

Abstract
Nitric oxide (NO) is a physiological mediator synthesized by NO synthases (NOS). Despite their structural similarity, endothelial NOS (eNOS) has a 6-fold lower NO synthesis activity and 6–16-fold lower cytochrome c reductase activity than neuronal NOS (nNOS), implying significantly different electron transfer capacities. We utilized purified reductase domain constructs of either enzyme (bovine eNOSr and rat nNOSr) to investigate the following three mechanisms that may control their electron transfer: (i) the set point and control of a two-state conformational equilibrium of their FMN subdomains; (ii) the flavin midpoint reduction potentials; and (iii) the kinetics of NOSr-NADP+ interactions. Although eNOSr and nNOSr differed in their NADP(H) interaction and flavin thermodynamics, the differences were minor and unlikely to explain their distinct electron transfer activities. In contrast, calmodulin (CaM)-free eNOSr favored the FMN-shielded (electron-accepting) conformation over the FMN-deshielded (electron-donating) conformation to a much greater extent than did CaM-free nNOSr when the bound FMN cofactor was poised in each of its three possible oxidation states. NADPH binding only stabilized the FMN-shielded conformation of nNOSr, whereas CaM shifted both enzymes toward the FMN-deshielded conformation. Analysis of cytochrome c reduction rates measured within the first catalytic turnover revealed that the rate of conformational change to the FMN-deshielded state differed between eNOSr and nNOSr and was rate-limiting for either CaM-free enzyme. We conclude that the set point and regulation of the FMN conformational equilibrium differ markedly in eNOSr and nNOSr and can explain the lower electron transfer activity of eNOSr.
Nitric oxide (NO)2 is a mediator of many cell functions (1). NO is synthesized from l-Arg by the NO synthases (NOS; EC 1.14.13.39), a family of homodimeric enzymes that are commonly known as neuronal NOS (nNOS), endothelial (eNOS), and inducible NOS in mammals (2). Each NOS monomer consists of an N-terminal oxygenase domain (NOSoxy) that contains binding sites for iron protoporphyrin IX (heme), (6R)-5,6,7,8,-tetrahydro-l-biopterin (H4B), and l-Arg, a flavoprotein subdomain that contains binding sites for FAD, FMN, and NADPH, and an intervening calmodulin (CaM)-binding sequence (3).
The NOSoxy domains and the NOS flavoprotein domains with their attached CaM-binding sequences (NOSr) can be expressed separately, and this has facilitated detailed studies of their structure and functions (3–5). Although the NOSoxy structure is unique, NOSr shares structural and catalytic features with a family of NADPH-utilizing dual-flavin enzymes, including cytochrome P450 reductase (CPR), methionine synthase reductase, sulfite reductase flavoprotein, and novel reductase-1 (6–8). The dual-flavin enzymes all consist of a subdomain that binds NADPH and FAD (FNR subdomain) and a subdomain that binds FMN (FMN subdomain). Their bound FAD receives electrons from NADPH via hydride transfer and then passes electrons to the bound FMN, from which electrons exit the reductase. All the dual-flavin enzymes exhibit cytochrome c and ferricyanide reductase activities (6–11). However, NOSr display unique properties in relation to CaM binding. CaM triggers NO synthesis by enabling the FMN subdomain to transfer electrons to the heme in NOSoxy (9, 12), and CaM also increases the reductase activities of NOSr, which indicates that it directly affects NOSr function (3, 13).
Despite their structural similarity, the three NOS isoforms each have a unique catalytic profile (14). The differences are greatest for eNOS and nNOS; eNOS has a 6-fold lower NO synthesis activity and a 6–16-fold lower cytochrome c reductase activity than nNOS (15, 16). The basis for these differences is unclear, but it appears to involve slower electron transfer by eNOSr. Previous studies have investigated structural elements that appear to regulate NOS activity. These include an autoinhibitory element in the FMN binding subdomain (a 40–50-amino acid loop) (17–19), a connecting hinge domain between the FMN and FAD binding subdomains (23–25-amino acid residues) (15, 20), and the C-terminal tail (21–42-amino acid extension) (16, 21, 22). It has also been shown that an FAD stacking residue (Phe-1160 in bovine eNOS and Phe-1395 in rat nNOS) and site-specific phosphorylation (Ser-1179 in bovine eNOS and Ser-1412 in rat nNOS) are involved in the catalytic regulation of NOS enzymes (21, 23–25).
