

Abstract
Mutations in human α-skeletal actin have been implicated in causing congenital nemaline myopathy, a disease characterized histopathologically by nemaline bodies in skeletal muscle and manifested in the patient as skeletal muscle weakness. Here we investigate the functional effects of three severe nemaline myopathy mutations (V43F, A138P, and R183G) in human α-skeletal actin. Wild-type and mutant actins were expressed and purified from the baculovirus/insect cell expression system. The mutations are located in different subdomains of actin; Val-43 is located in a flexible loop of subdomain 2, Ala-138 is near a hydrophobic cleft in the “hinge” region between subdomains 1 and 3, and Arg-183 is near the nucleotide-binding site. None of the three mutations affected the folding of the actin monomer, the velocity at which skeletal myosin moves actin in an in vitro motility assay, or the relative average isometric force supported by F-actin. Defects in fundamental actomyosin interactions are, therefore, unlikely to account for the muscle weakness observed in affected patients. There were, however, significant changes observed in the polymerization kinetics of V43F and A138P and in the rate of nucleotide release for V43F. No detectable defect was found for R183G. If these subtle changes in polymerization observed in vitro are amplified in the context of the sarcomere, it could in principle be one of the primary insults that triggers the development of nemaline myopathy.
Actin is an ubiquitous and highly conserved protein in muscle and non-muscle cells. There are greater than 90 mutations, including three recently discovered null mutations (1) in the human α-skeletal actin gene (ACTA1) that have been implicated in a number of skeletal muscle diseases (for review, see Refs. 2 and 3). These congenital myopathies are distinguished by the histopathological characteristics of muscle biopsies from affected patients. The presence of rod-like structures or “nemaline bodies” that appear to originate from the Z-discs in the sarcoplasm and contain deposits of actin and Z-line proteins (4–6) is the defining feature in the diagnosis of nemaline myopathy (NM).2 How or why these nemaline rods form is unknown. Their presence in NM patients with homozygous ACTA1 null mutations, which lack human α-skeletal actin but retain reduced levels of the α-cardiac fetal isoform, and under non-NM myopathic conditions (7, 8) suggests that the development of these rods may occur in any muscle tissue where an imbalance of sarcomeric proteins exists (1). Clinically, patients with NM present with non-progressive or slowly progressive skeletal muscle weakness resulting largely from dominant de novo point mutations. This means that the afflicted patients have a combination of a mutant actin with a single amino acid change and the wild-type actin in their muscles. Elucidation of the molecular mechanism of NM has been complicated by the genetic heterogeneity (mutations in five different thin filament genes lead to the same disease) and phenotypic heterogeneity (mutations in the same gene lead to a wide range of clinical severities) that have been observed in patients (9, 10).
Several approaches have been used to study the effects of actin NM mutations, including in vitro transcription/translation systems, C2C12 and NIH3T3 cell transfections, and analyses of actin purified from human muscle biopsies. A wide range of effects were observed, including but not limited to polymerization defects, folding defects, or actin-binding protein interaction defects in vitro and aggregation in vivo (11–15). Until recently, an in-depth study of actin mutations at the molecular level has been limited to yeast actin since a suitable in vitro expression system that yielded milligram quantities of human actin was unavailable. Now that the baculovirus/Sf9 cell system for expressing and purifying milligram amounts of pure human α-skeletal actin has been developed (16–19), analysis of human disease mutations are best studied in the appropriate context. This is the approach we have taken to investigate at the molecular level the primary defects of several actin NM mutations.
Three ACTA1 point mutations, V43F, A138P, and R183G (Fig. 1) were chosen for individual analysis. Any one of these mutations causes severe NM in patients, a phenotypic classification that indicates a high probability of death within the first year of life. Val-43 is located in a flexible loop of subdomain 2 that binds to DNase I (20), Ala-138 is located near a hydrophobic cleft in the hinge region between subdomains 1 and 3, and Arg-183, near the active site, adopts different conformations in the presence of ATP or ADP (21). We surveyed the effects of these three point mutations on several functional properties of actin and on the interaction of myosin with actin. None of the three mutations affected the folding of the actin monomer, the velocity at which skeletal myosin moves actin in an in vitro motility assay, or the relative average isometric force supported by F-actin. However, changes in the rate of nucleotide release, the polymerization kinetics, and the qualitative behavior of the actin mutants during the motility assays were observed. If these subtle changes observed in vitro are amplified in the context of the sarcomere, they could in principle trigger the development of NM. Additional work in a cellular context with a more extensive battery of mutants will be necessary to understand if there is a common molecular mechanism by which these mutations lead to a clinical manifestation of nemaline myopathy.
FIGURE 1.

Location of the ACTA1 mutations in the actin monomer. The four subdomains of actin are labeled 1–4, with subdomain 1 containing both the N and C termini. Mutated residues (Val-43, Ala-138, and Arg-183) are depicted by a space-filling representation in blue. The nucleotide, in the interior of the monomer, is shown in red. This figure was rendered with Swiss-PDBViewer 3.7 using PDB file 1J6Z.
EXPERIMENTAL PROCEDURES
Plasmid Construction—The coding sequence for wild-type α-skeletal actin (WT) was created by site-directed mutagenesis of the Drosophila 5C actin gene (gift from Loy Volkman). The untagged WT-actin was cloned into the baculovirus transfer vector pAcUW51 (BD Biosciences) behind the p10 promoter. N- or C-terminal FLAG-tagged versions of the WT actin were cloned into the baculovirus transfer vector pAcSG2 (BD Biosciences) behind the polyhedron promoter. The C-FLAG actin contained a thrombin cleavage site (amino acid sequence LVPRGS) between the protein and the FLAG tag. The FLAG tag at the N terminus was followed by a linker sequence (AMGAL) and either a thrombin or a tobacco etch virus (TEV) protease cleavage site (amino acid sequence ENLYFQD). For both N-FLAG actin constructs, a Kozak sequence (GCCGCCACC), which is thought to enhance expression (22), was added just before the start site. Actin mutants (V43F, A138P, and R183G) were created by site-directed mutagenesis of the untagged WT template. All constructs were sequenced to verify their correctness.
Expression and Purification of Recombinant Actins—Recombinant baculovirus were prepared according to established protocols (23). Untagged actins were expressed and purified using previously described methods (17) with a few minor changes. Notably, the elution gradient from the Q-Sepharose (Amersham Biosciences) column (50-ml bed volume, 1.5 × 48 cm) was 0.15–0.45 m NaCl. Also, the final F-actin was depolymerized by dialysis against G-buffer (5 mm Tris-HCl, pH 8, 0.2 mm CaCl2, 0.2 mm Na2ATP, 0.5 mm DTT, 1 μg/ml leupeptin) for 3 days and then clarified at 350,000 × g for 40 min.
