

Summary
Insulin resistance is a common disorder caused by a wide variety of physiological insults; some of these include poor diet, inflammation, anti-inflammatory steroids, hyperinsulinemia, and dyslipidemia. The common link between these diverse insults and insulin resistance is widely considered to involve impaired insulin signaling particularly at the level of the insulin receptor substrate (IRS). To test this model we utilized a heterologous system involving the PDGF pathway which recapitulates many aspects of insulin action independently of IRS. We comprehensively analyzed 6 models of insulin resistance in 3 experimental systems and consistently observed defects in both insulin and PDGF action despite a range of insult-specific defects within the IRS-Akt nexus. These findings indicate that while insulin resistance is associated with multiple deficiencies, the most deleterious defects and the origin of insulin resistance occur independently of IRS.
Keywords: GLUT4, diabetes, glucose transport, signaling
Introduction
Insulin plays an essential role in metabolic homeostasis in mammals by accelerating the disposal of post-prandial glucose into muscle and adipose tissue. This step is mediated via the insulin-regulated translocation of the glucose transporter GLUT4 from intracellular vesicles to the plasma membrane. This process is imperative as mice lacking GLUT4 in muscle or fat are insulin resistant (Minokoshi et al., 2003); a condition defined as an impairment in insulin-stimulated glucose entry into these tissues. Insulin resistance and the associated glucose intolerance are important metabolic markers that predispose individuals to more severe disorders including Type 2 diabetes. The origin and precise mechanism(s) mediating insulin resistance are unknown. However, since glucose entry into the cell represent the earliest rate-limiting defect in insulin resistant skeletal muscle (Petersen and Shulman, 2006), this suggests that the etiology of insulin resistance derives from a lesion in the pathway between the insulin receptor (IR) and GLUT4.
The precise path from the IR to GLUT4 is still unknown; however the PI3K/Akt pathway is essential for insulin-regulated glucose metabolism. The insulin-dependent phosphorylation of IRS proteins is the initial step in activation of PI3K which leads to the generation of PI-3,4,5-trisphosphate (PIP3), which serves as a docking site for pleckstrin homology (PH) domain containing-proteins such as PDK1 and Akt. Akt is the pleiotropic kinase essential for most of the metabolic actions of insulin and its activity is regulated by phosphorylation at Thr308 by PDK1 and Ser473 by the mTOR/Rictor complex. In many cases Akt substrates that directly control metabolic steps have been identified; some of these include glycogen synthase kinase 3 or GSK3 (Cross et al., 1995), the transcription factor FoxO (Biggs et al., 1999; Paradis and Ruvkun, 1998), the Rheb GAP TSC2 (Potter et al., 2002) the phosphodiesterase PDE3b (Kitamura et al., 1999), and the RabGAP TBC1D4/AS160 (Kane et al., 2002). The latter of these, TBC1D4, is the only Akt substrate shown to play an important role in insulin-stimulated GLUT4 translocation (Kane et al., 2002). It should be noted that Akt-independent pathways have also been implicated in insulin action (Baumann et al., 2000; Farese et al., 2007), however their role remains controversial (Huang and Czech, 2007). One possibility is that these pathways play a permissive rather than a regulatory role in insulin action. Regardless, their relevance to the development of insulin resistance in muscle and fat cells remains doubtful particularly since some of these pathways appear to be cell type specific (eg. c-Cbl).
While defects at any one of these loci between the IR and GLUT4 may contribute to insulin resistance, it is widely believed that defects at IRS-1 represent a central feature of this disorder (Aguirre et al., 2000; Dresner et al., 1999; Gao et al., 2002; Gao et al., 2003; Gual et al., 2005; Hotamisligil et al., 1996; Morino et al., 2005; Ozcan et al., 2004; Rice et al., 1993; Turnbow et al., 1994; Ueki et al., 2004; White, 2002; Yu et al., 2002; Zick, 2005; Zierath et al., 1997). To date, considerable evidence has focused on IRS proteins as a major target of insulin resistance under a broad range of conditions. Several mechanisms have been reported to modify the activity of IRS including protein/protein interactions, Ser/Thr hyperphosphorylation, tyrosine dephosphorylation, targeted degradation, and other post-translational modifications such as O-linked glycosylation and acetylation (reviewed in (Gual et al., 2005; Taniguchi et al., 2006)).
There are several caveats to the model describing a major role for IRS proteins in insulin resistance. First, many studies of IRS function in insulin resistance focus on elements in the signal transduction cascade as the major read out of insulin resistance rather than terminal actions of insulin like GLUT4 insertion into the plasma membrane. Second, there is evidence that insulin resistance is not always associated with defects in Akt activity (Kim et al., 1999; Nadler et al., 2001). Third, as originally shown by Kono and colleagues (Kono and Barham, 1971) for the insulin receptor there is often considerable spareness in various elements of the insulin signal cascade, and the same applies to IRS1 (Rice et al., 1992). This means that incomplete inhibition of IRS may not have a major impact on metabolism. Consistent with this, mice heterozygous for IRS-1 do not show defects in insulin action under normal conditions (Shirakami et al., 2002) and partial knockdown of IRS-1 in skeletal muscle has no effect on insulin action (Cleasby et al., 2007). Thus, it remains unclear if reduced insulin signaling through IRS is a primary cause of insulin resistance or simply a secondary consequence of an alternate primary disorder.
Results
Lack of linearity between Akt and GLUT4
Akt has been shown to play an essential role in many actions of insulin including GLUT4 translocation (Kohn et al., 1996). However, as shown in Figure 1 the dynamic range of Akt activity with respect to substrate phosphorylation and GLUT4 translocation is considerably lower than its maximal capacity. This is consistent with previous reports showing a similar non-linearity between insulin-stimulated Akt activation and glucose uptake (Whitehead et al., 2001). Strikingly, the dose response relationship of Akt substrate phosphorylation was discordant with that of insulin stimulated Akt phosphorylation and a further discordance was observed between distinct Akt substrates (Figure 1C). In adipocytes incubated with 0.1-1 nM insulin, a concentration within the high physiological range, GLUT4 translocation and TBC1D4 phosphorylation increased to ∼50% of their maximum values while Akt and GSK3 phosphorylation reached only 20% of their maximum output. The discordance between insulin-activated TBC1D4 phosphorylation is not likely due to an additive contribution of other insulin-stimulated upstream kinases on TBC1D4 because its phosphorylation was inhibited using a specific Akt inhibitor (Figure 1D). Furthermore, insulin had a maximal effect on GLUT4 translocation in L6 myotubes at concentrations where only 5% of the total Akt pool was phosphorylated (Figure 1E). Hence, these data indicate several important aspects of insulin signalling and insulin action not previously emphasised. First, there is considerable “spareness” in the activity of Akt in vivo with respect to insulin action as defined by GLUT4 translocation. Second, there is discordance in the efficiency of Akt substrate phosphorylation such that certain substrates such as TBC1D4 are more efficiently phosphorylated than others such as GSK3. One ramification of these data is that a modest defect in upstream elements of the insulin signalling cascade per se, as is often described in insulin resistance, may not translate into downstream defects due to spareness in the pathway. To test this hypothesis we conducted an extensive analysis of the insulin action pathway in a variety of insulin resistance models.
Figure 1. Insulin-stimulated GLUT4 translocation is not proportional to Akt phosphorylation.
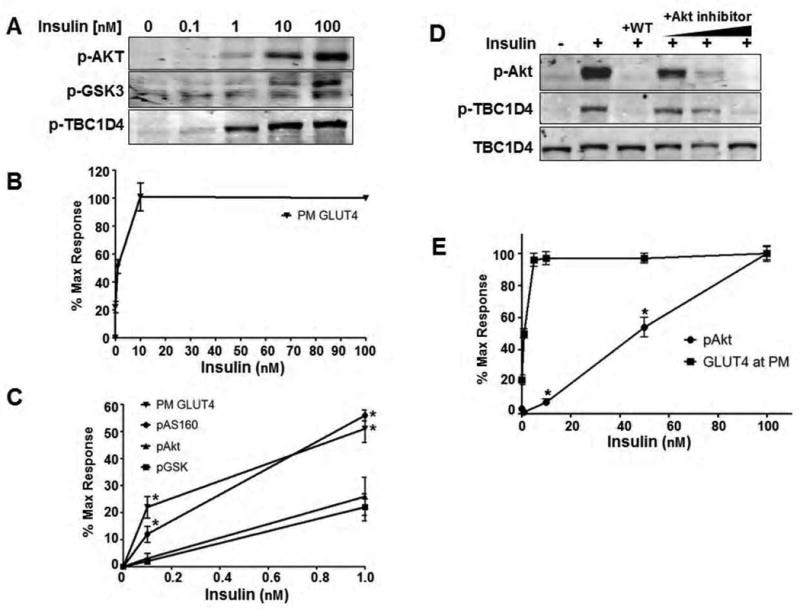
A) 3T3-L1 adipocytes were treated with a dose response of insulin. At each dose, the phosphorylation status of Akt at Ser473, GSK3α/β at Ser9/21, and TBC1D4 at Thr642 were detected by Western blot.
B) Insulin dose response for GLUT4 translocation to the plasma membrane in 3T3-L1 adipocytes.
