGFR3 transcripts found in these cell lines reflect those found in high-grade UCC.
The S249C mutation is predicted to induce disulphide bond formation by the introduction of an additional cysteine in the extracellular domain of FGFR3b. We demonstrated that this mutation both stabilised dimerization of FGFR3b and led to constitutive phosphorylation of the receptor in urothelial cells. Previous reports have described similar cysteine substitutions in FGFR3c that have also resulted in dimerization and ligand-independent activation (Adar et al., 2002; d'Avis et al., 1998). We achieved stable knockdown of both mutant and wildtype FGFR3 using retrovirally-delivered shRNA. This method has the advantage that the viruses infect a very high percentage of cells, yielding mass populations of transduced cells and so avoids potential problems with inter-clone differences that arise when using stable clones of transfected cells. This approach reduced FGFR3 transcript levels by up to 90%.
Importantly, we have shown that knockdown of S249C FGFR3b in 97-7 cells inhibits not only proliferation but also anchorage independent growth and clonogenicity at low density both of which are phenotypic markers of transformation. Knockdown also caused profound cell flattening. No increase in apoptosis was found following receptor downregulation. This contrasts with the situation in multiple myeloma cell lines where following inhibition of mutant FGFR3b by shRNAs, there was an increase in annexin V staining 5 days after transfection which was accompanied by decreased expression of BCL2 and MCL1 (Zhu et al., 2005). Similarly treatment of myeloma cells with a range of small molecule inhibitors that target FGFR3 was associated with an increase in apoptosis (Grand et al., 2004; Paterson et al., 2004; Trudel et al., 2004; Trudel et al., 2005). In these cells, apoptosis was markedly delayed compared with the rapid apoptosis seen in the same cells after treatment with chemotherapeutic drugs and FGFR3-associated apoptosis was associated with induction of markers of differentiation in the cells, including a significant increase in antibody light chain production (Trudel et al., 2004). The urothelium does not show rapid cell turnover and as cells move to the superficial cell layer, differentiation is not accompanied by cell shedding or death but by prolonged quiescence. Thus if mutant FGFR3 acts to prevent completion of a normal differentiation program as suggested in myeloma cells, then apoptosis would not be expected following inhibition of FGFR3 function. It will be important to examine markers of urothelial differentiation in bladder tumours with FGFR3b mutation and measure the effects of inhibition of FGFR3b on differentiation in vitro.
We have not been able to test the effects of S249C FGFR3b downregulation on the in vivo phenotype of 97-7 cells as they not produce tumours as subcutaneous xenografts in immunodeficient mice. This may reflect particular dependence of these tumour cells on tissue-specific micro-environmental factors. In the future it will be important to assess tumorigenicity in the orthotopic site, in order to develop a model in which the effects of mutant FGFR3b and its inhibition can be tested in vivo.
In contrast to the effect of mutant FGFR3b knockdown in tumour cell lines, TERT-NHUC showed no significant phenotypic effects. This implies a dependence of the tumour cells on continued FGFR3b signalling as described for oncogenic stimuli in other tumour cell types, so-called “oncogene addiction” (Weinstein, 2002). Significantly this also shows that although TERT-NHUC express FGFR3b, there is no dependence on this for either survival or proliferation. This differential sensitivity may allow non-toxic targeted therapies to be developed.
A major problem with shRNA is off-target gene-silencing (Birmingham et al., 2006) especially when only using one shRNA. To overcome this problem we re-expressed S249C FGFR3b using cDNA that did not contain the 3' UTR. Overexpression of S249C FGFR3b not only reversed the phenotypic changes but also dramatically increased the number of colonies formed in soft agar. This confirms that high levels of S249C FGFR3b expression may lead to increased phenotypic transformation of bladder cells. However, the high level of re-expressed S249C FGFR3b may have masked any off-target gene-silencing. To address this issue we expressed the shRNA in bladder cancer cell lines that express low levels of FGFR3. As we observed no changes in phenotype, we conclude that there were no significant off-target effects of the FGFR3 shRNA. Taken together, these results suggest that not only the presence of mutation, but also an increase in expression of mutant FGFR3b may contribute to tumour formation. If this relationship is confirmed in future studies, screening of bladder tumours by immunohistochemistry may have the potential to identify tumours that contain mutation, and are suitable for FGFR3-targeted therapy.