Tagged actins were harvested from infected Sf9 cells after 72 h and lysed in 1 m Tris-HCl, pH 7.5, 0.6 m KCl, 0.5 mm MgCl2, 4% Triton X-100, 1 mg/ml Tween 20, 0.5 mm Na2ATP, 1 mm DTT, 0.5 mm 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride, 5 mm benzamidine, and 5 μg/ml leupeptin for 1.5 h (50 ml/1 billion cells). After clarification at 178,000 × g for 1 h, the supernatant was dialyzed overnight into 10 mm imidazole, pH 7, 0.3 m NaCl, 0.2 mm MgCl2, 0.2 mm Na2ATP, 0.2 mm DTT, and 1 μg/ml leupeptin. The dialysate was clarified at 25,000 × g for 30 min and then incubated with FLAG resin (Sigma) for 1.5 h. The resin was washed with 10 mm imidazole, pH 7, 0.3 m NaCl, 0.2 mm MgCl2, 0.1 mm NaN3, and 0.2 mm Na2ATP until the A280 was negligible. Bound protein was eluted from the resin using 100 μg/ml FLAG peptide (Sigma) in 10 mm imidazole, pH 7, 0.3 m NaCl, 0.2 mm MgCl2, 0.1 mm NaN3, and 0.2 mm Na2ATP. Fractions containing measurable amounts of protein were pooled and dialyzed overnight into G-buffer. The protein was concentrated with an Amicon Ultra centrifugal filter device (Millipore Corp.). The typical yield of tagged-actin was ∼2–2.5 mg/3 billion cells.
Actin concentration was determined from the absorbance at 290 nm using an extinction coefficient of 0.63 ml mg–1 cm–1. Actin either was used within 5 days or was flash-frozen in liquid nitrogen in the G-actin state after the addition of 2 mg of sucrose per 1 mg of actin.
Chicken Skeletal Protein Purification—Chicken skeletal actin was used as a control in all experimental assays and was purified from chicken pectoralis acetone powder according to methods previously described (24). Chicken skeletal myosin for the in vitro motility and average force assays was purified using established methods (25) and then stored at –20 °C in 50% glycerol.
Preparation of Unlabeled and Labeled Actin for the Polymerization Assay—Recombinant human skeletal and chicken skeletal G-actins were polymerized by the addition of 2 mm MgCl2 and 0.1 m KCl for 1 h at room temperature. The F-actin was then depolymerized for 3 days with three changes of G buffer and clarified by centrifugation for 30 min at 350,000 × g. The supernatant was applied to a Superdex 200 gel filtration column (10/300 GL, GE Healthcare) to isolate pure monomeric actin, free of oligomers. The actin was stored on ice and used within 1 week.
Oregon Green (OG)-labeled actin was prepared by modification of previously described methods (26). Chicken skeletal F-actin was diluted to 1 mg/ml in 10 mm imidazole, pH 8, 10 mm NaCl, 1 mm MgCl2, 1 mm MgATP, 1 mm NaN3. The sulfhydryls were reduced by the addition of 2 mm DTT for 30 min at room temperature. The actin was then dialyzed overnight with 2 changes of buffer (10 mm imidazole, pH 7.6, 100 mm KCl, 1 mm MgCl2, 1 mm Na2ATP, 1 mm NaN3) to remove the DTT. Using a fresh 10 mm solution of OG 488 iodoacetamide (Molecular Probes) in N,N-dimethylformamide, a 10–15-fold molar excess of label was added dropwise to the actin with stirring and then gently stirred overnight at 4 °C. OG-labeled actin filaments were pelleted at 350,000 × g for 1.5 h and then depolymerized for 3 days against two changes of G buffer followed by dialysis into G6.5 buffer (5 mm PIPES-Tris, pH 6.5, 0.2 mm CaCl2, 0.2 mm Na2ATP, 0.5 mm DTT, 1 mm NaN3). The actin was clarified at 350,000 × g for 30 min and applied to a 1 × 5-cm column of DEAE-cellulose DE53 (Whatman) equilibrated in G6.5 buffer. OG and unlabeled actins were separated using a 100–350 mm KCl gradient in G6.5 buffer. The absorbance at 491 nm was used to determine the fractions containing labeled actin, which were pooled and dialyzed overnight against 3 changes of G buffer. The labeled actin was concentrated ∼20-fold using an Amicon Ultra centrifugal filter device (Millipore) and dialyzed overnight against 2 changes of G buffer. The labeled actin was clarified a final time at 350,000 × g for 30 min. The concentration was determined by measuring the absorbance at 290 and 491 nm using E491 = 77,800 m–1 cm–1 as the extinction coefficient for OG and a correction for OG absorbance at 290 nm (corrected A290 = A290 – 0.16991 × A491). The stoichiometry of labeling was 0.9 mol/mol of actin. Labeled actin was stored on ice at 4 °C and used within 4 weeks to visualize actin polymerization.
TEV Protease—The TEV protease (autoinactivation-resistant mutant S219V; see the website of Dr. D. Waugh at the National Cancer Institute, Frederick for information) was isolated from an Escherichia coli strain (BL21(DE3)-RIL/pRK793) using previously described methods (27) with minor modifications. Induced bacterial cells were resuspended in lysis buffer (50 mm potassium phosphate, pH 8, 100 mm NaCl, 5 mm imidazole, 10% glycerol) and sonicated for 10 min in pulse mode (0.5 s “on” and 0.5 s “off”). After clarification at 15,000 × g for 30 min, the supernatant was removed and applied to a prepared HIS6 column (HIS-Select, Sigma) equilibrated in lysis buffer. Proteins binding nonspecifically were removed by washing with 30 mm imidazole lysis buffer. The TEV protease was then eluted in lysis buffer using a linear gradient to 200 mm imidazole. Fractions containing the TEV protease were pooled, and EDTA and DTT were added to final concentrations of 1 mm. The protein was concentrated with an Amicon Ultra centrifugal filter device (Millipore Corp.) and then dialyzed overnight into 25 mm potassium phosphate, pH 8, 200 mm NaCl, 10% glycerol, 2 mm EDTA, and 10 mm DTT. After centrifugation at 350,000 × g for 15 min the protease was separated into aliquots, flash-frozen in liquid nitrogen, and stored at –80 °C.
Enzymatic Reactions—Digests to remove the FLAG tag from actin (100 μg) with thrombin (Haemtech, Essex Junction, VT, 0.1 units) were performed in 5 mm Tris, pH 8, 2 mm MgCl2, 1 mm EGTA, 30 mm NaCl at room temperature for 3 h. Reactions were stopped with 0.1 mg/ml leupeptin (Sigma) and 50 mm N-α-tosyl-l-lysine chloromethyl ketone hydrochloride (Sigma). TEV protease digests, to remove the N-terminal FLAG tag from actin (1:10 enzyme:substrate ratio), were performed at room temperature for 3 h in 5 mm Tris, pH 8, 0.2 mm CaCl2, 0.2 mm DTT, 0.2 mm ATP buffer. The trypsin (Sigma) digest (1:25 enzyme:substrate ratio) of Mg-G-actin (C-FLAG) was performed at room temperature for 1 h in 5 mm Tris, pH 8, 1 mm MgCl2, 0.2 mm EGTA, 0.2 mm DTT, 0.2 mm ATP buffer. The reaction was stopped with 3 mm N-α-tosyl-l-lysine chloromethyl ketone hydrochloride.
Thermal Denaturation—Unfolding of actin was assayed using a DNase-I inhibition assay according to the methods described in Blikstad et al. (28) and Schuler et al. (29) modified by (17). A plot of the percent of DNase I inhibition as a function of temperature (25–66 °C) was fitted with the curve (y = 0 + 100/(1 + exp(–(x – x0)/b))) to determine the melting temperature (Tm; x0 in the equation), defined as the 50% point of DNase-I inhibition.