C) To directly compare the insulin-stimulated response of each parameter in A) and B), the Western blots and translocation data were quantified and normalized to 0 and 100nM insulin representing minimal and maximal stimulation. An evaluation of submaximal stimulation reveals a distinct non-linearity between Akt phosphorylation and GLUT4 translocation to the plasma membrane. However, phosphorylation of TBC1D4 at submaximal insulin levels is more consistent with GLUT4 translocation than Akt, indicating an increased activity toward this substrate over others like GSK3. Error bars, ± s.e.m. of 3-4 experiments. *P<0.05 relative to Akt phosphorylation at the given dose of insulin, Student's t-test.
D) The PI3K inhibitor wortmannin (WT, 100 nM for 10 min prior to insulin) and an Akt inhibitor (at concentrations of 0.1, 1, and 10 μM) inhibit Akt phosphorylation and subsequent phosphorylation of TBC1D4 in 3T3-L1 adipocytes treated with 100 nM insulin for 12 minutes indicating that Akt is the sole insulin-regulated kinase acting on this site. Representative Western blots of 3 separate experiments are shown.
E) Insulin (20 minutes) was added to L6 myotubes at the concentrations indicated and the response of Akt phosphorylation and GLUT4 translocation were analyzed as a percent of maximal stimulation. Error bars, ± s.e.m., n= 3. *P<0.05, Student's t-test.
PDGF mimics insulin action independently of IRS
To dissect the specific role of IRS in insulin resistance we established an experimental system utilizing the PDGF pathway to bypass the requirement for IRS whilst preserving other elements of the insulin action cascade. Muscle and fat cells do not possess sufficient levels of the PDGF receptor to trigger a PDGF response in these cells ((Whiteman et al., 2003), and unpublished data). Therefore, the human PDGF-β receptor (PDGFR) was ectopically expressed in mouse 3T3-L1 preadipocytes and rat L6 myoblasts using recombinant retrovirus (Whiteman et al., 2003) (Figure 2A). Upon differentiation into adipocytes or myotubes we evaluated insulin and PDGF action to define the optimal doses and incubation times with ligand to achieve the most consistent amplification of signal transduction and GLUT4 translocation (data not shown). In both cell culture models, a maximum dose of insulin (100 nM) and PDGF (20 ng/mL) was utilized as this produced a consistent and similar response facilitating a direct comparison between both agonists (see Figure 2B). Both hormones activated PI3K without detectable cross-talk at the level of IRS-1 as indicated by the absence of PDGF-stimulated IRS-1 tyrosine phosphorylation at the PI3K binding site (tyrosine 612), compared to the robust phosphorylation observed with insulin. Conversely, whereas PDGF stimulated tyrosine phosphorylation of the PDGF receptor in both myotubes and adipocytes this was not observed in response to insulin stimulation (Figure 2B). Importantly, both insulin and PDGF stimulated Akt and TBC1D4 phosphorylation and GLUT4 translocation with similar amplitude (Figures 2B and 2C) and kinetics (data not shown) in both cell types. Consistent with previous studies (Whiteman et al., 2003) these data show that IRS proteins are not necessary for PDGF-induced GLUT4 translocation, but rather are a specific feature necessary for other aspects of insulin action. Hence this offers an ideal experimental system with which to assess the role of IRS proteins in insulin resistance.
Figure 2. PDGF mimics insulin action while circumventing the IR and IRS.
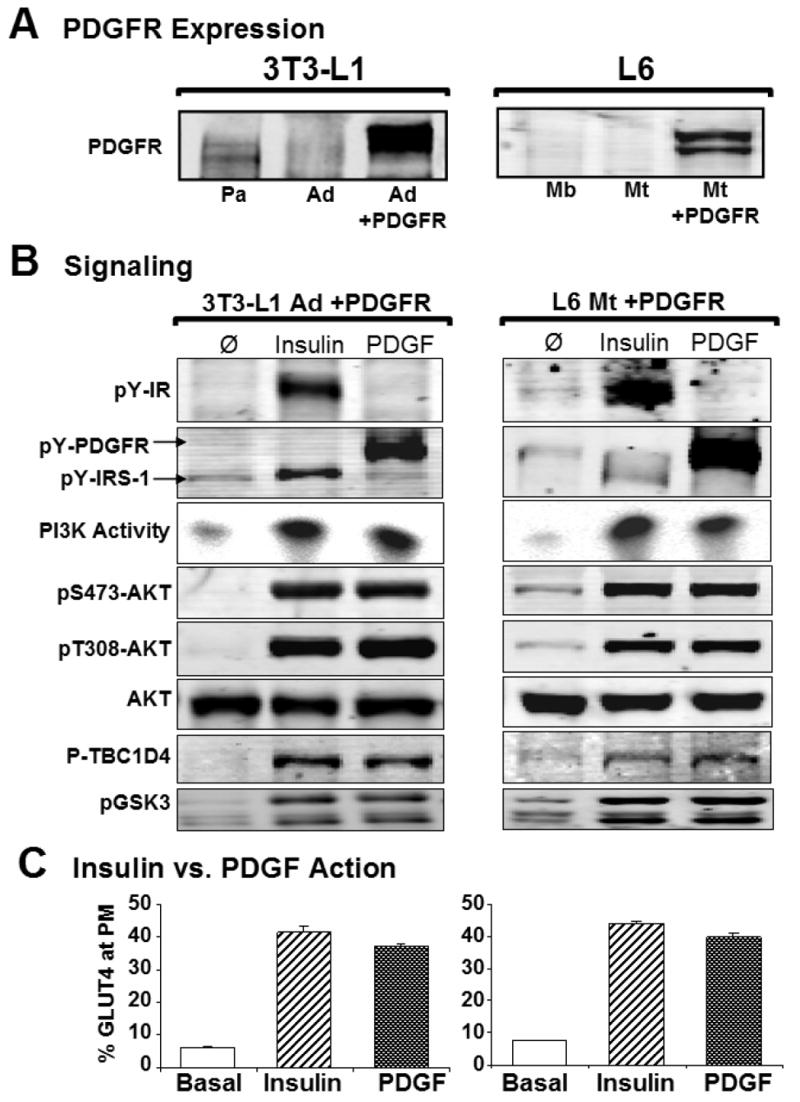
A) The human PDGF Receptor was ectopically over-expressed in mouse 3T3-L1 adipocytes and rat L6 myotubes. Representative Western blots of PDGFR expression are shown. Abbreviations: Pa, preadipocytes; Ad, adipocytes; Mb, myoblasts; and Mt, myotubes.
B) PDGFR expressing adipocytes (left column) and myotubes (right column) were serum starved for 90 min prior to acute stimulation with either 100 nM insulin or 20 ng/mL PDGF for 20 min. Representative Western blots of cell and tissue extracts show similar amplification of insulin- and PDGF-stimulated signaling intermediates.
C) Insulin- (hashed bars) and PDGF-stimulated (graded bars) GLUT4 translocation to the plasma membrane as a percentage of total HA-GLUT4 in adipocytes (left) and myotubes (right). In both cell types insulin was slightly more potent than PDGF (89±2% and 90±3% of maximal insulin for adipocytes and myotubes respectively). Error bars, ± s.e.m. (n = 5 experiments with >3 measurements each), Student's t-test.
Bypassing IRS does not override insulin resistance
To study the role of IRS in insulin resistance we next generated a range of metabolic disorders in L6 myotubes or 3T3-L1 adipocytes overexpressing the PDGFR using insults that recapitulate common causes of insulin resistance in mammals, including hyperinsulinemia (chronic insulin, CI), inflammation (tumor necrosis factor alpha, TNF), Cushing's syndrome/anti-inflammatory steroids (dexamethasone, DEX), and hyperlipidemia/dyslipidemia (palmitate, PALM). Oxidative stress, which has also been linked to insulin resistance (Houstis et al., 2006; Kaneto et al., 2005), was generated by incubating cells with glucose oxidase (GO). To avoid toxic or non-specific effects the treatment dose and/or duration for all insults was optimised to define minimal conditions required to elicit maximal insulin resistance (data not shown). GO, CI, TNF, DEX, and PALM inhibited insulin-stimulated GLUT4 translocation by 45-60% in both 3T3-L1 and L6 cells, indicating marked insulin resistance with each model (Figure 3). However, PDGF was unable to overcome these defects indicating that IRS-1 per se is not a major contributor to insulin resistance (Figure 3). Notably however, we did observe a consistent restoration of GLUT4 translocation with PDGF in myotubes, and to a lesser extent in adipocytes, treated with CI (Figure 3). This emphasizes the utility of this system clearly indicating that in the case of the CI model the major defect must occur at components unique to the insulin pathway involving either the IR or IRS-1.
Figure 3. Multiple insults antagonize insulin and PDGF action.
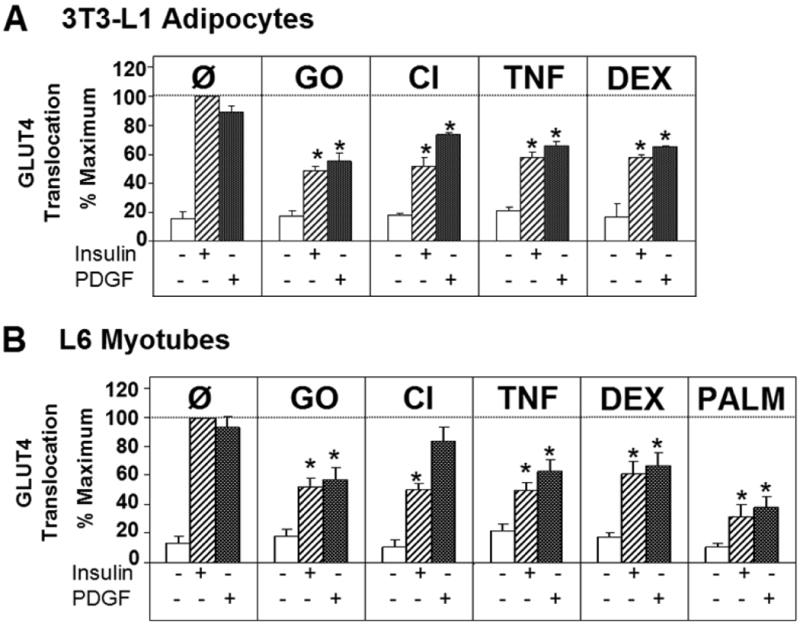
A) GLUT4 translocation to the plasma membrane was measured after glucose oxidase (GO, 50 mU/mL for 2 h), chronic insulin (CI, 10 nM for 24 h), TNFα (TNF, 2 ng/mL for 4 d), or dexamethasone (DEX, 20 nM for 8 d) treatment in 3T3-L1 adipocytes stimulated with insulin and PDGF.