The finding of FGFR3b mutations predominantly in low grade/stage bladder tumours and the more recent finding of mutations in human seborrheic keratoses but not malignant skin cancers, raises the possibility that in at least some tissue types mutant FGFR3 contributes only to the development of benign or non-aggressive tumours. This does not seem to be the case in malignant myeloma where the presence the t(4;14) translocation, of which approximately 2% contain an FGFR3 mutation, is associated with poor prognosis (Intini et al., 2001; Keats et al., 2005). In the urothelium where most FGFR3-mutant tumours are non-aggressive, it is possible that the oncogenic functions that confer a cellular selective advantage are subtle. It is known that FGFR functions show a high degree of tissue specificity with distinct cell type-specific effects (Eswarakumar et al., 2005), indicating the need to assess function in relevant cells. S249C and the Y375C mutations account for the majority of mutations identified in bladder cancer (∼80%) and evaluation of their oncogenic role is therefore of great interest. There is a recent report that Y375C FGFR3b plays a critical oncogenic role in the bladder cancer cell line MGH-U3, where siRNA or treatment with the small molecule inhibitor SU5402 inhibited proliferation and anchorage independent growth (Bernard-Pierrot et al., 2006). Our results now provide the first evidence for an oncogenic function of the more common mutation, S249C, in urothelial cells.
Understanding of the genetic alterations present in human cancers has led to the development of several highly successful targeted therapies (Strausberg et al., 2004). Already several potential approaches to targeting FGFR3 have been examined in multiple myeloma and promising responses have been obtained with small molecule inhibitors and antibodies both in vitro and in animal models (Chen et al., 2005; Grand et al., 2004; Trudel et al., 2004; Trudel et al., 2005; Trudel et al., 2006). It has also been clearly demonstrated that several of the agents tested, directly affect the function of both over-expressed wildtype and mutant FGFR3. In bladder cancer there is an urgent need for novel therapies for the large numbers of superficial tumours in which FGFR3b mutation is common (70-80%). There are also significant numbers of invasive bladder cancers with these mutations (∼15%). Our present findings indicate that mutant FGFR3b acts as an oncogene in these tumours, that targeting of FGFR3b may be a valid therapeutic approach and that detailed examination of the effects of available inhibitors is now merited.
Materials and Methods
Cell lines
Twenty seven bladder cancer cell lines were used; 97-7, RT4, RT112, 97-18, BFTC905, 97-29, SCaBER, DSH1, VMCUB3, SW1710, 96-1, VMCUB2, 97-24, J82, HT1376, 97-1, 647V, 253J, BFTC909, TCCSUP, SD, JO'N, UMUC3, VMCUB1, 5637, and T24. Cell lines were grown in standard growth media at 37°C in 5% CO2. Primary normal human urothelial cells (NHUC) or telomerase immortalised NHUC (TERT-NHU) (Chapman et al., 2006) were derived from urothelium stripped from human ureters obtained at nephrectomy (Southgate et al., 1994). NHUC and TERT-NHUC were maintained in KFSM keratinocyte medium (Life Technologies, Inc., Paisley, Scotland) supplemented with epidermal growth factor and bovine pituitary extract (Invitrogen, Paisley, Scotland).
Quantitative real-time reverse transcriptase-PCR for FGFR3
Total RNA was extracted from frozen tumour sections containing more than 90% tumour cells. RNA was extracted using Qiagen RNeasy Mini Kit (Qiagen, Crawley, United Kingdom) and 1 μg was reverse transcribed in the presence or absence of reverse transcriptase (Superscript II, Invitrogen, Paisley Scotland) according to the manufacturer's instructions. Real-time RT-PCR analysis was performed using SYBR Green I as reporter and ROX as reference dye (Perkin-Elmer, Applied Biosystems, Cheshire, United Kingdom). FGFR3 specific primers and SDHA (succinate dehydrogenase subunit A) control primers were used as previously described (Tomlinson et al., 2005).