Rate of Nucleotide Release—The rate of ATP dissociation from G-actin was measured by following the fluorescence decrease of etheno-ATP (ε-ATP; Molecular Probes) using methods previously described (21) with the following modifications. Unbound ATP was removed from 10 μm actin by the addition of 10% by volume of 50% Dowex AG-1 × 8 resin (Bio-Rad) equilibrated in assay buffer (5 mm HEPES, pH 7.5, 50 μm CaCl2, 1 mm DTT). The slurry was mixed for 1 min, and the resin was removed by centrifugation. This step was repeated twice more followed by high speed centrifugation (350,000 × g) for 20 min to remove remaining resin. After incubation of the actin with 100 μm ε-ATP for a minimum of 2 h on ice, the excess ε-ATP was removed by treatment with Dowex resin 3 times as before. A final high speed centrifugation (350,000 × g) step removed the remaining resin. G-actin was diluted to 2 μm with assay buffer just before measurement of nucleotide release at 25 °C. The decrease in fluorescence of ε-ATP as a function of time after the addition of 100 μm non-fluorescent ATP was fitted with a single exponential decay curve. t tests were used to determine significance (p < 0.05).
In Vitro Motility and Relative Isometric Force Assays—Chicken skeletal myosin was purified by hydrophobic interaction chromatography (30) immediately preceding the assay to remove myosin that was functionally compromised. Actin filament velocity was then measured using methods described previously (31). Briefly, skeletal myosin was added to nitrocellulose-coated flow cells at 100 μg/ml. The surface was then blocked by 0.5 mg/ml bovine serum albumin. Next, an unlabeled actin/ATP wash was used (1 μm vortexed actin followed by the addition of 1 mm ATP) to “block” myosin molecules unable to dissociate from actin in the presence of ATP. The velocity of rhodamine-phalloidin-labeled filaments at 30 °C was visualized upon the addition of ATP in motility buffer (25 mm imidazole, pH 7.5, 25 mm KCl, 1 mm EGTA, 4 mm MgCl2, 0.5% methylcellulose, 1 mm MgATP, 10 mm DTT, 2.9 mg/ml glucose, 0.125 mg/ml glucose oxidase (Sigma), and 0.023 mg/ml catalase (Sigma)). To determine the velocity for each actin species, a semi-automated filament tracking program (a gift from D. A. Winkelmann, Robert Wood Johnson Medical School, Piscataway, NJ) was used to determine a weighted probability of the actin filament velocities of at least 500 filaments as described previously (32, 33). A gaussian distribution fitted to a plot of probability versus velocity generated a mean ± S.D. t tests were used to determine significance (p < 0.05).
Isometric force in the motility assay was measured using previously described methods (17, 30). Varying concentrations of α-actinin, an actin-binding protein, were added to the motility assay to create an internal load on the actin filament until movement was stopped. T-tests were used to determine significance (p < 0.05).
Total Internal Reflection Fluorescence (TIRF) Assay for Actin
Polymerization—The kinetics of polymerization at the barbed end of
actin were measured using direct observation of polymerization with TIRF
microscopy, based on modifications to the methods developed by Kuhn and
Pollard (26). A 20-μl flow
cell chamber was created by using two different sized coverslips (bottom, 24
× 60 mm Corning no. 1; top, 22-mm square Corning no. 1) separated by
0.125 μm Mylar shim strips placed along the long axis of the bottom
coverslip. Just before assembly, the shims were dipped in an adhesive (Norland
no. 61, Cranbury, NJ) that can be cured under UV light to seal the flow cell
on two sides. The actin for each experiment (stored as a 10–15
μm Ca-ATP-actin stock) was prepared for polymerization by adding
a 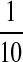 part of 10×
exchange buffer (10 mm EDTA, 1 mm MgCl2) and
then diluting with G buffer to 2× the final actin concentration
(2–6 μm) followed by incubation on ice for 5 min. The flow
cell was prepared at room temperature by addition of (a)20 μl of
0.2 μm N-ethylmaleamide-inactivated myosin for 1 min,
(b)20 μl of 1% bovine serum albumin (BSA) in high salt buffer (50
mm Tris-HCl, pH 7.6, 600 mm NaCl), and (c) 20
μl of 1% BSA in low salt buffer (50 mm Tris-HCl, pH 7.6, 50
mm NaCl). Excess solution was removed from the flow cell with a
Q-tip. To visualize polymerization of 1–3 μm actin at 30
°C, 15 μl of 2× actin (containing 30% OG labeled actin) was
quickly mixed with 15 μl of 2× TIRF buffer (20 mm
imidazole, pH 7.0, 100 mm KCl, 2 mm MgCl2, 2
mm EGTA, 200 mm DTT, 0.4 mm ATP, 30
mm glucose, 1% methylcellulose, 40 μg/ml catalase, and 200
μg/ml glucose oxidase), and 20 μl was flowed through the flow cell
chamber using a Q-tip or filter paper. If the time course for data collection
was longer than 5 min, the ends of the flow cell were sealed with vacuum
grease to prevent evaporation.
part of 10×
exchange buffer (10 mm EDTA, 1 mm MgCl2) and
then diluting with G buffer to 2× the final actin concentration
(2–6 μm) followed by incubation on ice for 5 min. The flow
cell was prepared at room temperature by addition of (a)20 μl of
0.2 μm N-ethylmaleamide-inactivated myosin for 1 min,
(b)20 μl of 1% bovine serum albumin (BSA) in high salt buffer (50
mm Tris-HCl, pH 7.6, 600 mm NaCl), and (c) 20
μl of 1% BSA in low salt buffer (50 mm Tris-HCl, pH 7.6, 50
mm NaCl). Excess solution was removed from the flow cell with a
Q-tip. To visualize polymerization of 1–3 μm actin at 30
°C, 15 μl of 2× actin (containing 30% OG labeled actin) was
quickly mixed with 15 μl of 2× TIRF buffer (20 mm
imidazole, pH 7.0, 100 mm KCl, 2 mm MgCl2, 2
mm EGTA, 200 mm DTT, 0.4 mm ATP, 30
mm glucose, 1% methylcellulose, 40 μg/ml catalase, and 200
μg/ml glucose oxidase), and 20 μl was flowed through the flow cell
chamber using a Q-tip or filter paper. If the time course for data collection
was longer than 5 min, the ends of the flow cell were sealed with vacuum
grease to prevent evaporation.
A Nikon TE2000-U microscope equipped with a PlanApo objective lens (×100, numerical aperture 1.45) for through-the-objective TIRF was used for all experiments. Actin filaments were visualized in TIRF mode through a ChromaHQ560/80 filter after excitation with an argon laser (488 nm, Spectra-Physics model 133). To decrease the amount of photobleaching, a neutral density filter was used to lower the laser intensity. Filaments were detected at 200× magnification using an image-intensified digital charge-coupled device camera (DVC Intensicam IV S, Austin, TX). Image-capturing software (QED Imaging, Pittsburgh, PA) allowed the recording of digital images (an average of 2–6 frames) every 2–10 s for 1.5–20 min depending upon the actin concentration. Barbed end growth rates (subunits/s) of 10 individual actin filaments at each actin concentration were determined manually from each TIFF stack using Image J (National Institutes of Health, rsb.info.nih.gov/ij) assuming a monomer length of 5.4 nm. The average growth rate at a minimum of four actin concentrations was plotted against actin concentration and fitted with a linear regression to determine the barbed end association rate (slope), dissociation rate (y intercept), and critical concentration (x intercept) for each experiment.