B) GLUT4 translocation to the plasma membrane was analyzed in L6 myotubes treated with the above conditions or palmitate (PALM, 150 μM for 18 h). All data were normalized to maximum stimulation with 100nM insulin for each cell type. Results are displayed as means ± s.e.m., *P<0.05 vs. respective insulin- or PDGF-stimulated control (Ø) (n=3-5 experiments for each data set), Student's t-test.
Upstream signal transduction in insulin resistance
We next examined IRS-1 expression and phosphorylation since defects in both of these parameters have been reported in many insulin resistance models. A significant reduction in IRS-1 protein levels were observed in adipocytes treated with CI, TNF and DEX, consistent with previous studies (Figure 4A). However, this did not consistently correlate with changes in insulin-stimulated IRS-1 tyrosine phosphorylation as exemplified in cells treated with DEX. Moreover, we observed uncoupling between IRS-1 tyrosine phosphorylation and downstream signaling in adipocytes treated with CI. In this instance we observed persistent tyrosine phosphorylation of IRS-1 following chronic insulin treatment despite an extensive insulin wash-out period. The insulin wash-out was clearly effective because Akt phosphorylation had returned to basal levels (see CI, Figure 4A). Hence this suggests that additional aspects of IRS-1 function, such as subcellular localization (Clark et al., 1998; Inoue et al., 1998) may play an important role in its coupling to downstream signalling elements. Finally, GO treatment had no effect on IRS1 tyrosine phosphorylation or abundance despite significant insulin resistance (Figure 3A) indicating that this insult likely functions downstream of IRS as suggested recently (JeBailey et al., 2007).
Figure 4. Signal transduction in 3T3-L1 adipocytes and L6 myotubes treated with multiple models of insulin resistance. Cells were left in unstimulated basal conditions (open bars) or stimulated with Insulin (hashed bars) or PDGF (graded bars).
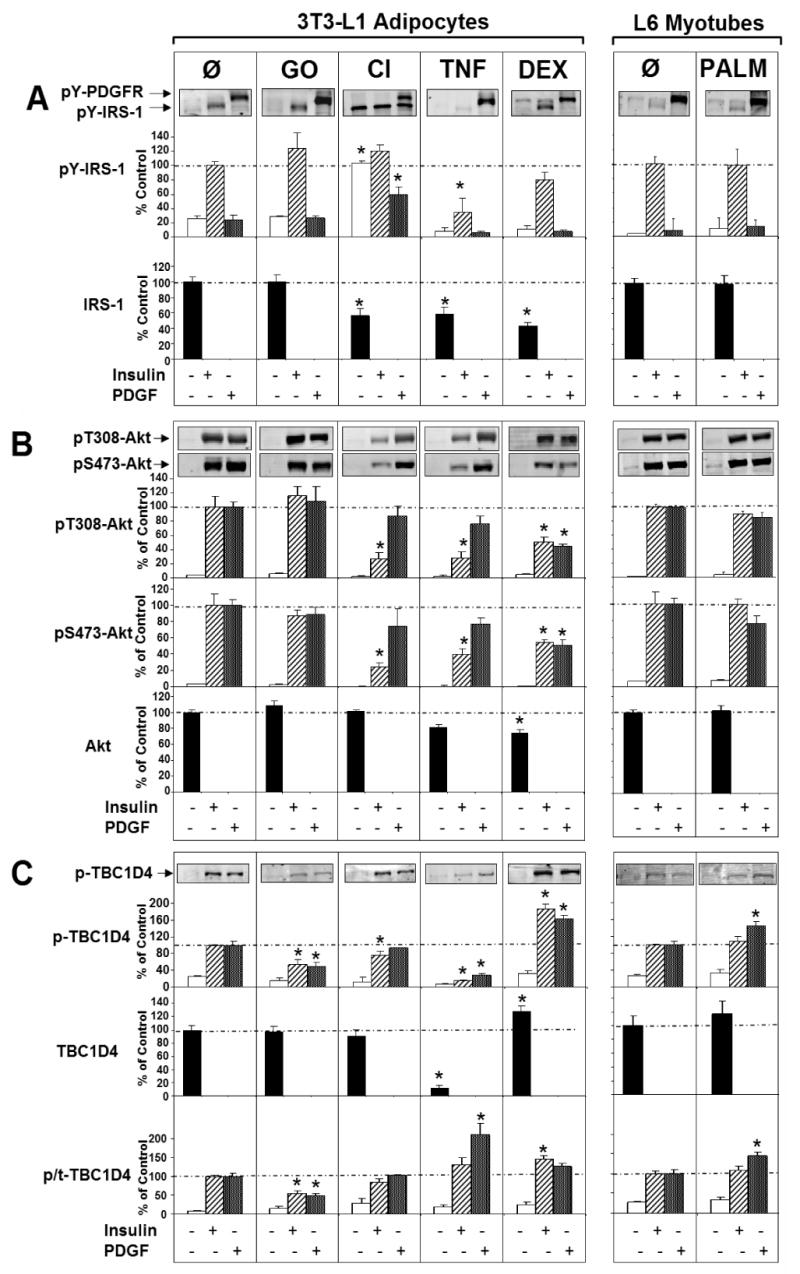
A) Representative Western blots and quantitative bar graphs are shown for the detection of PDGFR and IRS-1 tyrosine phosphorylation and total IRS-1 protein levels. The pY612-IRS-1 antibody detects phosphorylation of the PI3 kinase binding site in both molecules.
B) Representative Western blots and quantification of Akt Ser473 and Thr308 phosphorylation and total Akt levels are shown in this section.
C) Cell lysates were also probed for TBC1D4 phosphorylation at Thr642 and total TBC1D4 expression. The lower panel represents a quantification of phosphorylated to total (p/t)-TBC1D4. Western blots are representative and statistical values are displayed as means ± s.e.m., *P<0.05 vs. respective control (Ø), Student's t-test, n=3-5 experiments.
We next focused on Akt to determine if the changes in IRS function described above were transmitted distally throughout the insulin signaling cascade. Phosphorylation of Akt at both Thr308 and Ser473 was measured as an index of its activation by upstream regulators. To determine the in vivo activity state of Akt the phosphorylation of TBC1D4, a key target of Akt involved in GLUT4 translocation (Kane et al., 2002; Larance et al., 2005), was measured. We and others have shown that Thr642 is one of the key Akt substrate sites in TBC1D4 that plays an essential role in its function (Kane et al., 2002; Ramm et al., 2006; Sano et al., 2003). Therefore, a phosphorylation-specific antibody targeting this site was produced and used to probe cell lysates.
Consistent with changes in IRS function we observed a significant reduction in Akt phosphorylation in cells treated with CI, TNF and DEX, but not in cells treated with either GO or PALM (Figure 4B). These data are consistent with previous reports showing upstream signaling defects upon treatment with CI, TNF, and DEX (Aguirre et al., 2000; Ball et al., 2005; Hotamisligil, 1999; Houstis et al., 2006; Ishizuka et al., 2007; Paz et al., 1997; Saad et al., 1993; Sun et al., 1999; Turnbow et al., 1994). Interestingly, PDGF treatment overcame the defect in Akt activation in cells treated with CI and TNF, but not in cells treated with DEX. To determine if these changes in Akt phosphorylation resulted in defective Akt signalling we next examined TBC1D4 phosphorylation (Figure 4C). Surprisingly we observed a complete discordance between these parameters. For example, there was no effect of GO on either insulin or PDGF-stimulated Akt phosphorylation (Figure 4B), whereas the specific activity of TBC1D4 phosphorylation was significantly diminished (Figure 4C). Conversely, with CI, despite reduced insulin-stimulated Akt phosphorylation, TBC1D4 phosphorylation was unimpaired (Figure 4C). This highlights a degree of “spareness” at the level of Akt as described in Figure 1. TNF-treated cells exhibited a distinct phenotype at the level of TBC1D4 compared with either GO or CI-treated cells. Here we observed a dramatic reduction in TBC1D4 protein levels. Despite this, the specific activity of TBC1D4 phosphorylation at Thr642 was unchanged in response to either insulin or PDGF. Again this emphasizes the “spareness” in these signalling intermediates because TBC1D4 is believed to be a negative regulator of GLUT4 translocation under basal conditions and phosphorylation overcomes this negative regulation (Kane et al., 2002; Larance et al., 2005; Ramm et al., 2006). Hence, it is notable that cells were able to tolerate this substantial reduction in TBC1D4 levels with only a mild increase in basal cell surface levels of GLUT4 (Figure 3A). Intriguingly, DEX yielded changes at the level of TBC1D4 that were diametrically opposed to TNF. TBC1D4 total protein levels and Thr642 phosphorylation in response to insulin and PDGF were significantly enhanced in these cells. Considering that both TNF and DEX caused significant insulin resistance at the level of GLUT4 translocation these data suggest that intermediates such as TBC1D4 may operate as regulated switches with an intrinsic threshold such that modest defects upstream of this molecule may not directly translate downstream.