shRNA constructs
The shRNAs were cloned into pGEM-T easy containing a U6 promoter (kind gift from Dr. D. Takai (Matsukura et al., 2003)) flanked with EcoRI and XhoI restriction sites. The plasmid was digested with SphI (New England Biolabs, Hertfordshire, United Kingdom) and treated with Klenow fragment (New England Biolabs). The linear blunt-ended plasmid was ethanol precipitated and digested with ApaI (New England Biolabs). Oligonucleotides targeting FGFR3 or a non-specific target (for the successful FGFR3 shRNA – forward oligo 5' -GTTTATTCCGGAAACTAGTTTCAAGAGAACTAGTTTCCGGAATAAACTTTTTGGGCC - 3', reverse oligo 5' – CAAAAAGTTTATTCCGGAAACTAGTTCTCTTGAAACTAGTTTCCGGAATAAAC - 3', and non-specific control – forward oligo 5' -CTTCAGCCGTTACGCTCGGTTCAAGAGACCGAGCGTAACGGCTGAAGTTTTTGGGCC - 3', reverse oligo 5' -CAAAAACTTCAGCCGTTACGCTCGGTCTCTTGAACCGAGCGTAACGGCTGAAG - 3') were annealed, resulting in a fragment containing a 5' blunt end and a 3' ApaI restriction site overhang, and cloned into the restriction digested plasmid, to create pGEM-shRNA-U6. pRetroSuper-puro (pRS-puro) was generated by replacing the hygromycin resistance gene from pRS-hyg (a kind gift from Reuven Agami (Voorhoeve & Agami, 2003)) with a puromycin resistance gene. The H1 promoter was removed from pRS-puro using EcoRI and XhoI restriction enzymes, and replaced with the shRNA-U6 restriction digested from pGEM-shRNA-U6 with the same enzymes.
Cloning of S249C FGFR3b, production of retroviruses and transductions
We used site directed mutagenesis on FGFR3b cDNA to create S249C FGFR3b. The mutation was verified by sequencing and S249C FGFR3b was subcloned into a retroviral expression vector (pFB-neo, Stratagene, La Jolla, CA). FGFR3b, S249C FGFR3b and shRNA constructs were transfected into Phoenix A packaging cells (ATCC), using siPOR™ XP-1 transfection agent (Ambion, Huntingdon, Cambridgeshire, UK). After 48 hours, medium was harvested, 0.4μm filtered and mixed in equal amounts with fresh medium containing 8μg/ml of polybrene (Sigma, Poole, UK). Cells were incubated with retroviral supernatants for 8 hours. Forty-eight hours after transduction, cells were transferred into selection medium containing puromycin or neomycin.
Western blotting and immunoprecipitation
Cells were lysed in RIPAE buffer [1% Triton-X 100, 1mM EDTA, 0.1% SDS, 0.5% sodium deoxycholate, 10% glycerol and protease inhibitor cocktail (Sigma) in PBS] and lysates cleared by centrifugation at 10000 rpm at 4°C. The protein concentrations were determined using the BCA (bicinchonic acid) assay (Pierce, Rockford, IL, USA). Immunoprecipitation of FGFR3 was carried out at 4°C. Lysates were incubated overnight with the extracellular (E) FGFR3 antibody (Sigma) and mixed by rotation. Lysates were mixed with protein A sepharose beads (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK) by rotation for 2 h, centrifuged at 1000 rpm for 1 min and the beads washed twice in lysis buffer. Beads were resuspended in sample buffer (Laemmli, β-mercaptoethanol), boiled for 3 min, centrifuged for 1 min at 13000 rpm and proteins resolved in a 7% SDS-polyacrylamide gel. Proteins were transferred to PVDF membranes (Amersham Biosciences) and incubated in blocking buffer (5% BSA in PBS 0.1% Tween) followed by incubation with 4G10 anti-phosphotyrosine antibody (Cell Signaling, Danvers, MA, USA) or FGFR3 B9 antibody (Autogen Bioclear, Calne, Wiltshire, UK). Bound antibody was detected using anti-mouse horseradish peroxidase-conjugated antibody and chemiluminescence (ECL Plus Kit, Amersham Biosciences). Blots were stripped in 50mM Tris pH 7.5, 10M urea at 55°C for 1 hr, prior to re-probing with FGFR3 B9 antibody as a loading control.
Immunofluorescence
Cells were cultured on sterile coverglasses and fixed for immunohistochemistry with ice-cold methanol. Cells were blocked in 1 % milk, and incubated with rat anti-alpha-tubulin (Serotec UK, Oxford, England) diluted in 1 % milk for 1 hr. Coverglasses were washed in PBS, and incubated with AlexaFluor® 488 goat anti-rat IgG (H+L) secondary antibody (Molecular Probes, Oregan, USA). Immunolocalisation of alpha-tubulin was performed using a Zeiss AxioVert 200 microscope.