RESULTS
Expression of Human α-Skeletal Actins—Our original strategy was to engineer a FLAG tag at the N or C terminus of actin to facilitate purification of expressed human α-skeletal actin by affinity chromatography (Fig. 2A). Because the location and the type of tag (FLAG, His6 or Myc) have been shown to perturb actin function (34–36), a thrombin cleavage site was engineered to allow for tag removal. Typical yields before tag cleavage were ∼0.75 mg of actin/billion Sf9 cells. Surprisingly, thrombin was incapable of removing the FLAG tag at either the N or C terminus (Fig. 2C), suggesting that the FLAG tag or the structure of the tagged actin interfered with thrombin recognizing its cleavage site. The C-terminal FLAG tag could, however, be removed by digestion of Mg-G-actin with trypsin, which cleaves after Arg-372 and Lys-373 (37). The resulting actin was not a viable alternative for our structure/function studies because removal of the C-terminal residues has significant effects on actin function (37, 38).
FIGURE 2.

Purification of wild-type human α-skeletal actin. A, total expression of N-terminal FLAG tagged actin in Sf9 cells (asterisk, lane 2). The presence of the tag decreased the solubility of the actin, so that little remained in the supernatant following a high speed spin (lane 3). Most proteins did not bind to the FLAG affinity column (lane 4) thereby yielding purified N-FLAG tagged actin (lane 5). Purification of C-FLAG-tagged actin is similar and not shown. Lane 1, molecular weight markers. B, untagged actin is more soluble in Sf9 cells than FLAG-tagged actin (lane 2, total expression; lane 3, soluble fraction). Use of chromatographic ion exchange columns (lane 4) (see “Experimental Procedures”) followed by a polymerization and depolymerization step yielded relatively pure expressed human α-skeletal actin (lane 5). Lane 1, molecular weight markers. C, thrombin did not cleave at its recognition site when it was located between the FLAG tag (N or C terminus) and actin. Lane 1, N-FLAG-tagged actin was resolved from untagged actin by SDS-PAGE. Lane 2, the FLAG tag was not removed after a 3-h incubation with thrombin. Control experiments (not shown) confirmed that the thrombin was active. D, TEV protease cleaved at its recognition site located between the N-terminal FLAG tag and actin (lane 2, 10 min digestion; lane 3, 60 min digestion). Lane 1, undigested N-terminal FLAG tagged actin. Samples were run on 12% SDS-polyacrylamide gels.
We next engineered a TEV protease cleavage site between the N-terminal FLAG tag and Asp-1 of actin such that no additional amino acids would remain after proteolytic digestion. Removal of the tag with TEV protease was successful (Fig. 2D). WT actin produced by this method polymerized normally and could be moved by skeletal myosin in the motility assay at velocities similar to tissue-purified skeletal actin (tissue-purified, 5.4 ± 1.1 μm/s; expressed WT, 5.3 ± 0.8 μm/s). However, yields of WT actin after removal of the N-terminal FLAG tag were too low (∼0.5 mg/billion Sf9 cells) for many of the experimental assays that we wished to perform.
Untagged WT and three mutant human α-skeletal actins (V43F, A138P, and R183G) were, therefore, purified from infected Sf9 cells to near homogeneity (Fig. 2B) by techniques that we previously used with cardiac and cytoplasmic actins (17, 18). Protein yields were comparable with human α-cardiac actin (∼3 mg/billion Sf9 cells) (17). All experimental assays were performed on actin from two independent preparations unless otherwise indicated.
Thermal Unfolding of G-actin—The effect of each mutation on the stability of the G-actin monomer was assessed by thermal unfolding. The endonuclease activity of DNase-I is inhibited when bound to native, folded actin. As the temperature increases and actin denatures, DNase-I cannot bind and becomes active. A plot of the percentage of DNase-I inhibition as a function of temperature is fitted to determine the temperature at which there is 50% inhibition of DNase-I activity, defined as the melting temperature (Tm). The DNase inhibition curves for all four isoforms were very similar (Fig. 3 and Table 1), showing that the mutations had no affect on the stability of the actin monomer. As was seen previously for the cardiac actins (17), the melting temperatures for WT skeletal actin remained stable for 5 days before starting to decrease (data not shown). All experiments were, therefore, performed within 5 days after purification or after thawing frozen aliquots. The thermal denaturation assay depends upon the proper binding of actin and DNase-I, of which Val-43 is directly involved (20). However, the melting temperature of V43F actin was similar to WT actin, and thus, mutation of this residue has no significant effect on DNase I binding in this assay.
FIGURE 3.

Thermal unfolding of G-actin. The melting temperature (Tm) of the WT and mutant actins was determined after 1–2 days of storage on ice. The endonuclease activity of DNase was inhibited when bound to native actin. The Tm is defined as the temperature at which DNase I activity is inhibited by 50%. All three actin mutants (V43F, open squares; R183G, open triangles; A138P, open circles) were similar to WT (filled circles) and gave melting temperatures of ∼60 °C (Table 1). Experimental conditions: 1.2 μm actin, 40 μg/ml DNA, 100 mm Tris, pH 7.6, 4 mm MgCl2, 1.8 mm CaCl2, 2 mm NaN3, and 0.6 μg/ml DNase-I at 25 °C.
TABLE 1.
Summary of G-actin properties
Measurements were made in duplicate or triplicate using a minimum of two independent actin preparations unless otherwise indicated. The data are presented as mean ± S.E. Tm, the temperature at which DNase I activity is 50% inhibited.
| Assay | WT actin | V43F actin | R183G actin | A138P actin |
|---|---|---|---|---|
| Tm (°C) | 60.0 ± 0.7 | 60.2a | 60.6a | 60.8 ± 0.6 |
| Exchange rate (s-1) | 0.029 ± 0.001 | 0.022 ± 0.002b | 0.030 ± 0.001 | 0.030 ± 0.002 |
Only a single measurement was performed.
Significant at p < 0.05 compared to WT actin.
Nucleotide Release Rates—Mutation or modification of actin distant from the nucleotide pocket can perturb nucleotide binding in the actin monomer (39, 40). To assess possible changes at the active site, the rate of release of the fluorescent nucleotide ε-ATP from the active site was determined. Of the three actin mutants, only the V43F mutation slowed the nucleotide release rate (Fig. 4 and Table 1). This 24% decrease as compared with WT actin suggests that a point mutation in subdomain 2 propagates a structural change to the active site that slows the rate of nucleotide release.
FIGURE 4.

Rate of dissociation of ε-ATP from the active site of G-actin. The rate of release of ε-ATP was measured from the decrease in fluorescence as a function of time after the addition of non-fluorescent ATP (100 μm) to 2 μm G-actin. The dissociation rate from V43F (open squares) actin decreased 24% relative to WT (Table 1). A138P (open circles) and R183G (open triangles) actin were similar to WT (filled circles). Experimental conditions: 5 mm Hepes, pH 7.5, 50 μm CaCl2, and 1 mm DTT at 25 °C.