Effects of Palmitate on Insulin Action
The most striking discrepancy between Akt activation and GLUT4 translocation was observed in L6 myotubes incubated with palmitate. In the presence of 150 μM palmitate, insulin- and PDGF-stimulated GLUT4 translocation were inhibited by >50% (Figure 3B) without any observed signaling defects at IRS-1, Akt, or TBC1D4 (Figure 4). This was surprising as other investigators have described defects in insulin signalling following incubation of either adipocytes or myotubes with palmitate (Powell et al., 2004; Sinha et al., 2004). However, many of these studies utilize much higher doses of palmitate to achieve these effects than used in the present study. In our preliminary investigations we observed that high palmitate doses (>300 μM) were toxic to cells resulting in morphological changes and even detachment from the substratum. Hence, we further characterised the dose response relationship between palmitate and insulin action. We observed a significant reduction in insulin-stimulated GLUT4 translocation with palmitate doses as low as 50 μM reaching a maximum diminution at 150 μM (Figure 5A). No defects in either IRS-1 expression or insulin-stimulated IRS-1 tyrosine phosphorylation, Akt phosphorylation, or TBC1D4 phosphorylation were observed using palmitate concentrations between 0-150 μM (Figure 5B and 5C). Whereas at concentrations exceeding 300 μM, substantial defects in each of these signalling parameters were observed. Again this model of insulin resistance points to a major node of insulin resistance distal to IRS/PI3K/Akt and suggests that signalling defects may arise either as a consequence of insulin resistance per se (i.e. reduced GLUT4 translocation) or independently of insulin resistance possibly due to toxic “off-target” effects.
Figure 5. Palmitate antagonizes insulin action at GLUT4 without disrupting signal transduction through TBC1D4 in L6 myotubes.
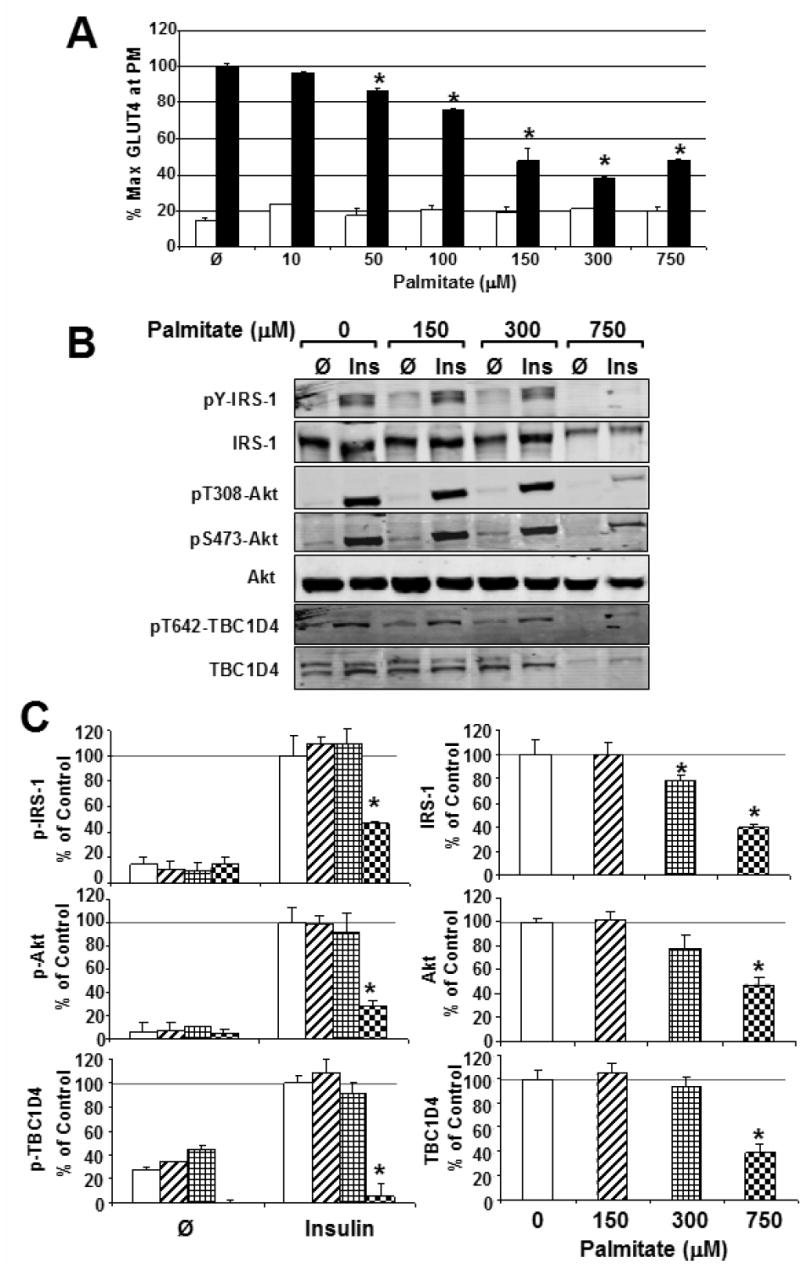
A) Basal (white bars) and Insulin-stimulated (black bars) GLUT4 translocation was measured in L6 myotubes incubated with a dose curve of palmitate for 18 h. Error bars, ± s.e.m., (n = 3 independent experiments of >3 measurements each) *P<0.05 between palmitate and carrier control, Student's t-test.
B) Representative Western blots of phosphorylated and total IRS-1, Akt, and TBC1D4 in palmitate-treated myotubes.
C) No significant difference was observed in the phosphorylation of IRS-1, Akt or TBC1D4 between control myotubes and those treated with up to 300 μM palmitate. However, higher concentrations severely antagonized insulin action at these molecules as shown.
The Role of IRS /Akt in high fat fed Mice
The above studies indicate that while a range of signaling defects can be observed in some (GO, CI, TNF, and DEX), but not all models of insulin resistance (PALM), it is apparent that there is sufficient “spareness” in many of these elements such that they may neither represent the sole nor primary defect. While such cell systems have been invaluable in dissecting the molecular basis of insulin resistance it is important to determine if such findings can be translated to a more physiological setting. Hence, we next performed a series of studies in transgenic mice overexpressing the PDGFR in muscle. PDGF stimulated signal transduction and glucose transport to a similar extent as insulin in isolated soleus muscle from these mice (Figures 6A-C). Thus these animals provide a unique system with which to assess the role of IRS and downstream signalling elements in insulin resistance in vivo.
Figure 6. Modulation of Insulin and PDGF action in high fat fed PDGFR transgenic mice.
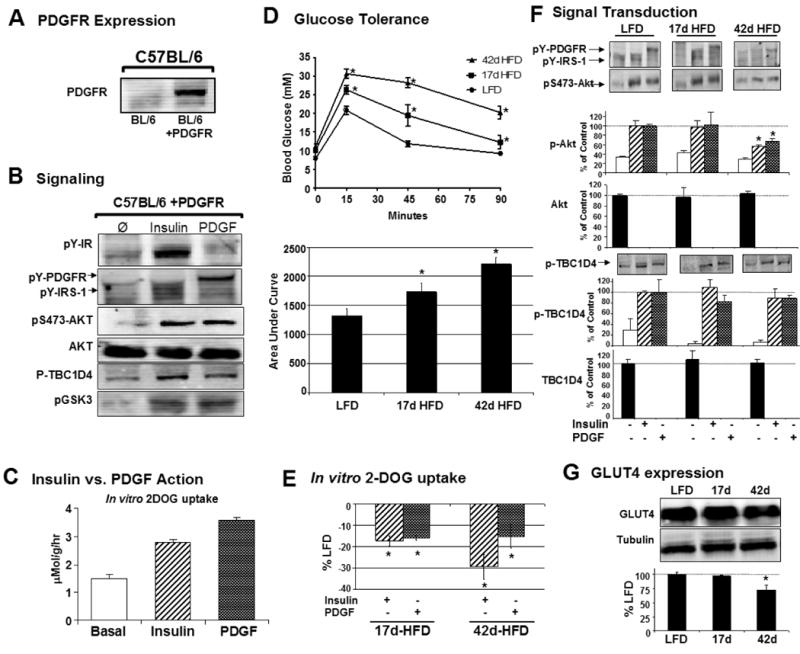
A) Intact soleus muscles from transgenic mice were blotted for expression of the PDGFR.
B) Isolated solei were incubated in vitro and stimulated with 2 mU/mL insulin or 500 ng/mL PDGF for 30 min and Western blots were performed on whole cell lysates. Representative blots are shown. Phosphorylation of T308-Akt was undetectable.
C) PDGF (shaded bars) had a more potent effect on 3H-2-deoxyglucose uptake into soleus muscle than insulin (hashed bars) in transgenic mice (3.59 ± 0.09 vs. 2.78 ± 0.12 μMol 3H-2-deoxy-D-glucose/g/hr) consistent with previous studies (Yuasa et al., 2004).