Phenotypic assays
For growth curves, 3×104 cells per well were plated in six well dishes. Cells were counted in triplicate wells on day 1 and at three other time points up to day 8 (n=3). The ability of cells to form colonies at low-density was assayed by plating 3,000 or 500 cells in 10 cm dishes and culturing them for twelve days (n=3). Colonies were fixed and visualised using 1% methylene blue (Sigma) in 50% ethanol. For assessment of anchorage independent growth, 1×105, 5×104 or 1×104 cells, in duplicate, were suspended in 0.3% agar in medium and cultured for 21 days (n=2). Cultures were fed weekly with 0.3% agar. Viable colonies were identified by overnight staining with p-iodonitrotretrazolium violet (Sigma).
Acknowledgements
We thank Dr Catherine Reznikoff for generously providing the cell lines 97-7, 97-18, 97-29, 96-1, 97-24 and 97-1, Dr D Podolsky for providing FGFR3b cDNA and Dr Eva Pitt for excellent tissue culture support. This work was funded by Cancer Research UK.
References
- Adar R, Monsonego-Ornan E, David P, Yayon A. Differential activation of cysteine-substitution mutants of fibroblast growth factor receptor 3 is determined by cysteine localization. J Bone Miner Res. 2002;17:860–868. doi: 10.1359/jbmr.2002.17.5.860. [DOI] [PubMed] [Google Scholar]
- Bakkar AA, Wallerand H, Radvanyi F, Lahaye JB, Pissard S, Lecerf L, et al. FGFR3 and TP53 gene mutations define two distinct pathways in urothelial cell carcinoma of the bladder. Cancer Research. 2003;63:8108–8112. [PubMed] [Google Scholar]
- Bernard-Pierrot I, Brams A, Dunois-Larde C, Caillault A, Diez de Medina SG, Cappellen D, et al. Oncogenic properties of the mutated forms of fibroblast growth factor receptor 3b. Carcinogenesis. 2006;27:740–747. doi: 10.1093/carcin/bgi290. [DOI] [PubMed] [Google Scholar]
- Billerey C, Chopin D, Aubriot-Lorton MH, Ricol D, Gil Diez de Medina S, Van Rhijn B, et al. Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol. 2001;158:1955–1959. doi: 10.1016/S0002-9440(10)64665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, et al. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods. 2006;3:199–204. doi: 10.1038/nmeth854. [DOI] [PubMed] [Google Scholar]
- Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- Chapman EJ, Hurst CD, Pitt E, Chambers P, Aveyard JS, Knowles MA. Expression of hTERT immortalises normal human urothelial cells without inactivation of the p16/Rb pathway. Oncogene. 2006;25:5037–5045. doi: 10.1038/sj.onc.1209513. [DOI] [PubMed] [Google Scholar]
- Chen J, Lee BH, Williams IR, Kutok JL, Mitsiades CS, Duclos N, et al. FGFR3 as a therapeutic target of the small molecule inhibitor PKC412 in hematopoietic malignancies. Oncogene. 2005 doi: 10.1038/sj.onc.1208989. [DOI] [PubMed] [Google Scholar]
- Chesi M, Bergsagel PL, Kuehl WM. The enigma of ectopic expression of FGFR3 in multiple myeloma: a critical initiating event or just a target for mutational activation during tumor progression. Current Opinion in Hematology. 2002;9:288–293. doi: 10.1097/00062752-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Chesi M, Brents LA, Ely SA, Bais C, Robbiani DF, Mesri EA, et al. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;97:729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92:3025–3034. [PubMed] [Google Scholar]
- d'Avis PY, Robertson SC, Meyer AN, Bardwell WM, Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth Differ. 1998;9:71–78. [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Gomez-Roman JJ, Saenz P, Molina M, Cuevas Gonzalez J, Escuredo K, Santa Cruz S, et al. Fibroblast growth factor receptor 3 is overexpressed in urinary tract carcinomas and modulates the neoplastic cell growth. Clin Cancer Res. 2005;11:459. [PubMed] [Google Scholar]
- Grand EK, Chase AJ, Heath C, Rahemtulla A, Cross NC. Targeting FGFR3 in multiple myeloma: inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia. 2004;18:962–966. doi: 10.1038/sj.leu.2403347. [DOI] [PubMed] [Google Scholar]