Actin Polymerization—The effect of each point mutation on polymerization was quantified by following the polymerization of Mg-ATP-actin in real time using TIRF microscopy, a recently developed and powerful assay (26, 41). This approach has significant advantages over following light scattering changes, which depend on the polymer concentration and the size of the filaments, or following changes in pyrene fluorescence, which measure only polymer concentration, leaving the direction of growth and number of filaments to be inferred.
Here, a series of images were collected at defined intervals after the addition of Mg2+ and KCl (Fig. 5A). Using ImageJ software, the increase in length at the barbed end of 10 filaments at each actin concentration was followed as a function of time. A plot of growth rate versus actin concentration, fitted with a linear regression, yielded the barbed end association rate (slope of the line), the dissociation rate (y intercept), and the critical concentration (x intercept, Fig. 5B). Two of the mutants showed significant differences (Table 2). Both the V43F and A138P mutations showed a ∼25% higher overall association rate at the barbed end relative to WT skeletal actin. In addition, the A138P actin showed a 2–2.5-fold higher critical concentration and rate of dissociation compared with WT actin. The R183G mutation did not affect the measured kinetic parameters.
FIGURE 5.

Rate of actin polymerization by TIRF microscopy. A, growth of a single WT actin filament at 1 μm actin using 30% Oregon Green-labeled chicken skeletal actin to visualize the filaments. Four minutes has elapsed between the first and last image, and the scale bar represents 1 μm. B, the rate of filament growth was measured at a minimum of four actin concentrations and plotted and fitted with a linear regression. A representative experiment is shown for each actin, with the error bars representing the deviation in growth rate at each actin concentration for a single experiment. See Table 2 for the derived kinetic parameters. Polymerization rates were measured in 10 mm imidazole, pH 7.0, 50 mm KCl, 1 mm MgCl2, 1 mm EGTA, 100 mm DTT, 0.2 mm ATP, 0.5% methylcellulose, 15 mm glucose, 20 μg/ml catalase, and 100 μg/ml glucose oxidase at 30 °C.
TABLE 2.
Summary of barbed end polymerization kinetics
Polymerization kinetic parameters were measured in duplicate using a minimum of two independent actin preparations. All parameters were calculated from a linear regression fitted to the data (the slope yielded the association rate, the y intercept yielded the dissociation rate, and the x intercept yielded the critical concentration). The data are presented as the mean ± S.E.
| Kinetic parameter | WT actin | V43F actin | R183G actin | A138P actin |
|---|---|---|---|---|
| Association rate (μm s-1) | 6.70 ± 0.68 | 8.56 ± 0.62a | 6.49 ± 0.64 | 8.42 ± 0.23a |
| Dissociation rate (s-1) | 0.83 ± 0.29 | 1.42 ± 0.57 | 0.71 ± 0.38 | 2.09 ± 0.40b |
| Critical concentration (μm) | 0.11 ± 0.03 | 0.16 ± 0.06 | 0.12 ± 0.06 | 0.25 ± 0.04b |
Significant at p < 0.10 compared to WT actin.
Significant at p < 0.05 compared to WT actin.
The measurement of pointed end kinetic parameters was not pursued because the slow rate of growth at this end results in large deviations in the growth rates, as also seen by Kuhn and Pollard (26). Differences would have to be extremely large to be statistically significant.
In Vitro Motility and Average Force—The effect of each actin mutation on actomyosin interactions was assessed by measuring unloaded actin filament velocity in the in vitro motility assay. No significant differences in the average velocity were observed for the three actin mutants relative to WT actin (Fig. 6, A–D, and Table 3). Motility of the A138P actin filaments resulted in a broader distribution of probabilities than all other actins, suggesting that the mutation affected the movement of the actin filaments that was not reflected in the mean velocity.
FIGURE 6.

In vitro motility and relative isometric force of WT and mutant skeletal actins. A–D, The plots of probability against speed (greater than 500 filaments) were fitted with a gaussian curve to determine the mean velocity. A representative experiment is shown for each of the actins. Note the peak of the distribution is similar for all three mutants as compared with the WT (see Table 3). E, the concentration of α-actinin that stops movement is defined as the relative isometric force. No significant differences were observed for all three mutants as compared with WT. The bars represent the mean ± S.D. All measurements were made with 100 μg/ml chicken skeletal myosin at 30 °C in motility buffer: 25 mm imidazole, pH 7.5, 25 mm KCl, 1 mm EGTA, 4 mm MgCl2, 0.5% methylcellulose, 1 mm MgATP, 10 mm DTT, 2.9 mg/ml glucose, 0.125 mg/ml glucose oxidase, and 0.023 mg/ml catalase.
TABLE 3.
Summary of in vitro motility and force experiments
Chicken skeletal myosin was purified by hydrophobic interaction chromatography and then used at 100 μg/ml to measure velocity and relative isometric force. To measure the isometric force, α-actinin was added to the in vitro motility assay to create a load, and the concentration of α-actinin required to completely stop movement of the actin filaments was determined. No differences in velocity or force were observed for any of the actin mutants. Measurements were made on a minimum of two independent actin preparations. The data for velocity are presented as the mean ± S.D. for each experiment, and the data for average force is the mean ± S.D.
| Assay | WT actin | V43F actin | R183G actin | A138P actin |
|---|---|---|---|---|
| Velocity (μm/s) | 4.9 ± 0.9 | |||
| 5.3 ± 0.8 | 4.9 ± 0.8 | 5.4 ± 1.0 | 5.0 ± 1.2 | |
| 5.0 ± 0.7 | 4.9 ± 0.7 | 5.1 ± 0.7 | 5.1 ± 0.9 | |
| 4.9 ± 0.6 | ||||
| α-Actinin (μg/ml) | 36 ± 4 | 42 ± 12 | 37 ± 5 | 35 ± 2 |
The binding of increasing concentrations of α-actinin to actin was used to generate an internal load against which myosin must work to move actin in the in vitro motility assay (42). The α-actinin concentration at which movement stops is a measure of the relative average force generating capacity of actomyosin. This assay gives the same results as those obtained with an ultracompliant glass microneedle, a more direct measure of average force (30). No significant differences were observed in the measured isometric force for all three mutant actin filaments relative to WT actin (Fig. 6E and Table 3). Collectively, these results indicate either the mutations have negligible effects on the fundamental actomyosin interactions necessary for generating motion and force or are too small to be detected by these assays.
DISCUSSION
We anticipated finding large changes in the in vitro functional properties of the mutant actins that cause severe nemaline myopathies; this turned out not to be the case. One could argue that large functional perturbations to such a conserved protein as actin, which is critical to skeletal muscle function, would be incompatible with survival. The mutations that cause NM are not clustered in a particular region of actin but are interspersed throughout the molecule. This observation alone implies that it is unlikely that all mutations will affect the same underlying function of actin. Hence, we surveyed the functional effects of three mutations (V43F, A138P, and R183G) located in different subdomains of actin with a variety of biochemical techniques. The three mutant actins did not show any intrinsic folding problems, as assessed from their melting temperature compared with WT actin. To date, only two mutations that lead to severe NM have caused actin to fold improperly when expressed in reticulocyte lysates, thus producing nonfunctional actin (12). The interaction of our mutant actins with skeletal myosin was also unaffected in the motility assays, suggesting that actomyosin interactions are not significantly perturbed by these mutations. Nearly all NM mutations, including V43F, A138P, and R183G, are not directly located in a region known to bind myosin, so any effects would have to occur via propagated changes in actin. Because muscle weakness is a hallmark symptom of NM, a more comprehensive study of the interactions of the mutant actins with myosin under load may still yield additional phenotypes that we were unable to detect.