D) Glucose tolerance tests were performed on mice in the 4h fasted state to predict insulin resistance. Both diet durations of 17 and 42 days produced significant reductions in glucose tolerance indicated by differences blood glucose levels of high fat fed mice versus the low fat diet group, n=7-9 mice per curve, individual points are displayed as means ± s.e.m., *P<0.05 between lean diet and test diet groups by students t-test.
E) Reduction in insulin- (hashed bars) and PDGF-stimulated (shaded bars) 2DOG uptake in isolated soleus muscles from mice fed a high fat diet for 17 days (-17±6% and -16±5% of control insulin- and PDGF-stimulation, respectively) or 42 days (-29±5% and -15±2% of control insulin- and PDGF-stimulation, respectively). n= 3-7 solei per data set.
F) Representative Western blots and bar graphs are shown for soleus muscles stimulated with insulin (hashed bars) or PDGF (shaded bars) for 30 min. Total protein levels are shown in full bars. n = 3-5 solei per experiment.
G) Representative Western blots and quantification of GLUT4 expression in mice fed a LFD and HFD for 17 and 42 days. n=4 per group.
E-G) Results are displayed as means ± s.e.m., *P<0.05 between lean diet and test diet groups, Student's t-test.
We created insulin resistance in this model by dietary modification to reflect the physiological impact of poor diet. The diets consisted of either standard lab chow (8% calories from fat, 21% calories from protein, 71% calories from carbohydrate, 2.6 kcal/g) or a high saturated fat diet (45% of calories from fat, of which 87% was lard and 13% safflower oil by weight, 20% calories from protein, and 35% calories from carbohydrates; 4.7 kcal/g). Consistent with other reports (Bonnard et al., 2008) we observed glucose intolerance within weeks of high fat feeding (Figure 6D). To study the mechanism of this effect, we examined the capacity of insulin and PDGF to increase glucose uptake and activate signal transduction in this model (Figures 6E and 6F). High fat feeding for as little as 17 days resulted in a significant reduction in both insulin- and PDGF-stimulated 2DOG uptake, without any detectable decrease in GLUT4 levels (Figure 6G) or Akt or TBC1D4 phosphorylation. As was the case with the cell models described above, impaired insulin- and PDGF-stimulated Akt phosphorylation was apparent only with longer term dietary treatment (42 days HFD; Figure 6F). It should also be noted that we observed a significant reduction in total muscle GLUT4 levels by 30% in the 42 day high fat fed animals. Intriguingly, this was accompanied by an additional decrease in insulin, but not PDGF-stimulated, 2DOG uptake compared to that observed in animals fed HFD for 17 days. This suggests that IRS-dependent defects might contribute to a worsening or maintenance of the condition. These data are consistent with the in vitro PALM data in myotubes indicating that early defects leading to insulin resistance are not due to impaired IRS/Akt function and while defects in Akt phosphorylation are observed with prolonged high fat diet there is sufficient “spareness” in the system such that these defects are not necessarily transmitted further down the pathway since phosphorylation of TBC1D4 remained intact.
Discussion
Defining the mechanism(s) of insulin resistance is challenging. Numerous insults can cause insulin resistance yet it has been difficult to establish if there is a common link between these disparate triggers. A common dogma in the field is that IRS proteins represent a major node of insulin resistance for most of these models (Aguirre et al., 2000; Dresner et al., 1999; Gao et al., 2002; Gao et al., 2003; Gual et al., 2005; Hotamisligil et al., 1996; Morino et al., 2005; Ozcan et al., 2004; Rice et al., 1993; Turnbow et al., 1994; Ueki et al., 2004; White, 2002; Yu et al., 2002; Zick, 2005; Zierath et al., 1997). In the present study we provide evidence that while defects in upstream elements of the insulin cascade may occur in various insulin resistance models it is unlikely that this is a major cause of impaired glucose metabolism, the defining feature of insulin resistance.
We have utilized a system enabling us to test the role of IRS proteins in insulin resistance by comparing the effects of insulin and PDGF on signaling and metabolism. For example, with chronic insulin a significant reduction in insulin-stimulated GLUT4 translocation was observed whereas PDGF-stimulated GLUT4 translocation was similar to that observed in control cells. This indicates that with chronic insulin the major impairment likely involves a target that is specific to the IR most likely IRS1. This is consistent with previous literature and acts as an important control for the present model system. In contrast, in all other models, which more closely parallel physiologic states of insulin resistance we observed a similar impairment in GLUT4 translocation with both ligands. The most extreme discordance between upstream signaling and GLUT4 translocation was observed in palmitate treated L6 myotubes and high fat fed mice. Here using either low doses of palmitate or relatively short exposure to a HFD we observed impaired insulin and PDGF-stimulated GLUT4 translocation and 2DOG uptake, respectively, without any detectable defect within the IRS/PI3K/Akt nexus. Collectively these studies suggest that while defects in IRS/PI3K/Akt may occur in insulin resistance it is unlikely that such defects contribute to its early development. This raises the possibility that upstream signaling defects are either corollary or a consequence rather than a cause of insulin resistance possibly exacerbating the insulin resistance state once it has become established and promote progression to metabolic disease. Alternatively, these signaling defects may be completely independent of the metabolic milieu possibly involving cytotoxicity, a potential consequence of chronic exposure to cytokines, lipids and other metabolic insults that in many cases lead to cellular stress (Bloch-Damti and Bashan, 2005; Evans et al., 2005; Kaneto et al., 2005).
A major problem with many studies that have implicated an important role for upstream signaling elements in insulin resistance is that they assume that such pathways operate stoichiometrically such that a modest defect in one component will be linearly transmitted all the way down the pathway. However, our analysis of insulin action under control and insulin resistant conditions indicates that these pathways are governed more by a stochastic operating principle. This can be illustrated by the observation that only a finite amount of Akt is required to be phosphorylated in order to orchestrate a maximal biological response (Figure 1). Moreover, once activated there is discordance in the ability of Akt to activate discrete downstream substrates such as TBC1D4 and GSK3. The nature of this differential transmission downstream of Akt may involve a role for discrete Akt isoforms, the unique localization of certain Akt substrates, or the assembly of unique signaling subcomplexes involving scaffolding proteins and this certainly requires further investigation. Careful analysis of our insulin resistance models as summarized in Table 1 reveals a somewhat fragmented response pattern in upstream elements of the pathway which again is consistent with stochastic regulation involving many negative feedback loops. However, a unifying feature that emerges from this analysis is that despite this fragmented pattern upstream both insulin and PDGF stimulated TBC1D4 phosphorylation responded quite normally in the face of insulin resistance at the level of GLUT4 translocation.
Table 1.
Summary of defects in insulin and PDGF-stimulated signal transduction and GLUT4 translocation (for cell culture models) or 2DOG uptake (for animal model). Down arrows indicate the severity of defect for each variable and tilde represents the absence of a defect.
| IRS-1 Defect | p-Akt | P-TBC1D4 | GLUT4/2-DOG | ||||||
|---|---|---|---|---|---|---|---|---|---|
| pY | Expression | Insulin | PDGF | Insulin | PDGF | Insulin | PDGF | Primary Defect | |
| Glucose Oxidase | ∼ | ∼ | ∼ | ∼ | ↓↓ | ↓↓ | ↓↓ | ↓↓ | IRS Independent |
| Chronic Insulin | ∼ | ↓↓ | ↓↓ | ∼ | ∼ | ∼ | ↓↓ | ↓ | IRS |
| TNFα | ↓↓ | ↓↓ | ↓↓ | ∼ | ∼ | ∼ | ↓↓ | ↓↓ | IRS + IRS Ind. |
| Dexamethasone | ∼ | ↓↓ | ↓↓ | ↓↓ | ∼ | ∼ | ↓↓ | ↓↓ | IRS + IRS Ind. |
| Palmitate | ∼ | ∼ | ∼ | ∼ | ∼ | ∼ | ↓↓ | ↓↓ | IRS Independent |
| 17d HFD | ∼ | ∼ | ∼ | ∼ | ∼ | ∼ | ↓↓ | ↓↓ | IRS Independent |
How do insulin sensitive cells achieve graded outputs in response to different concentrations of the relevant agonist given that the active range of Akt with respect to glucose transport appears to be extremely finite? One possibility is that signaling through Akt is a digital rather than an analog response. A similar mechanism has been described for Ras signaling (Tian et al., 2007). In addition, we have recently presented a model whereby Akt may control GLUT4 translocation via a quantal release mechanism again consistent with a digital or threshold response (Larance et al., 2007). Such a mechanism likely denotes signaling subcompartments that are not resolved by the types of analyses employed in this and many other studies. For example, it has been suggested that there are critical pools of IRS-1 that may be localized close to the plasma membrane (Clark et al., 1998) in which case one can envisage that defects in these compartments may disrupt signaling through the pathway in an apparent non-linear fashion. While we cannot dismiss these possibilities, it seems likely that such adaptations would merely account for the non-linearity in the system thus emphasizing the need to exercise caution when interpreting analyses involving simple changes in total levels or the phosphorylation status of such intermediates. With this in mind it is important therefore to consider the output from signaling nodes such as Akt substrate phosphorylation or GLUT4 translocation, which in both cases was discordant with the flux through these upstream elements.