- Hart KC, Robertson SC, Donoghue DJ. Identification of tyrosine residues in constitutively activated fibroblast growth factor receptor 3 involved in mitogenesis, Stat activation, and phosphatidylinositol 3-kinase activation. Molecular Biology of the Cell. 2001;12:931–942. doi: 10.1091/mbc.12.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart KC, Robertson SC, Kanemitsu MY, Meyer AN, Tynan JA, Donoghue DJ. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene. 2000;19:3309–3320. doi: 10.1038/sj.onc.1203650. [DOI] [PubMed] [Google Scholar]
- Holmang S, Hedelin H, Anderstrom C, Johansson SL. The relationship among multiple recurrences, progression and prognosis of patients with stages Ta and T1 transitional cell cancer of the bladder followed for at least 20 years. J Urol. 1995;153:1823–1826. discussion 1826-1827. [PubMed] [Google Scholar]
- Intini D, Baldini L, Fabris S, Lombardi L, Ciceri G, Maiolo AT, et al. Analysis of FGFR3 gene mutations in multiple myeloma patients with t(4;14) Br J Haematol. 2001;114:362–364. doi: 10.1046/j.1365-2141.2001.02957.x. [DOI] [PubMed] [Google Scholar]
- Jebar AH, Hurst CD, Tomlinson DC, Johnston C, Taylor CF, Knowles MA. FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene. 2005;24:5218–5225. doi: 10.1038/sj.onc.1208705. [DOI] [PubMed] [Google Scholar]
- Keats JJ, Maxwell CA, Taylor BJ, Hendzel MJ, Chesi M, Bergsagel PL, et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood. 2005;105:4060–4069. doi: 10.1182/blood-2004-09-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Suzuki H, Ohashi T, Asano K, Kiyota H, Eto Y. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92:2555–2561. doi: 10.1002/1097-0142(20011115)92:10<2555::aid-cncr1607>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Knowles MA. Molecular subtypes of bladder cancer: Jekyll and Hyde or chalk and cheese? Carcinogenesis. 2006;27:361–373. doi: 10.1093/carcin/bgi310. [DOI] [PubMed] [Google Scholar]
- Kurth KH, Denis L, Bouffioux C, Sylvester R, Debruyne FM, Pavone-Macaluso M, et al. Factors affecting recurrence and progression in superficial bladder tumours. Eur J Cancer. 1995;31A:1840–1846. doi: 10.1016/0959-8049(95)00287-s. [DOI] [PubMed] [Google Scholar]
- Logie A, Dunois-Larde C, Rosty C, Levrel O, Blanche M, Ribeiro A, et al. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Human Molecular Genetics. 2005;14:1153. doi: 10.1093/hmg/ddi127. [DOI] [PubMed] [Google Scholar]
- Martinez-Torrecuadrada J, Cifuentes G, Lopez-Serra P, Saenz P, Martinez A, Casal JI. Targeting the extracellular domain of fibroblast growth factor receptor 3 with human single-chain Fv antibodies inhibits bladder carcinoma cell line proliferation. Clin Cancer Res. 2005;11:6280–6290. doi: 10.1158/1078-0432.CCR-05-0282. [DOI] [PubMed] [Google Scholar]
- Matsukura S, Jones PA, Takai D. Establishment of conditional vectors for hairpin siRNA knockdowns. Nucleic Acids Research. 2003;31:e77. doi: 10.1093/nar/gng077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Ohtsuki Y, Ochii K, Seike Y, Iseda N, Sasaki T, et al. Fibroblast growth factor receptor 3 protein expression in urothelial carcinoma of the urinary bladder, exhibiting no association with low-grade and/or non-invasive lesions. Oncol Rep. 2004;12:967. [PubMed] [Google Scholar]
- Mhawech-Fauceglia P, Cheney RT, Fischer G, Beck A, Herrmann FR. FGFR3 and p53 protein expressions in patients with pTa and pT1 urothelial bladder cancer. Eur J Surg Oncol. 2006;32:231–237. doi: 10.1016/j.ejso.2005.11.018. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- Paterson JL, Li Z, Wen XY, Masih-Khan E, Chang H, Pollett JB, et al. Preclinical studies of fibroblast growth factor receptor 3 as a therapeutic target in multiple myeloma. British Journal of Haematology. 2004;124:595–603. doi: 10.1111/j.1365-2141.2004.04814.x. [DOI] [PubMed] [Google Scholar]