Significant changes were detected in the polymerization kinetics of two of the mutants studied here (V43F and A138P). The association of monomers at the barbed end was enhanced for V43F, with no change in critical concentration. A138P showed a faster rate of both association and dissociation and an increase in critical concentration. A survey of a larger number of actin mutants expressed in reticulocyte lysates (12) showed that 11 of 19 mutants copolymerized with WT actin filaments to less than 50%, using a simple sedimentation assay to measure net polymer formed. If any common underlying mechanism is suggested from the relatively few studies assessing the functional properties of the mutant actins, it may be in polymerization properties. Polymerization can be affected by altering direct interaction sites or by perturbing nucleotide dynamics, as monomers predominantly add onto the barbed end in the ATP state and dissociate from the pointed end in the ADP state. Many actin point mutations have been shown to affect polymerization, including several residues not directly implicated in actin-actin monomer interactions (12, 14, 43–45).
Functional Effects of the V43F Mutation—Val-43 is part of the DNase binding loop (His-40 to Gly-48) located in subdomain 2 (Fig. 1). Crystallographic structures show that this region can adopt a variety of conformations, ranging from an extended β-hairpin loop when bound to DNase to a disordered loop to an α-helix, consistent with this region being structurally plastic (for review, see Ref. 46). Crystallographic studies have not conclusively shown if this region undergoes a nucleotide-dependent change in conformation (21, 47, 48). Proteolytic digestion studies in solution show an altered rate of cleavage of this region in the presence of ADP versus ATP (49), suggesting that changes at the active site can be propagated outward to subdomain 2. Our data provide evidence for communication in the reverse direction; a mutation in this region causes a 24% decrease in the rate of ATP release from the active site. Modeling of the F-actin structure also implicates this region in forming intermolecular contacts with specific residues of subdomains 1 and 3 of the neighboring monomer along the long pitch of the F-actin helix (Ref. 50; for review, see Ref. 51). The increase in the rate of polymerization at the fast-growing barbed end of V43F actin that we observed is consistent with the involvement of this region in making intermolecular contacts.
Functional Perturbations of A138P Actin—Ala-138 is located near a hydrophobic cleft in the hinge region between subdomains 1 and 3 (Fig. 1), a region important for binding of depolymerizing and capping proteins (for review, see Ref. 51) as well as small molecule toxins (for review, see Ref. 52). This region has been proposed to function as a hinge when the actin cleft between subdomains 2 and 4 opens (ADP-bound) and closes (ATP-bound) to allow nucleotide exchange. The only crystallographic structure of monomeric actin to show an open cleft, however, is an actin-profilin complex (53). Modeling of actin crystal structures into electron microscopic images of yeast F-actin suggests that an open cleft model best fits filaments in the ADP state (54). Despite a prediction that the A138P mutation would affect nucleotide exchange (3), we observed no change in the rate of ATP release.
The A138P mutation increased the association rate, dissociation rate, and critical concentration at the barbed end of A138P actin filaments, suggesting a perturbation in monomer-monomer interactions. Structural models of F-actin (50) do not suggest that this residue is directly involved in binding to a neighboring monomer, but a proline residue in place of an alanine is a significant change and could alter the local structure and thereby affect the strength of nearby interactions. In support of this hypothesis, we observed a broader distribution of filament velocities in the motility assay (Fig. 6D), which might be related to a subtle difference in the filament that causes more variable interactions with myosin. A more obvious example of altered inter-monomer interactions inhibiting motility and force generation, without affecting the kinetic properties of myosin, was previously observed when monomers in F-actin were cross-linked (55).
An A138V mutation in actin from the indirect flight muscle of Drosophila (56) behaves dominantly in the background of the WT protein and results in a reduced flight ability (57). It is not known if the mutant actin interferes with assembly or function of the thin filaments in muscle, but this observation provides additional evidence that mutations at this residue can have significant affects on actin function. It also supports the hypothesis that small perturbations in in vitro actin function may be amplified in the context of the sarcomere, thereby producing the disease phenotype.
R183G Actin Functions Normally—Arg-183 in subdomain 4 is near the nucleotide pocket but does not interact with the nucleotide directly (Fig. 1). This side chain adopts different conformations in the presence of ATP or ADP. The bonds made by the side chain of Arg-183 with Glu-72 and the backbone carbonyl between residues 72 and 73 in the ADP-state are abolished when ATP binds, allowing Arg-183 to preferentially occupy an alternate conformation (21). Based on these data, mutation of Arg-183 might be expected to affect the rate of nucleotide exchange, but we did not observe any effect. In fact, the R183G actin behaved normally in every experimental assay, which indicates either that the perturbations caused by this mutation are subtle and buried within the error of the experiments or that different assays are needed to detect the changes caused by this mutation.
Two earlier studies detected an effect of R183G on polymerization. Using a pelleting assay, Costa et al. (12) showed a decreased extent of co-polymerization of in vitro translated mutant actin with skeletal muscle actin. Also, transfection of C2C12 myoblast cells with a C-terminal enhanced green fluorescent protein construct resulted in abnormal cytoplasmic and intranuclear aggregates, although the R183G patient did not show intranuclear bodies (14). In the same study, a transfected C-terminal FLAG tagged R183G actin construct showed less of an ability to localize to the insoluble actin filament pool, although with time, the more mature myotubes showed increased incorporation (14). Of note in the latter study is the use of a C-terminal FLAG tagged actin, which we have shown perturbs actin function. The amount of overexpression may also have affected the actin monomer population and influenced the amount of actin that incorporated into filaments.
Other mutations at this residue lead to varied affects on actin function. R183C and R183S mutations produce the same clinically severe phenotype in NM patients as seen with R183G actin, but the R183C mutation showed no differences compared with WT when assayed for actin folding, binding to several actin-binding proteins, and co-polymerization with skeletal actin (12). Nothing is known about the effects of the R183S mutation on actin function. An R183A mutation in yeast (58) did not produce any obvious functional perturbations. In a β-cytoplasmic actin backbone, the R183W mutation causes deafness, and abnormalities of the actin cytoskeleton were observed in a cell line derived from an affected patient (59).
Effect of Tags on Actin—Methods for expressing high yields of human actin isoforms in the baculovirus/insect cell system were developed recently (16–18). Although we have successfully purified untagged expressed cytoplasmic and α-cardiac actins using conventional chromatography (17, 18), here we first tried to express FLAG-tagged α-skeletal actin constructs to facilitate purification by affinity chromatography. Unfortunately, the FLAG tag significantly reduced the solubility of actin in the Sf9 cells, resulting in low yields of pure protein. The presence of a tag, especially at the C terminus, has been shown to perturb actin function (34–36), and we also observed a significant decrease in the extent of polymerization of C-FLAG actin (data not shown). C-terminal tags should, therefore, be avoided unless the tag will be removed. Small N-terminal tags are optimal, but if they are too large they also perturb function.