How can the present data be interpreted in light of numerous studies suggesting that IRS1 is the major node of insulin resistance? First, it is noteworthy that many studies have focused on signaling rather than metabolic endpoints as their index of insulin resistance. Here we utilized GLUT4 translocation or glucose transport as our index of insulin resistance. This is important because not only is this process rate limiting for many of the metabolic actions of insulin, but defects in glucose transport appear to represent some of the earliest and most significant defects observed in insulin resistance (Petersen and Shulman, 2006). Moreover, our analysis of GLUT4 translocation has been normalized to the total expression of GLUT4 in the cell thus overcoming the contribution of altered GLUT4 expression in response to insulin resistance treatments (Hajduch et al., 1995; Hajduch et al., 1992; Kaestner et al., 1991; Lundgren et al., 2004; Stephens et al., 1997; Stephens and Pekala, 1992). We also suggest that this assay is superior to the measurement of cellular glucose transport or glycogen synthesis as this is easily influenced by changes in the expression of alternate transporter isoforms such as GLUT1, a common feature of in vitro cell culture models of insulin resistance (Hosaka et al., 1999; Mei et al., 2003; Sakoda et al., 2000) but not in vivo insulin resistance (Kahn et al., 1991). Second, many studies have also overlooked the temporal and/or dose response characteristics of the metabolic insult with respect to the onset of insulin resistance. In the present study we carefully characterized minimal conditions required to achieve maximal insulin resistance as defined by inhibition of GLUT4 translocation to the plasma membrane in cell culture or glucose uptake in mice. This was essential as in at least two models (150 μM palmitate in L6 myotubes or 17 d HFD in mice) we observed marked insulin resistance without any significant change in signaling, whereas under more extreme conditions signaling defects became apparent. Finally, many studies implicating IRS proteins in insulin resistance rely on correlative changes between metabolism and IRS Ser/Thr phosphorylation in response to various reversal strategies. For instance inhibition of mTOR with rapamycin, or JNK with peptide and small molecule drugs have been used in this way, yet these agents likely impact upon many cellular processes due to their effect on protein synthesis and gene expression, respectively. Thus if insulin resistance is independent of the signaling defects as suggested here, then any manipulation that corrects insulin resistance might also potentially override defects in signaling that derive from the same insult.
A major question arising from these studies is what are the molecular targets that contribute to insulin resistance? Based on the present studies we conclude that these defects likely occur downstream of IRS and in some cases possibly downstream of Akt. However, in relation to the latter it is important to emphasize that the regulation of Akt is proving to be quite complex as indicated by recent studies showing a role for the Akt binding protein APPL1 in insulin stimulated GLUT4 translocation (Saito et al., 2007). Hence, this may invoke some type of nano-regulation that will be challenging to dissect. This could involve the assembly of signaling modules close to or at the plasma membrane. In this regard it is interesting that reduced levels of PIP2 at the cell surface may disrupt the ability to recruit GLUT4 vesicles to this site possibly due to disruptions in the actin cytoskeleton (McCarthy et al., 2006; Strawbridge and Elmendorf, 2005). This poses yet another important question which is, does insulin resistance represent a uniform disruption in all of the metabolic actions of insulin or is there a temporal progression of defects whereby an initial defect in glucose transport for example could subsequently lead to defects in other metabolic parameters such as glycogen and lipid synthesis? Consistent with such a hypothesis is the observation that deletion of GLUT4 in adipose tissue results in insulin resistance in other tissues in the intact animal (Abel et al., 2001). Hence, this potentially places GLUT4 translocation and glucose transport at the center of this disorder.
In conclusion, these data reveal that upstream elements of the insulin signaling cascade are not a central feature of the origin of insulin resistance as commonly thought. While defects in these upstream signaling elements may be frequently observed in insulin resistance in view of the stochastic nature of the pathway such defects may or may not be transmitted further downstream. Moreover, such defects may occur as a consequence of an alternate initial defect possibly to sustain the insulin resistant state. The nature of these initial defect(s) remains unknown at the present time. It is likely that a search for additional Akt substrates and other downstream regulators of this pathway will be revealing. The identification of such targets will be of enormous utility in the design of targeted therapies for prevention of the disorder. This study provides new molecular insight into the mechanisms and origins of insulin resistance and should facilitate a renewed focus in the pursuit of new molecular targets downstream of IRS.
Experimental Procedures
Models of insulin resistance
3T3-L1 preadipocytes or L6 myoblasts were infected with replication incompetent retroviruses expressing HA-GLUT4 and human PDGF β-Receptor and selected with puromycin and geneticin. Cells were differentiated into adipocytes or myotubes as described previously (Lee et al., 2006). Oxidative stress was induced in 3T3-L1 adipocytes by washing cells 3 times in PBS and incubating with serum free DMEM supplemented with 0.1% BSA (stepdown media) containing 50 mU/mL glucose oxidase or carrier (0.16μM sodium acetate, pH 7.4) for 2 h. This generated a final H2O2 concentration of 25.8 -/+ 0.1 μM, while total glucose levels (measured with an Accu-check II glucometer (Roche, NSW, Australia)) decreased slightly from 24.7 -/+ 1.6mM to 23.3 -/+ 1.9mM. Chronic low-dose Inflammation was mimicked in 3T3-L1 adipocytes by incubation with 2 ng/mL TNFα (Calbiochem) for 4 days. Media was changed every 24 h. On the fourth day cells were washed 3× with PBS and incubated in stepdown media in the absence of TNFα for 90 min prior to acute insulin or PDGF stimulation. Hyperinsulinemia was created by addition of 10 nM insulin to adipocytes at 1200, 1600 and 2000 h on day 1 and 0800 h the following day. At 1200 h on the second day cells were washed 4-5 times with PBS and cultured in stepdown media for 90 min before acute insulin or PDGF stimulation. Glucocorticoid-induced insulin resistance was created with 20 nM dexamethasone (0.01% ethanol carrier as control) changed every other day for 8 d. On day 8 the cells were washed 3× with PBS and incubated for 90 min in stepdown media before acute stimulation. Palmitate was conjugated to 20% BSA in culture media at a concentration 5 fold of final. BSA/palmitate or BSA/ethanol (control) were heated to 50C for 20 minutes, cooled to 37C and diluted 5× with culture media before filter sterilizing. The flow through was delivered to cells at the specified doses with a final BSA concentration of 4% in all treatments. The complete treatment lasted 18 h, and consisted of two additions of palmitate. The first treatment was performed in the presence of 2% fetal calf serum for 16.5 h, after which the myotubes were washed 3× with PBS, and the second incubation was identical to the first except without serum and for 90 min prior to acute stimulation with insulin or PDGF. All insulin resistance treatment regimens were initiated in 3T3-L1 adipocytes between 7-12 days post-differentiation, and in L6 myotubes between 6-9 days post-differentiation. After the insulin resistance treatment, 100 nM insulin or 20 ng/mL PDGF was added to the cells for 20 min before harvest or analysis of GLUT4 translocation. Total GLUT4 protein levels were measured by Western blot following each treatment condition. While we did not observe a significant change in GLUT4 levels following GO, CI, or PALM treatments, GLUT4 levels were increased by 22% and 94% after treatment with TNF and DEX, respectively (data not shown). Therefore all measures of GLUT4 translocation were normalized to total GLUT4 and represented as the percent of GLUT4 at the plasma membrane.
Animals
Male C57BL/6 mice overexpressing the PDGFR in muscle were obtained from Yuasa et al (Yuasa et al., 2004). Homozygous transgenic mice (clone #2) were fed a chow diet until 8 wks of age when they were split into two groups. One group was maintained on a standard lab chow (8% calories from fat, 21% calories from protein, 71% calories from carbohydrate, 2.6 kcal/g) and the other was switched to a high saturated fat diet (45% of calories from fat (of which 87% was lard and 13% safflower oil by weight), 20% calories from protein, and 35% calories from carbohydrates; 4.7 kcal/g; based on Rodent Diet #D12451 (Research Diets, Inc., New Brunswick, NJ)) for a period of 17 d or 6 wks. Mice were housed in a temperature-controlled room (22±1°C) on a 12-h light/dark (0700/1900) cycle.
Materials
Rat recombinant PDGF-BB, puromycin, dexamethasone, biotin, isobutyl-methylxanthine, pyruvate, mannitol, TLC plates, potassium oxalate, and glucose oxidase were from Sigma (Sigma, St. Louis, MO). Geneticin, 100× penicillin/streptomycin/glutamine solution, NuPAGE precast gels, DMEM, and alpha-MEM were from Invitrogen (Mount Waverly, VIC, Australia). [1-14C] mannitol, 2-[2-6-3H]-deoxyglucose, and 32P-gamma ATP were from GE Health Care (Buckinghamshire, England). Fetal calf serum was from Thermo (Melbourne, Australia). Standard rodent chow was from Gordon's Specialty Stock Feeds (Yanderra, New South Wales, Australia). The Akt1/2 inhibitor was obtained from Symansis (Auckland, NZ) and details on its characterization can be found here (DeFeo-Jones et al., 2005).
Antibodies
Phospoho-S473 Akt, p-T308 Akt, total Akt, and p-GSK3 were from Cell Signaling Technology (Danvers, MA). Antibodies detecting the Insulin Receptor, PDGFR, IRS-1, and pY-99 were from Santa Cruz Biotechnology (Santa Cruz, CA). pY-IR and pY-IRS antibodies were from Millipore (formerly Upstate Biotechnology, Billerica, MA). The pThr642-TBC1D4 antibody was created for us by Symansis (Auckland, NZ).
Animal Experiments
Intraperitoneal glucose tolerance tests (GTT) were performed on mice the morning of sacrifice. Mice were fasted for 4 hours (0730-1130) before i.p. injection of 25% glucose solution at a dose of 2g/kg. Blood glucose was measured by sampling blood from the tail tip with an Accu-check II glucometer. After the GTT mice were killed by cervical dislocation and solei muscles were rapidly dissected tendon to tendon and in vitro 2DOG uptake was measured as described previously (Turner et al., 2007).