- Richelda R, Ronchetti D, Baldini L, Cro L, Viggiano L, Marzella R, et al. A novel chromosomal translocation t(4; 14)(p16.3; q32) in multiple myeloma involves the fibroblast growth-factor receptor 3 gene. Blood. 1997;90:4062–4070. [PubMed] [Google Scholar]
- Ronchetti D, Greco A, Compasso S, Colombo G, Dell'Era P, Otsuki T, et al. Deregulated FGFR3 mutants in multiple myeloma cell lines with t(4;14): comparative analysis of Y373C, K650E and the novel G384D mutations. Oncogene. 2001;20:3553–3562. doi: 10.1038/sj.onc.1204465. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Julicher KP, Burger MS, Della Valle V, Larsen CJ, Yeager TR, et al. Different combinations of genetic/epigenetic alterations inactivate the p53 and pRb pathways in invasive human bladder cancers. Cancer Res. 2000;60:3862–3871. [PubMed] [Google Scholar]
- Scotet E, Houssaint E. The choice between alternative IIIb and IIIc exons of the FGFR-3 gene is not strictly tissue-specific. Biochim Biophys Acta. 1995;1264:238–242. doi: 10.1016/0167-4781(95)00156-b. [DOI] [PubMed] [Google Scholar]
- Sibley K, Bell S, Knowles MA. Redefining a critical region of LOH on 4p16.3 in bladder cancer. Genes Chromosomes Cancer. 2000;29:378–379. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1045>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Southgate J, Hutton KA, Thomas DF, Trejdosiewicz LK. Normal human urothelial cells in vitro: proliferation and induction of stratification. Laboratory Investigation. 1994;71:583–594. [PubMed] [Google Scholar]
- Strausberg RL, Simpson AJ, Old LJ, Riggins GJ. Oncogenomics and the development of new cancer therapies. Nature. 2004;429:469–474. doi: 10.1038/nature02627. [DOI] [PubMed] [Google Scholar]
- Tomlinson DC, L'Hote CG, Kennedy W, Pitt E, Knowles MA. Alternative splicing of fibroblast growth factor receptor 3 produces a secreted isoform that inhibits fibroblast growth factor-induced proliferation and is repressed in urothelial carcinoma cell lines. Cancer Research. 2005;65:10441–10449. doi: 10.1158/0008-5472.CAN-05-1718. [DOI] [PubMed] [Google Scholar]
- Trudel S, Ely S, Farooqi Y, Affer M, Robbiani DF, Chesi M, et al. Inhibition of fibroblast growth factor receptor 3 induces differentiation and apoptosis in t(4;14) myeloma. Blood. 2004;103:3521–3528. doi: 10.1182/blood-2003-10-3650. [DOI] [PubMed] [Google Scholar]
- Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, et al. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105:2941. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
- Trudel S, Stewart AK, Rom E, Wei E, Li ZH, Kotzer S, et al. The inhibitory anti-FGFR3 antibody, PRO-001, is cytotoxic to t(4;14) multiple myeloma cells. Blood. 2006;107:4039–4046. doi: 10.1182/blood-2005-10-4179. [DOI] [PubMed] [Google Scholar]
- van Rhijn BW, Lurkin I, Chopin DK, Kirkels WJ, Thiery JP, van der Kwast TH, et al. Combined microsatellite and FGFR3 mutation analysis enables a highly sensitive detection of urothelial cell carcinoma in voided urine. Clin Cancer Res. 2003;9:257–263. [PubMed] [Google Scholar]
- van Rhijn BW, Lurkin I, Radvanyi F, Kirkels WJ, van der Kwast TH, Zwarthoff EC. The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res. 2001;61:1265–1268. [PubMed] [Google Scholar]
- Voorhoeve PM, Agami R. The tumor-suppressive functions of the human INK4A locus. Cancer Cell. 2003;4:311–319. doi: 10.1016/s1535-6108(03)00223-x. [DOI] [PubMed] [Google Scholar]
- Webster MK, Donoghue DJ. FGFR activation in skeletal disorders: too much of a good thing. Trends Genet. 1997;13:178–182. doi: 10.1016/s0168-9525(97)01131-1. [DOI] [PubMed] [Google Scholar]
- Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Wu XR. Urothelial tumorigenesis: a tale of divergent pathways. Nat Rev Cancer. 2005;5:713–725. doi: 10.1038/nrc1697. [DOI] [PubMed] [Google Scholar]
- Zhu L, Somlo G, Zhou B, Shao J, Bedell V, Slovak ML, et al. Fibroblast growth factor receptor 3 inhibition by short hairpin RNAs leads to apoptosis in multiple myeloma. Mol Cancer Ther. 2005;4:787. doi: 10.1158/1535-7163.MCT-04-0330. [DOI] [PubMed] [Google Scholar]