Removal of the tag is necessary for studying mutant actin function in vitro. Surprisingly, thrombin would not cleave at its optimal cleavage site between the FLAG tag and actin, although the TEV protease was successful when its recognition site was cloned between the FLAG tag and actin. Overall, the significant reduction in yield of actin after purification and removal of the FLAG tag was prohibitive for some of our experiments, and we chose to isolate untagged actin for this study. Therefore, the use of a tag for purifying actin should only be exploited when small quantities of actin (<1 mg) are needed.
Implications for the Disease Mechanism of NM—The findings from this study and all other studies to date are unable to provide a clear molecular mechanism for the development of NM. Even more puzzling is the recent result that the complete lack of α-skeletal muscle actin also causes NM (1). The patient compensates by retaining α-cardiac actin, the skeletal muscle fetal actin isoform. This observation suggests that a potential mutation-independent therapeutic intervention for skeletal myopathies is to up-regulate cardiac actin in skeletal muscle (60). This strategy would, however, probably be ineffective with dominant NM actin mutations, where the mutant actin acts as a “poison” protein and interferes with the normal function of WT actin in muscle cells.
Can any generalizations be drawn about possible molecular defects in the mutated actins from existing studies? Most mutant actins do not appear to have folding defects, and those that have severe folding problems are probably degraded and never become incorporated into the actin filament. The three mutants we studied did not alter actomyosin interactions, suggesting that this may not be a common mechanism underlying the muscle weakness observed in patients. This result does not, however, eliminate possible dysfunction when myosin interacts with a regulated thin filament (actin-tropomyosin-troponin) containing the mutant actin. Future experimentation in this direction would reveal whether calcium-dependent thin filament regulation is affected by any of the actin mutations. Changes in polymerization were seen with two of our mutants, and a number of mutants assayed by Costa et al. (12), which may be an avenue to pursue if similar perturbations are observed with additional mutations. There is also the possibility that a given mutation could alter the dynamic interactions between actin and an actin-binding protein, such as CapZ, α-actinin, or tropomodulin, thereby leading to changes in muscle function. If such changes occur, they would probably not provide a common general mechanism for all nemaline mutations, if one does exist.
Because the phenotypes observed in vitro are quite subtle, it is also desirable to complement in vitro studies with more ordered systems because phenotypes may be exacerbated in the context of the sarcomere. Along these lines, Bathe et al. (11) have recently examined the phenotypes of myopathy-causing actin mutants in differentiated C2C12 myotubes. Future progress may also require additional mouse models of nemaline myopathy; one exists with a point mutation in slow α-tropomyosin (61). Data from this model indicate the presence of a repair process and possibly a delayed maturation of nemaline muscles. Fully understanding this disease and ultimately designing therapeutic interventions will undoubtedly require considerable additional in vivo studies and animal models.
Acknowledgments
We thank Alex Hodges, Carol Bookwalter, and Susan Lowey for advice during the course of this work and helpful comments on the manuscript. Guy Kennedy provided excellent technical assistance with microscopy. We also thank the laboratories of David Warshaw and Peter VanBuren for use of their microscopes.
This work was supported, in whole or in part, by National Institutes of Health Grant HL059408 (to K. T.) and National Institutes of Health Institutional Training Grants T32 HL007944 (to D. Warshaw) and T32 HL07647 (to M. LeWinter). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: NM, nemaline myopathy; WT, wild type; TEV, tobacco etch virus; DTT, dithiothreitol; TIRF, total internal reflection fluorescence; Tm, melting temperature; OG, Oregon Green; PIPES, 1,4-piperazinediethanesulfonic acid.
References
- 1.Nowak, K. J., Sewry, C. A., Navarro, C., Squier, W., Reina, C., Ricoy, J. R., Jayawant, S. S., Childs, A. M., Dobbie, J. A., Appleton, R. E., Mountford, R. C., Walker, K. R., Clement, S., Barois, A., Muntoni, F., Romero, N. B., and Laing, N. G. (2007) Ann. Neurol. 61 175–184 [DOI] [PubMed] [Google Scholar]
- 2.Clarkson, E., Costa, C. F., and Machesky, L. M. (2004) J. Pathol. 204 407–417 [DOI] [PubMed] [Google Scholar]
- 3.Sparrow, J. C., Nowak, K. J., Durling, H. J., Beggs, A. H., Wallgren-Pettersson, C., Romero, N., Nonaka, I., and Laing, N. G. (2003) Neuromuscul. Disord. 13 519–531 [DOI] [PubMed] [Google Scholar]
- 4.Conen, P. E., Murphy, E. G., and Donohue, W. L. (1963) Can. Med. Assoc. J. 89 983–986 [PMC free article] [PubMed] [Google Scholar]
- 5.Shy, G. M., Engel, W. K., Somers, J. E., and Wanko, T. (1963) Brain 86 793–810 [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi, M., Robson, R. M., Stromer, M. H., Dahl, D. S., and Oda, T. (1982) J. Neurol. Sci. 56 35–56 [DOI] [PubMed] [Google Scholar]
- 7.Karpati, G., Carpenter, S., and Eisen, A. A. (1972) Arch. Neurol. 27 237–251 [DOI] [PubMed] [Google Scholar]
- 8.Kawabuchi, M. (1982) J. Neuropathol. Exp. Neurol. 41 298–314 [DOI] [PubMed] [Google Scholar]
- 9.Agrawal, P. B., Strickland, C. D., Midgett, C., Morales, A., Newburger, D. E., Poulos, M. A., Tomczak, K. K., Ryan, M. M., Iannaccone, S. T., Crawford, T. O., Laing, N. G., and Beggs, A. H. (2004) Ann. Neurol. 56 86–96 [DOI] [PubMed] [Google Scholar]
- 10.Sanoudou, D., and Beggs, A. H. (2001) Trends Mol. Med. 7 362–368 [DOI] [PubMed] [Google Scholar]
- 11.Bathe, F. S., Rommelaere, H., and Machesky, L. M. (2007) BMC Cell Biol. 8: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costa, C. F., Rommelaere, H., Waterschoot, D., Sethi, K. K., Nowak, K. J., Laing, N. G., Ampe, C., and Machesky, L. M. (2004) J. Cell Sci. 117 3367–3377 [DOI] [PubMed] [Google Scholar]
- 13.D'Amico, A., Graziano, C., Pacileo, G., Petrini, S., Nowak, K. J., Boldrini, R., Jacques, A., Feng, J. J., Porfirio, B., Sewry, C. A., Santorelli, F. M., Limongelli, G., Bertini, E., Laing, N., and Marston, S. B. (2006) Neuromuscul. Disord. 16 548–552 [DOI] [PubMed] [Google Scholar]