Studies with nNOSr suggest that it exists in a two-state conformational equilibrium that may determine the movement and electron transfer functions of its FMN subdomain (4, 22, 24, 26–30). In this model the nNOSr structure shifts between an “FMN-shielded” conformation that has the FMN subdomain interacting with the FNR subdomain to receive electrons, and an “FMN-deshielded” conformation that has the FMN subdomain moved away from the FNR subdomain and exposing the FMN surface so it may transfer electrons to protein acceptors like the NOSoxy domain or cytochrome c (Fig. 1).
FIGURE 1.

Conformational equilibrium of the nNOSr FMN subdomain. The diagram illustrates a two-state conformational equilibrium that may control the electron transfer reactions of the FMN subdomain (triangle). A connecting hinge links the FMN subdomain to the FNR subdomain (upper left). The FMN subdomain is expected to move back and forth to contact the FNR subdomain (Shielded) and an electron acceptor like cytochrome c (cyto c) or NOSoxy (Deshielded). The C-terminal tail (CT) is known to regulate the equilibrium by stabilizing the FMN-shielded conformation. This figure has been adapted from Ref. 27.
The conformational equilibrium of nNOSr is known to be regulated by CaM and NADPH binding (27, 28, 31), through a mechanism involving the nNOSr C-terminal tail regulatory element (16, 22), which holds the FMN subdomain against the FNR subdomain in the nNOSr crystal structure (4). One residue located just upstream in sequence from the C-terminal tail (Phe-1395 in nNOS) is also critical for regulation of the nNOSr conformational equilibrium. The presence of this Phe is required to shift the nNOSr conformational equilibrium toward its FMN-shielded form in the absence of bound CaM (24). Phe-1395 is also required to facilitate NADP+ dissociation so that this step does not become rate-limiting during catalysis by the CaM-bound form of nNOSr.
CaM binding destabilizes the FMN-shielded state of nNOSr, and this has been proposed to be a primary mechanism by which CaM may increase the cytochrome c reductase activity of nNOSr (28, 32). However, CaM binding is also proposed to speed intermolecular electron transfer between the bound FAD and FMN cofactors (33), and recently it was reported to perturb the midpoint reduction potentials of the FAD and FMN cofactors in nNOSr (32). Conceivably, all these effects could help determine the catalytic activity of NOSr. In addition, although the properties and electron transfer reactions of nNOSr have been studied in depth (11, 28, 31–37), the amount of data available for eNOSr is much less (20, 38, 39).
To address this issue we characterized an Escherichia coli-expressed bovine eNOSr, focusing on three mechanisms that could possibly cause its low activity. These mechanisms are a slow NADP+ release, altered flavin midpoint potentials relative to nNOSr, and a different native FMN-shielded:FMN-deshielded ratio (or equilibrium “set point”) plus differential effects of CaM and NADPH binding on the eNOSr set point relative to nNOSr (i.e. regulation of the conformational equilibrium). We ran identical experiments with nNOSr to enable a direct comparison. Our results show there are significant differences in the set point and regulation of the conformational equilibrium of eNOSr and nNOSr that can explain their individual catalytic activities.
EXPERIMENTAL PROCEDURES
General Methods and Materials—All reagents and materials were obtained from Sigma, Amersham Biosciences, or other sources as reported previously (24). Absorption spectra and steady-state kinetic data were obtained using a Shimadzu UV-2401PC or Cary 50 Biospectrometer. Single wavelength stopped-flow kinetic experiments were performed using a Hi-Tech Scientific (Salisbury, UK) SF-61 instrument equipped with anaerobic setup and photomultiplier detection. Full-spectra stopped-flow experiments were performed using a Hi-Tech Scientific KinetAsyst SF-61DX2 stopped-flow system equipped with anaerobic setup and rapid-scanning photodiode array detector. Data from multiple identical stopped-flow experiments were averaged to improve the signal-to-noise ratio. The spectral traces were fit according to single or multiple exponential equations using software provided by the instrument manufacturer. The best fit was determined when adding further exponentials did not improve the fit as judged from the residuals. All plots and some additional curve-fitting were done using Origin® 7.5 (OriginLab, Northampton, MA). All experiments were repeated two or more times with at least two independently prepared batches of proteins to ensure consistent reproducibility of the results. Data were analyzed and are expressed as mean ± S.D. For all experiments and protein purifications, the buffer used contained 40 mm EPPS (pH 7.6), 10% glycerol, and 150 mm NaCl (buffer A), unless noted otherwise. Anaerobic samples were prepared in an air-tight cuvette using repeated cycles of vacuum followed by a positive pressure of catalyst-deoxygenated nitrogen gas. All NOSr proteins samples were fully oxidized by adding potassium ferricyanide, and the excess potassium ferricyanide was removed by passing through a Sephadex G-25 column (PD-10, GE Healthcare).