Solei (approx 10mg) were processed for immunoblotting by homogenizing at 4C with 250uL of 1× phosphate buffered saline pH 7.4 containing 1 mM EDTA, 30 mM sodium pyrophosphate, 10 mM NaF, 150 mM NaCl, 2 mM Na3VO4, 1% NP-40, 10% glycerol, and 1× protease inhibitor cocktail (Roche) using a Polytron instrument (Kinematica, Littau-Lucerne, Switzerland). The lysates were rotated at 4C for 1 hour before being centrifuged at 16,000×g for 15 minutes at 4C. Supernatant protein concentration was determined by the bicinchoninic acid method (BCA, Pierce, Rockford, IL). All experiments were carried out with the approval of the Garvan Institute/St. Vincent's Hospital Animal Experimentation Ethics Committee, following guidelines issued by the National Health and Medical Research Council of Australia.
Immunoblots
3T3-L1 and L6 cultures were washed with cold PBS 2× before harvesting with HES buffer, pH 7.4, containing 2% SDS. Lysates were sonicated for 15 seconds and centrifuged at 15,000×g for 20 minutes at 15C. Supernatants were then assayed for protein content by BCA assay. Homogenates from soleus muscle were prepared as described above. Clarified lysates were then diluted with 4× Laemmli buffer and heated to 65C for 3 minutes. Equal amounts of protein were electrophoresed through self-made 8% polyacrylamide gels or NuPage pre-cast gradient gels, transferred to PVDF membranes, and blocked with 5% BSA in tris buffered saline (TBS) for 1 hour. Blocked membranes were incubated overnight at 4C in primary antibodies diluted in TBS-0.1% Tween-20 (TBST) containing 5% BSA and 0.05% sodium azide. The following morning membranes were incubated for 0-2 more hours at room temperature and washed for 30 minutes in TBST. Membranes were then incubated with their respective fluorescently labeled secondary antibodies diluted in TBST+5% BSA+0.1% SDS in the dark at room temperature for 1 hour and washed 3× for >30 minutes in the dark. Membranes were then developed and quantified using a LICOR Odyssey infrared imaging instrument and software (LICOR, Lincoln, NE). 14-3-3 expression and Ponceau staining were utilized as loading controls for these experiments.
PI3 Kinase Activity Assays
These assays were performed as described previously (Wang and Summers, 2003).
GLUT4 Translocation Assays
These assays were performed as we have described previously (Govers et al., 2004).
Statistical Analyses
Data are expressed as means ± standard error. For GLUT4 translocation analysis, unless otherwise specified, each experiment was normalized to 100% of maximal insulin before comparison across experiments to decrease inter-assay variation and allow direct comparison across all insults of the same cell type. P-values were calculated by two-tailed Student's t-test using Microsoft excel (Microsoft Corp, Seattle, WA) or Graph Pad Prism (San Diego, CA). Statistical significance was set at P<0.05.
Acknowledgments
This work was supported by the US National Institute of Health (DK067509 to DEJ and DK075249 to KLH), National Health and Medical Research Council of Australia (DEJ), and Diabetes Australia Research Trust (DEJ). We thank Dr Roger Daly and Dr Greg Cooney for discussions as well as other members of the James lab. The Akt inhibitor and pT642-TBC1D4 antibody were kind gifts from Dr. Peter Shephard (Symansis, Auckland, NZ), and the PDGFR plasmid was kindly provided by Dr. J.A. Cooper (Fred Hutchinson Cancer Research Center, Seattle, Washington).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- Ball LE, Berkaw MN, Buse MG. Identification of the major site of O-GlcNAc modification in the C-terminus of IRS-1. Mol Cell Proteomics. 2005 doi: 10.1074/mcp.M500314-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann CA, Ribon V, Kanzaki M, Thurmond DC, Mora S, Shigematsu S, Bickel PE, Pessin JE, Saltiel AR. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–207. doi: 10.1038/35025089. [see comments] [DOI] [PubMed] [Google Scholar]
- Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch-Damti A, Bashan N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid Redox Signal. 2005;7:1553–1567. doi: 10.1089/ars.2005.7.1553. [DOI] [PubMed] [Google Scholar]
- Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, Vidal H, Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SF, Martin S, Carozzi AJ, Hill MM, James DE. Intracellular localization of phosphatidylinositide 3-kinase and insulin receptor substrate-1 in adipocytes: potential involvement of a membrane skeleton. J Cell Biol. 1998;140:1211–1225. doi: 10.1083/jcb.140.5.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleasby ME, Reinten TA, Cooney GJ, James DE, Kraegen EW. Functional studies of Akt isoform specificity in skeletal muscle in vivo; maintained insulin sensitivity despite reduced insulin receptor substrate-1 expression. Mol Endocrinol. 2007;21:215–228. doi: 10.1210/me.2006-0154. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- DeFeo-Jones D, Barnett SF, Fu S, Hancock PJ, Haskell KM, Leander KR, McAvoy E, Robinson RG, Duggan ME, Lindsley CW, et al. Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol Cancer Ther. 2005;4:271–279. [PubMed] [Google Scholar]
- Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. 2005;7:1040–1052. doi: 10.1089/ars.2005.7.1040. [DOI] [PubMed] [Google Scholar]
- Farese RV, Sajan MP, Wang H, Li P, Mastorides S, Gower WR, Nimal S, Choi CS, Kim S, Shulman GI, et al. Muscle-specific knockout of PKC-lambda impairs glucose transport and induces metabolic and diabetic syndromes. J Clin Invest. 2007 doi: 10.1172/JCI31408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon MJ, Ye J. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. 2002;277:48115–48121. doi: 10.1074/jbc.M209459200. [DOI] [PubMed] [Google Scholar]
- Gao Z, Zuberi A, Quon MJ, Dong Z, Ye J. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases. J Biol Chem. 2003;278:24944–24950. doi: 10.1074/jbc.M300423200. [DOI] [PubMed] [Google Scholar]
- Govers R, Coster AC, James DE. Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol Cell Biol. 2004;24:6456–6466. doi: 10.1128/MCB.24.14.6456-6466.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Hajduch E, Hainault I, Meunier C, Jardel C, Hainque B, Guerre-Millo M, Lavau M. Regulation of glucose transporters in cultured rat adipocytes: synergistic effect of insulin and dexamethasone on GLUT4 gene expression through promoter activation. Endocrinology. 1995;136:4782–4789. doi: 10.1210/endo.136.11.7588207. [DOI] [PubMed] [Google Scholar]
- Hajduch EJ, Guerre-Millo MC, Hainault IA, Guichard CM, Lavau MM. Expression of glucose transporters (GLUT1 and GLUT4) in primary cultured rat adipocytes: differential evolution with time and chronic insulin effect. Journal of Cellular Biochemistry. 1992;49:251–258. doi: 10.1002/jcb.240490307. [DOI] [PubMed] [Google Scholar]
- Hosaka T, Yaga K, Oka Y. Regulation of insulin-stimulated glucose transport by chronic glucose exposure in 3T3-L1 adipocytes. Endocr J. 1999;46:349–357. doi: 10.1507/endocrj.46.349. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Mechanisms of TNF-alpha-induced insulin resistance. Exp Clin Endocrinol Diabetes. 1999;107:119–125. doi: 10.1055/s-0029-1212086. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Inoue G, Cheatham B, Emkey R, Kahn CR. Dynamics of insulin signaling in 3T3-L1 adipocytes. Differential compartmentalization and trafficking of insulin receptor substrate (IRS)-1 and IRS-2. J Biol Chem. 1998;273:11548–11555. doi: 10.1074/jbc.273.19.11548. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Usui I, Kanatani Y, Bukhari A, He J, Fujisaka S, Yamazaki Y, Suzuki H, Hiratani K, Ishiki M, et al. Chronic tumor necrosis factor-alpha treatment causes insulin resistance via insulin receptor substrate-1 serine phosphorylation and suppressor of cytokine signaling-3 induction in 3T3-L1 adipocytes. Endocrinology. 2007;148:2994–3003. doi: 10.1210/en.2006-1702. [DOI] [PubMed] [Google Scholar]
- JeBailey L, Wanono O, Niu W, Roessler J, Rudich A, Klip A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes. 2007;56:394–403. doi: 10.2337/db06-0823. [DOI] [PubMed] [Google Scholar]
- Kaestner KH, Flores RJR, McLenithan JC, Janicot M, Lane MD. Transcriptional repression of the mouse insulin-responsive glucose transporter (GLUT4) gene by cAMP. Proc Natl Acad Sci U S A. 1991;88:1933–1937. doi: 10.1073/pnas.88.5.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn BB, Rossetti L, Lodish HF, Charron MJ. Decreased in vivo glucose uptake but normal expression of GLUT1 and GLUT4 in skeletal muscle of diabetic rats. J Clin Invest. 1991;87:2197–2206. doi: 10.1172/JCI115254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane S, Sano H, Liu SC, Asara JM, Lane WS, Garner CC, Lienhard GE. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277:22115–22118. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- Kaneto H, Matsuoka TA, Nakatani Y, Kawamori D, Miyatsuka T, Matsuhisa M, Yamasaki Y. Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J Mol Med. 2005;83:429–439. doi: 10.1007/s00109-005-0640-x. [DOI] [PubMed] [Google Scholar]