- 14.Ilkovski, B., Nowak, K. J., Domazetovska, A., Maxwell, A. L., Clement, S., Davies, K. E., Laing, N. G., North, K. N., and Cooper, S. T. (2004) Hum. Mol. Genet. 13 1727–1743 [DOI] [PubMed] [Google Scholar]
- 15.Marston, S., Mirza, M., Abdulrazzak, H., and Sewry, C. (2004) Neuromuscul. Disord. 14 167–174 [DOI] [PubMed] [Google Scholar]
- 16.Anthony Akkari, P., Nowak, K. J., Beckman, K., Walker, K. R., Schachat, F., and Laing, N. G. (2003) Biochem. Biophys. Res. Commun. 307 74–79 [DOI] [PubMed] [Google Scholar]
- 17.Bookwalter, C. S., and Trybus, K. M. (2006) J. Biol. Chem. 281 16777–16784 [DOI] [PubMed] [Google Scholar]
- 18.Joel, P. B., Fagnant, P. M., and Trybus, K. M. (2004) Biochemistry 43 11554–11559 [DOI] [PubMed] [Google Scholar]
- 19.Rutkevich, L. A., Teal, D. J., and Dawson, J. F. (2006) Can J. Physiol. Pharmacol. 84 111–119 [DOI] [PubMed] [Google Scholar]
- 20.Kabsch, W., Mannherz, H. G., Suck, D., Pai, E. F., and Holmes, K. C. (1990) Nature 347 37–44 [DOI] [PubMed] [Google Scholar]
- 21.Rould, M. A., Wan, Q., Joel, P. B., Lowey, S., and Trybus, K. M. (2006) J. Biol. Chem. 281 31909–31919 [DOI] [PubMed] [Google Scholar]
- 22.Kozak, M. (1987) Nucleic Acids Res. 15 8125–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Reilly, D. R., Miller, L. K., and Luckow, V. A. (1992) Baculovirus Expression Vectors, A Laboratory Manual, W. H. Freeman and Co., New York
- 24.Pardee, J. D., and Spudich, J. A. (1982) Methods Enzymol. 85 164–181 [DOI] [PubMed] [Google Scholar]
- 25.Margossian, S. S., and Lowey, S. (1982) Methods Enzymol. 85 55–71 [DOI] [PubMed] [Google Scholar]
- 26.Kuhn, J. R., and Pollard, T. D. (2005) Biophys. J. 88 1387–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kapust, R. B., Tozser, J., Fox, J. D., Anderson, D. E., Cherry, S., Copeland, T. D., and Waugh, D. S. (2001) Protein Eng. 14 993–1000 [DOI] [PubMed] [Google Scholar]
- 28.Blikstad, I., Markey, F., Carlsson, L., Persson, T., and Lindberg, U. (1978) Cell 15 935–943 [DOI] [PubMed] [Google Scholar]
- 29.Schuler, H., Lindberg, U., Schutt, C. E., and Karlsson, R. (2000) Eur. J. Biochem. 267 476–486 [DOI] [PubMed] [Google Scholar]
- 30.Malmqvist, U. P., Aronshtam, A., and Lowey, S. (2004) Biochemistry 43 15058–15065 [DOI] [PubMed] [Google Scholar]
- 31.Trybus, K. M. (2000) Methods 22 327–335 [DOI] [PubMed] [Google Scholar]
- 32.Kinose, F., Wang, S. X., Kidambi, U. S., Moncman, C. L., and Winkelmann, D. A. (1996) J. Cell Biol. 134 895–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang, Q., Moncman, C. L., and Winkelmann, D. A. (2003) J. Cell Sci. 116 4227–4238 [DOI] [PubMed] [Google Scholar]
- 34.Brault, V., Sauder, U., Reedy, M. C., Aebi, U., and Schoenenberger, C. A. (1999) Mol. Biol. Cell 10 135–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buzan, J., Du, J., Karpova, T., and Frieden, C. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 2823–2827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rommelaere, H., Waterschoot, D., Neirynck, K., Vandekerckhove, J., and Ampe, C. (2004) Biol. Proced. Online 6 235–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mossakowska, M., Moraczewska, J., Khaitlina, S., and Strzelecka-Golaszewska, H. (1993) Biochem. J. 289 897–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strzelecka-Golaszewska, H., Mossakowska, M., Wozniak, A., Moraczewska, J., and Nakayama, H. (1995) Biochem. J. 307 527–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kudryashov, D. S., and Reisler, E. (2003) Biophys. J. 85 2466–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKane, M., Wen, K. K., Meyer, A., and Rubenstein, P. A. (2006) J. Biol. Chem. 281 29916–29928 [DOI] [PubMed] [Google Scholar]
- 41.Fujiwara, I., Takahashi, S., Tadakuma, H., Funatsu, T., and Ishiwata, S. (2002) Nat. Cell Biol. 4 666–673 [DOI] [PubMed] [Google Scholar]
- 42.Bing, W., Knott, A., and Marston, S. B. (2000) Biochem. J. 350 693–699 [PMC free article] [PubMed] [Google Scholar]
- 43.Buzan, J. M., and Frieden, C. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen, X., Cook, R. K., and Rubenstein, P. A. (1993) J. Cell Biol. 123 1185–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen, K. K., and Rubenstein, P. A. (2003) J. Biol. Chem. 278 48386–48394 [DOI] [PubMed] [Google Scholar]
- 46.Dominguez, R., and Graceffa, P. (2003) Biophys. J. 85 2073–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graceffa, P., and Dominguez, R. (2003) J. Biol. Chem. 278 34172–34180 [DOI] [PubMed] [Google Scholar]
- 48.Otterbein, L. R., Graceffa, P., and Dominguez, R. (2001) Science 293 708–711 [DOI] [PubMed] [Google Scholar]
- 49.Strzelecka-Golaszewska, H., Moraczewska, J., Khaitlina, S. Y., and Mossakowska, M. (1993) Eur. J. Biochem. 211 731–742 [DOI] [PubMed] [Google Scholar]
- 50.Holmes, K. C., Popp, D., Gebhard, W., and Kabsch, W. (1990) Nature 347 44–49 [DOI] [PubMed] [Google Scholar]
- 51.Dominguez, R. (2004) Trends Biochem. Sci. 29 572–578 [DOI] [PubMed] [Google Scholar]
- 52.Allingham, J. S., Klenchin, V. A., and Rayment, I. (2006) Cell. Mol. Life Sci. 63 2119–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chik, J. K., Lindberg, U., and Schutt, C. E. (1996) J. Mol. Biol. 263 607–623 [DOI] [PubMed] [Google Scholar]
- 54.Belmont, L. D., Orlova, A., Drubin, D. G., and Egelman, E. H. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim, E., Bobkova, E., Hegyi, G., Muhlrad, A., and Reisler, E. (2002) Biochemistry 41 86–93 [DOI] [PubMed] [Google Scholar]
- 56.Drummond, D. R., Hennessey, E. S., and Sparrow, J. C. (1991) Mol. Gen. Genet. 226 70–80 [DOI] [PubMed] [Google Scholar]
- 57.Sparrow, J. C., Drummond, D. R., Hennessey, E. S., Clayton, J. D., and Lindegaard, F. B. (1992) Symp. Soc. Exp. Biol. 46 111–129 [PubMed] [Google Scholar]
- 58.Wertman, K. F., Drubin, D. G., and Botstein, D. (1992) Genetics 132 337–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Procaccio, V., Salazar, G., Ono, S., Styers, M. L., Gearing, M., Davila, A., Jimenez, R., Juncos, J., Gutekunst, C. A., Meroni, G., Fontanella, B., Sontag, E., Sontag, J. M., Faundez, V., and Wainer, B. H. (2006) Am. J. Hum. Genet. 78 947–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laing, N. G., and Nowak, K. J. (2005) BioEssays 27 809–822 [DOI] [PubMed] [Google Scholar]
- 61.Sanoudou, D., Corbett, M. A., Han, M., Ghoddusi, M., Nguyen, M. A., Vlahovich, N., Hardeman, E. C., and Beggs, A. H. (2006) Hum. Mol. Genet. 15 2603–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]