Molecular Biology, Protein Expression, Purification, and Analysis—A recombinant bovine eNOSr construct that contains an intact CaM -binding site (amino acids 445–1204) was created using a PCR-based approach. The PCR primers were used to amplify a fragment from eNOS that introduced an NdeI and a start codon at the 5′ end (5′-TTTT CAT ATG GTC AAC TAC ATC CTG TCC CCT-3′; reverse 5′-AAT GCC GCC TTT GCC CAA CCA C-3′). The PCR product was gel-purified and digested with NdeI and SacI. The resulting 673-bp fragment was cloned into the pCWori vector that had been similarly digested with NdeI and SacI. After confirming the DNA sequence, the vector was transformed into E. coli Rosetta cells and selected using ampicillin and chloramphenicol. The rat nNOSr construct (695–1429) was prepared as described previously with modifications (35). The nNOSr proteins were overexpressed in E. coli BL21(DE3) cells transformed with a pACYC plasmid containing human CaM and selected with chloramphenicol to co-express CaM with the protein. The eNOSr and nNOSr proteins were purified using 2′,5′-ADP-Sepharose affinity chromatography followed by CaM-Sepharose affinity chromatography as described previously (24). The purified proteins were concentrated, buffer-exchanged (buffer A), and stored at -80 °C. The purity of protein was verified by SDS-PAGE and UV-visible spectroscopy. The concentration of NOSr proteins was estimated using an extinction coefficient of 22,900 m-1 cm-1 at 457 nm for the fully oxidized form. To quantify protein-bound flavins, a known amount of NOSr was boiled in a sealed vial shielded from light to release the flavins followed by centrifugation to remove the denatured protein. The concentration of free flavins in the supernatant was determined by absorbance spectroscopy (λmax = 447 nm; ε447 nm = 12.2 mm-1 cm-1).
Steady-state Cytochrome c Reduction Assays—The cytochrome c reductase activity was determined at 10 and 25 °C by monitoring the absorbance increase at 550 nm and using an extinction coefficient ε550 = 21 mm-1cm-1 as described previously (40).
Interaction between the NOSr Proteins and NADP+ or thio-NADP+—UV-visible spectral changes resulting from the association of NADP+ or thio-NADP+ (S-NADP+) with the eNOSr and nNOSr proteins were determined by calculating a difference spectrum between the protein (40 μm) before and after treatment with a 3-fold molar excess of NADP+ or S-NADP+ (120 μm). For these experiments, the eNOSr and nNOSr proteins were incubated with 1 mm of 2′-AMP and then passed through a PD-10 desalting column prior to use to remove any NADP+ that remained bound to the proteins (22, 24). The observed rate constant (koff) values for the dissociation of either NADP+ or S-NADP+ from the eNOSr and nNOSr proteins were determined in the stopped-flow instrument at 10 °C. A solution containing eNOSr or nNOSr (40 μm) and NADP+ or S-NADP+ (120 μm) was rapidly mixed with a solution of 2′,5′-ADP (4 mm). The absorbance changes were monitored at 300–700 nm spectral range. The kinetic traces were fit to a single-exponential decay function to yield the observed NADP+ or S-NADP+ dissociation rate (koff).