- Kim YB, Nikoulina SE, Ciaraldi TP, Henry RR, Kahn BB. Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J Clin Invest. 1999;104:733–741. doi: 10.1172/JCI6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, Konishi H, Matsuzaki H, Kikkawa U, Ogawa W, Kasuga M. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol Cell Biol. 1999;19:6286–6296. doi: 10.1128/mcb.19.9.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- Kono T, Barham FW. The relationship between the insulin-binding capacity of fat cells and the cellular response to insulin. Studies with intact and trypsin-treated fat cells. J Biol Chem. 1971;246:6210–6216. [PubMed] [Google Scholar]
- Larance M, Ramm G, James DE. The GLUT4 Code. Mol Endocrinol. 2007 doi: 10.1210/me.2007-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larance M, Ramm G, Stockli J, van Dam EM, Winata S, Wasinger V, Simpson F, Graham M, Junutula JR, Guilhaus M, James DE. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, Ye JM, Lee CH, Oh WK, Kim CT, et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes. 2006;55:2256–2264. doi: 10.2337/db06-0006. [DOI] [PubMed] [Google Scholar]
- Lundgren M, Buren J, Ruge T, Myrnas T, Eriksson JW. Glucocorticoids down-regulate glucose uptake capacity and insulin-signaling proteins in omental but not subcutaneous human adipocytes. J Clin Endocrinol Metab. 2004;89:2989–2997. doi: 10.1210/jc.2003-031157. [DOI] [PubMed] [Google Scholar]
- McCarthy AM, Spisak KO, Brozinick JT, Elmendorf JS. Loss of cortical actin filaments in insulin-resistant skeletal muscle cells impairs GLUT4 vesicle trafficking and glucose transport. Am J Physiol Cell Physiol. 2006;291:C860–868. doi: 10.1152/ajpcell.00107.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei J, Wang CN, O'Brien L, Brindley DN. Cell-permeable ceramides increase basal glucose incorporation into triacylglycerols but decrease the stimulation by insulin in 3T3-L1 adipocytes. Int J Obes Relat Metab Disord. 2003;27:31–39. doi: 10.1038/sj.ijo.0802183. [DOI] [PubMed] [Google Scholar]
- Minokoshi Y, Kahn CR, Kahn BB. Tissue-specific ablation of the GLUT4 glucose transporter or the insulin receptor challenges assumptions about insulin action and glucose homeostasis. J Biol Chem. 2003;278:33609–33612. doi: 10.1074/jbc.R300019200. [DOI] [PubMed] [Google Scholar]
- Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005;115:3587–3593. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler ST, Stoehr JP, Rabaglia ME, Schueler KL, Birnbaum MJ, Attie AD. Normal Akt/PKB with reduced PI3K activation in insulin-resistant mice. Am J Physiol Endocrinol Metab. 2001;281:E1249–1254. doi: 10.1152/ajpendo.2001.281.6.E1249. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Paradis S, Ruvkun G. Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 1998;12:2488–2498. doi: 10.1101/gad.12.16.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz K, Hemi R, LeRoith D, Karasik A, Elhanany E, Kanety H, Zick Y. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem. 1997;272:29911–29918. doi: 10.1074/jbc.272.47.29911. [DOI] [PubMed] [Google Scholar]
- Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med. 2006;119:S10–16. doi: 10.1016/j.amjmed.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- Powell DJ, Turban S, Gray A, Hajduch E, Hundal HS. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J. 2004;382:619–629. doi: 10.1042/BJ20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramm G, Larance M, Guilhaus M, James DE. A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem. 2006;281:29174–29180. doi: 10.1074/jbc.M603274200. [DOI] [PubMed] [Google Scholar]
- Rice KM, Lienhard GE, Garner CW. Regulation of the expression of pp160, a putative insulin receptor signal protein, by insulin, dexamethasone, and 1-methyl-3-isobutylxanthine in 3T3-L1 adipocytes. J Biol Chem. 1992;267:10163–10167. [PubMed] [Google Scholar]
- Rice KM, Turnbow MA, Garner CW. Insulin stimulates the degradation of IRS-1 in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 1993;190:961–967. doi: 10.1006/bbrc.1993.1143. [DOI] [PubMed] [Google Scholar]
- Saad MJA, Folli F, Kahn JA, Kahn CR. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J Clin Invest. 1993;92:2065–2072. doi: 10.1172/JCI116803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Jones CC, Huang S, Czech MP, Pilch PF. The interaction of Akt with APPL1 is required for insulin-stimulated Glut4 translocation. J Biol Chem. 2007;282:32280–32287. doi: 10.1074/jbc.M704150200. [DOI] [PubMed] [Google Scholar]
- Sakoda H, Ogihara T, Anai M, Funaki M, Inukai K, Katagiri H, Fukushima Y, Onishi Y, Ono H, Fujishiro M, et al. Dexamethasone-induced insulin resistance in 3T3-L1 adipocytes is due to inhibition of glucose transport rather than insulin signal transduction. Diabetes. 2000;49:1700–1708. doi: 10.2337/diabetes.49.10.1700. [DOI] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- Shirakami A, Toyonaga T, Tsuruzoe K, Shirotani T, Matsumoto K, Yoshizato K, Kawashima J, Hirashima Y, Miyamura N, Kahn CR, Araki E. Heterozygous knockout of the IRS-1 gene in mice enhances obesity-linked insulin resistance: a possible model for the development of type 2 diabetes. J Endocrinol. 2002;174:309–319. doi: 10.1677/joe.0.1740309. [DOI] [PubMed] [Google Scholar]
- Sinha S, Perdomo G, Brown NF, O'Doherty RM. Fatty acid-induced insulin resistance in L6 myotubes is prevented by inhibition of activation and nuclear localization of nuclear factor kappa B. J Biol Chem. 2004;279:41294–41301. doi: 10.1074/jbc.M406514200. [DOI] [PubMed] [Google Scholar]
- Stephens JM, Lee J, Pilch PF. Tumor necosis factor-α-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. Journal of Biological Chemistry. 1997;272:971–976. doi: 10.1074/jbc.272.2.971. [DOI] [PubMed] [Google Scholar]
- Stephens JM, Pekala PH. Transcriptional repression of the C/EBP-alpha and GLUT4 genes in 3T3-L1 adipocytes by tumor necrosis factor-alpha. Regulations is coordinate and independent of protein synthesis. J Biol Chem. 1992;267:13580–13584. [PubMed] [Google Scholar]
- Strawbridge AB, Elmendorf JS. Phosphatidylinositol 4,5-bisphosphate reverses endothelin-1-induced insulin resistance via an actin-dependent mechanism. Diabetes. 2005;54:1698–1705. doi: 10.2337/diabetes.54.6.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XJ, Goldberg JL, Qiao LY, Mitchell JJ. Insulin-induced insulin receptor substrate-1 degradation is mediated by the proteasome degradation pathway. Diabetes. 1999;48:1359–1364. doi: 10.2337/diabetes.48.7.1359. [DOI] [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol. 2007;9:905–914. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- Turnbow MA, Keller SR, Rice KM, Garner CW. Dexamethasone down-regulation of insulin receptor substrate-1 in 3T3-L1 adipocytes. J Biol Chem. 1994;269:2516–2520. [PubMed] [Google Scholar]
- Turner N, Bruce CR, Beale SM, Hoehn KL, So T, Rolph MS, Cooney GJ. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes. 2007;56:2085–2092. doi: 10.2337/db07-0093. [DOI] [PubMed] [Google Scholar]
- Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol. 2004;24:5434–5446. doi: 10.1128/MCB.24.12.5434-5446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LP, Summers SA. Measuring insulin-stimulated phosphatidyl-inositol 3-kinase activity. Methods Mol Med. 2003;83:127–136. doi: 10.1385/1-59259-377-1:127. [DOI] [PubMed] [Google Scholar]
- White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- Whitehead JP, Molero JC, Clark S, Martin S, Meneilly G, James DE. The role of Ca2+ in insulin-stimulated glucose transport in 3T3-L1 cells. J Biol Chem. 2001;276:27816–27824. doi: 10.1074/jbc.M011590200. [DOI] [PubMed] [Google Scholar]
- Whiteman EL, Chen JJ, Birnbaum MJ. Platelet-derived growth factor (PDGF) stimulates glucose transport in 3T3-L1 adipocytes overexpressing PDGF receptor by a pathway independent of insulin receptor substrates. Endocrinology. 2003;144:3811–3820. doi: 10.1210/en.2003-0480. [DOI] [PubMed] [Google Scholar]
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- Yuasa T, Kakuhata R, Kishi K, Obata T, Shinohara Y, Bando Y, Izumi K, Kajiura F, Matsumoto M, Ebina Y. Platelet-derived growth factor stimulates glucose transport in skeletal muscles of transgenic mice specifically expressing platelet-derived growth factor receptor in the muscle, but it does not affect blood glucose levels. Diabetes. 2004;53:2776–2786. doi: 10.2337/diabetes.53.11.2776. [DOI] [PubMed] [Google Scholar]
- Zick Y. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci STKE. 2005;2005:4. doi: 10.1126/stke.2682005pe4. [DOI] [PubMed] [Google Scholar]
- Zierath JR, Houseknecht KL, Gnudi L, Kahn BB. High-fat feeding impairs insulin-stimulated GLUT4 recruitment via an early insulin-signaling defect. Diabetes. 1997;46:215–223. doi: 10.2337/diab.46.2.215. [DOI] [PubMed] [Google Scholar]