Redox Potentiometry—Sample preparation and redox titrations were carried out in a glove box (Bell Technology) under nitrogen atmosphere with oxygen levels below 5 ppm as described previously (41). The NOSr protein concentration was 30–40 μm containing either EDTA (1 mm) or CaCl2 (2 mm) + CaM (60–80 μm) in buffer A. Absorption spectra were recorded in Cary 50 using a dip probe detector, and the potentials were monitored using Accumet AB15 coupled to a silver/silver chloride electrode saturated with 4 m KCl. Measurements were done in a custom-made glass beaker kept in a water bath at 15 ± 1 °C. A reductive titration was performed by stepwise addition of sodium dithionite, whereas an oxidative titration was done by adding potassium ferricyanide. The redox mediators with midpoint potentials in the range of flavin potentials used were 0.5–1 μm of phenazine methasulfate (+80 mV), indigo carmine (-125 mV), 2-hydroxy-1,4-naphthoquinone (-152 mV), anthraquinone-2,6-disulfide (-184 mV), anthraquinone-2-sulfonate (-225 mV), phenosafranin (-252 mV), safranin O (-280 mV), benzyl viologen (-348 mV), and methyl viologen (-443 mV). The electrode was calibrated against phenosafranin (-252 mV) and the potential of a 5 mm solution of ferricyanide/ferrocyanide in 0.1 m potassium phosphate (pH 7.0), at 25 °C (+425 mV). A correction factor of (+199 mV) was obtained, which is in good agreement with the reported potential of the electrode. Based on the temperature dependence of the electrode potential, a correction factor of (+209 mV) at 15 °C was used. The absorption changes at 457 and at 600 nm were plotted with electrochemical potentials (mV). The midpoint potentials were calculated using the four-electron Nernst Equation 1,
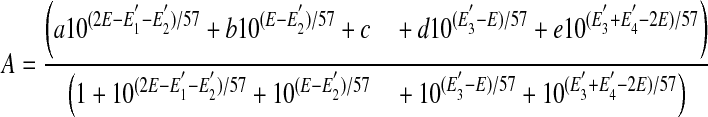 |
(Eq. 1) |
where A is the absorbance; a–e are the relative absorbance values contributed by the diflavin in each of five nondegenerate oxidation states; E is the observed system potential, and E′1 – E′4 are the four midpoint potentials, two for each flavin.
Fluorescence Spectroscopy—Flavin fluorescence was measured at room temperature using a Hitachi model F-2000 spectrofluorometer. Different concentrations from 1–5 μm of the oxidized eNOSr and nNOSr proteins were dissolved in buffer A. The samples were placed in a 1-ml quartz cuvette with a path length of 1 cm. Both proteins were excited at 457 nm wave-length, and the fluorescence spectra were recorded from 480 to 650 nm. The isolated FNR subdomain was prepared as described previously (41). The fluorescence intensities of the FNR subdomain in buffer A were also measured under similar conditions. A correction factor was generated from a linear regression fit of the FNR subdomain fluorescence intensities versus its concentration to obtain only the FMN subdomain fluorescence intensity in the eNOSr and nNOSr samples. This correction factor was subtracted from the total flavin fluorescence intensity of each protein. The maximum fluorescence at 525 nm of eNOSr and nNOSr proteins was plotted versus protein concentration. Linear regression analysis of the data gave the slope of the line for each sample.
EPR Spectroscopy—EPR sample preparation and power saturation experiments were carried out essentially as described previously (24). Briefly, solutions (40 μm) of eNOSr and nNOSr were prepared in 20 mm HEPES buffer (pH 7.4) containing 25% glycerol (v/v). The proteins were initially treated with a slight molar excess of NADPH and allowed to air-oxidize to generate the flavin semiquinone radical species before being frozen in EPR tubes. For CaM-bound samples, CaM (80 μm) and CaCl2 (2 mm) were added. Each sample contained either 0, 3.3, 6.7, 10, or 13 mm dysprosium(III)-HEDTA complex (DyIII-HEDTA). EPR spectra were recorded on a Bruker ESP 300 EPR spectrometer equipped with an ER 035 NMR gauss meter and a Hewlett-Packard 5352B microwave frequency counter. All spectra were obtained at a temperature of 150 K and a microwave frequency of 9.45 GHz. Each run was done with modulation amplitude of 4.01 G at 100-kHz modulation frequency, a center field of 3375 G, and a sweep width of 400 G. Five scans were accumulated and averaged for each spectrum. A total of 17 power settings, ranging from 0.020 to 200.0 milliwatts, were used for the saturation experiments. The resulting microwave power saturation data were fit to the following Equation (2) as described previously (24),
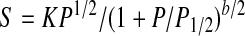 |
(Eq. 2) |
where S is the signal height; K is a proportionality factor; P is the microwave power; P½ is the power required for half-saturation, and b is the inhomogeneity parameter, which was set at 1 in this case. The values of P½ obtained in the absence of DyIII-HEDTA were subtracted from all other corresponding P½ values to yield the parameter ΔP½ for each sample. The ΔP½ values were plotted versus the corresponding concentrations of DyIII-HEDTA to reveal a linear relationship. Linear regression analysis of these data gave the slopes of the lines, m (milliwatts/mm).
Anaerobic Pre-steady-state Cytochrome c Reduction—A solution of eNOSr or nNOSr protein (15–18 μm), glycine (3 mm), 5-deazariboflavin (∼1 μm), and either EDTA (1 mm) or CaCl2 (2 mm) + CaM (30 μm) was completely photo-reduced in an anaerobic cuvette using a commercial slide projector bulb until no changes in the UV-visible spectrum of the sample were observed upon further irradiation of the sample. The fully reduced protein sample was rapidly mixed in the stopped-flow instrument with a solution of cytochrome c (3 μm) at 10 °C. The absorbance change at 550 nm was recorded. In some cases, 1 mm of NADPH was added to the fully reduced protein sample, and the mixture was incubated at 10 °C for 15 min prior to mixing. The eNOSr to cytochrome c ratio was varied to 4, 8, and 12 to determine the effect on the kinetic rates. The concentration of fully reduced eNOSr was changed in the absence of NADPH and CaM and mixed in the stopped-flow with a fixed concentration of cytochrome c (2.5 μm). The reaction was monitored using the absorbance change at 550 nm.
Reaction of Fully Reduced NOSr Proteins with Excess Cytochrome c—The rate of reduction of excess cytochrome c by fully reduced eNOSr or nNOSr protein was measured in the stopped-flow instrument under anaerobic conditions at 10 °C. The eNOSr (20 μm) or nNOSr (5 μm) protein in buffer A containing 5-deazariboflavin and either EDTA (1 mm) or CaCl2 (2 mm) + CaM (three times the protein concentration) was photo-reduced in an anaerobic cuvette. A solution of fully reduced NOS protein and NADPH (200 μm) was mixed with cytochrome c (100 μm) while monitoring the absorbance changes at 550 nm. Initially, a solution of cytochrome c and NADPH was mixed with anaerobic buffer to obtain the initial absorbance reading at time 0. The steady-state trace was fit to linear regression, and the slope was used to determine the cytochrome c reductase activity.
Reaction of 1-Electron Reduced NOSr Proteins with Excess Cytochrome c—1-Electron reduced eNOSr (20 μm) or nNOSr (5 μm) was obtained by adding a slight molar excess of NADPH in a cuvette and then allowed to air-oxidize until the FMN semiquinone (FMNsq) was stable for 1 h. The 1-electron reduced protein was rapidly mixed with a solution of cytochrome c (100 μm) and NADPH (200 μm) in the stopped-flow at 10 °C. The absorbance changes were monitored at 550 nm, and the linear fit of the kinetic trace was measured. The slope of the linear fit was used to calculate the cytochrome c reductase activity. The delay time was determined from the residual and defined as the time point where there is at least 5–10% deviation from the linear fit.
RESULTS
Purification and Properties of eNOSr and nNOSr—The bovine eNOSr and rat nNOSr proteins were purified by tandem affinity chromatography on 2′,5′-ADP-Sepharose and CaM-agarose resins as described previously (24). Prior to use, we verified that both proteins contained an ∼2:1 stoichiometric flavin-to-protein ratio and a 1:1 FAD-to-FMN ratio (data not shown).
Steady-state Cytochrome c Reductase Activities—The NADPH-dependent cytochrome c reductase activity is used to measure electron flux through NOSr proteins and the ability of CaM to increase this rate. Table 1 shows that the steady-state cytochrome c reductase activity of eNOSr (either at 25 or 10 °C) was ∼6–8-fold lower than the activity of nNOSr in the CaM-free state and was ∼15–16-fold lower than the activity of nNOSr in the CaM-bound state. This confirms that eNOSr has a much lower electron transfer activity than nNOSr at both temperatures and in the presence or absence of CaM.
TABLE 1.
Steady-state cytochrome c reductase activities of NOSr proteins The cytochrome c reductase activities were determined at 25 and 10 °C as described under “Experimental Procedures.” The values are the mean ± S.D. from triplicate experiments representative of two independent protein preparations and are expressed as mole of product formed per mol of protein per min.
| Protein | –CaM at 25 °C | +CaM at 25 °C | –CaM at 10 °C | +CaM at 10 °C | ||
|---|---|---|---|---|---|---|
| min–1 | min–1 | |||||
| eNOSr | 64 ± 3 | 264 ± 11 | 26 ± 4 | 67 ± 9 | ||
| nNOSr | 430 ± 81 | 4272 ± 191 | 227 ± 18 | 1179 ± 70 | ||
Interaction of NADP+ with eNOSr and nNOSr—One possible mechanism for low eNOS activity might involve an altered interaction with NADP(H). For example, a slower and rate-limiting dissociation of product NADP+ can explain the lower reductase activities of certain nNOSr and CPR mutants (22, 24, 42–45). We first characterized the eNOSr interaction with NADP(H) using visible spectroscopy. When NADPH binds to enzymes like CPR and nNOSr, a stacking interaction must occur between the nicotinamide ring and the FAD isoalloxazine ring in order for hydride transfer to be possible (22, 24, 41, 46, 47). This stacking phenomenon can be observed by UV-visible spectroscopy, because the NADP+-free versus NADP+-bound difference spectrum typically shows an absorbance increase centered near 510 nm (22, 24, 47–50). The magnitude of the absorbance increase correlates with the extent of nicotinamide-FAD ring stacking and thus reflects the percentage of the NADP+-bound enzyme that is in a “competent” conformation for electron transfer at equilibrium (48). We thus examined NADP+ binding in eNOSr by recording its difference spectrum (NADP+-saturated minus NADP+-free), and compared it with data obtained for nNOSr under identical conditions (Fig. 2A). For nNOSr, there were two positive difference peaks at 508 and 406 nm, consistent with previous findings (22, 24). The difference spectrum of eNOSr also had two positive peaks, but their wave-lengths were shifted (500 and 375 nm), and the peak magnitudes were about half as large as those for nNOSr. Similar differences were observed between eNOSr and nNOSr when we used the structural analog thio-NADP+ (S-NADP+)3 in place of NADP+ (Fig. 2B). For example, with S-NADP+ the major difference peak was at 508 nm for eNOSr and at 512 nm for nNOSr. These results indicate subtle but measurable differences between eNOSr and nNOSr regarding their NADP+ interaction and a lesser degree of nicotinamide-FAD stacking in eNOSr.
FIGURE 2.

Binding interaction of NADP+ and S-NADP+ with NOSr proteins. A and B, given difference spectra were calculated by subtracting the spectrum of each fully oxidized protein from the spectra of the NADP+ or S-NADP+-saturated protein. C and D, kinetics of dissociation of NADP+ or S-NADP+ from NOSr proteins. The NADP+- or S-NADP+-saturated NOSr proteins were rapidly mixed with an excess of 2′,5′-ADP in the stopped-flow instrument at 10 °C. Data are representative of at least two experiments.
The spectral changes we observed upon NADP+ binding to eNOSr were used to estimate the dissociation rate of NADP+. Our previous work showed that rapid mixing of an NADP+-bound nNOSr with an excess of 2′,5′-ADP resulted in a time-dependent absorbance decrease near 500 nm that estimates the dissociation rate (koff) for NADP+ (24). We utilized the same approach to obtain koff estimates for NADP+ and S-NADP+ binding to eNOSr and nNOSr in this study. Representative traces of the absorbance decrease versus time during NADP+ or S-NADP+ dissociation from eNOSr and nNOSr are shown in Fig. 2, C and D. All dissociation curves were fit to a single exponential function, and the resulting koff estimates are given in the figure. For both eNOSr and nNOSr, the koff values for NADP+ were approximately double the corresponding koff values for S-NADP+. This suggests that S-NADP+ binds more tightly to the NOSr proteins, as was also reported for related flavoproteins (51, 52). For nNOSr, the koff values of NADP+ and S-NADP+ were 1.5 times faster than for eNOSr. This suggests that eNOSr may have greater binding affinity toward NADP+ than nNOSr, assuming they have similar association rates for binding. However, the NADP+ koff values for eNOSr are still 40–100 times faster than its catalytic turnover rates for cytochrome c reductase activity measured at 10 °C (Table 1). Thus, the NADP+ koff does not likely limit the activity of eNOSr.
Thermodynamics of Flavin Reduction—It is possible that the low electron transfer activity of eNOSr relative to nNOSr could involve thermodynamic differences among flavin equilibrium midpoint potentials that diminish the driving force for FMNH2 formation in eNOSr. To test this, we carried out potentiometric titrations of the eNOSr flavins in the presence and absence of bound CaM. We performed an initial reductive titration using sodium dithionite followed by an oxidative titration of the same sample using ferricyanide. Representative spectra for CaM-bound eNOSr collected during a redox titration are shown in Fig. 3A. We obtained similar spectra when titrating the CaM-free eNOSr (data not shown). In the reductive direction there was a first phase that involved an absorbance decrease around 457 nm (general flavin reduction) and a broad absorbance increase around 600 nm (flavin semiquinone formation), with an isosbestic point near 510 nm as the first electron equivalent was added. A second phase then took place that was characterized by a decrease in absorbance at all wavelengths from 400 to 700 nm until the enzyme reached the fully reduced (4 e-) state. The absorbance changes at 457 and 600 nm were plotted versus the measured equilibrium potential (mV) obtained in both the reductive and oxidative4 directions for each set of conditions (CaM-bound or free), and the two sets of data obtained at both wavelengths were fit simultaneously to the four-electron Nernst equation (Fig. 3, B and C) as described under “Experimental Procedures.” The fits gave the equilibrium midpoint potentials (Em) of each flavin redox couple (oxidized/semiquinone (ox/sq); semiquinone/hydroquinone (sq/hq) as listed in Table 2.
FIGURE 3.

Potentiometric titration of eNOSr flavins in the presence and absence of CaM at 15 °C. A, representative set of visible spectra obtained during potentiometric titration of CaM-bound eNOSr with sodium dithionite. B and C, plots of absorbance at 457 nm (solid circles) and at 600 nm (solid triangles) versus the electrochemical potential (mV) for CaM-free and CaM-bound eNOSr along with the lines of best fit as calculated using the fourelectron Nernst equation described under “Experimental Procedures.” Data obtained at 457 nm (open circles) and at 600 nm (open triangles) from oxidative titrations of reduced protein samples using potassium ferricyanide are also plotted. Data are representative of at least two experiments.
TABLE 2.
Equilibrium midpoint potentials (Em) versus standard hydrogen electrode of NOSr proteins Midpoint potentials of each flavin (ox/sq and sq/hq) were determined for the NOSr proteins by potentiometric titration at 15 ± 1 °C as described under “Experimental Procedures.”
|
Sample
|
Condition
|
FMN
|
FAD
|
||
|---|---|---|---|---|---|
| ox/sq | sq/hq | ox/sq | sq/hq | ||
| eNOSr | –CaM | –25 ± 2 | –270 ± 10 | –226 ± 18 | –275 ± 7 |
| +CaM | –43 ± 2 | –267 ± 5 | –264 ± 16 | –283 ± 30 | |
| nNOSr | –CaM | –49 ± 2 | –273 ± 6 | –269 ± 23 | – 286 ± 13 |
eNOSr exhibited one high potential couple (FMN ox/sq) and three low potential couples, as also observed in nNOSr (32, 34, 53) and related flavoproteins (6, 7, 54–56). For reference, we also carried out a potentiometric titration of our CaM-free nNOSr, and the flavin midpoint potentials we obtained (Table 2) are in good agreement with previously published data (34). Our measured flavin midpoint potentials for eNOSr and nNOSr were generally similar and were generally unaffected by CaM, with the single exception of the FAD ox/sq couple in eNOSr, which appeared to be 40 mV more positive in CaM-free relative to the CaM-bound form. This single difference among the otherwise similar midpoint potentials is relatively minor, and may not significantly affect the driving force for electron transfer from eNOSr to cytochrome c.
NOSr Conformational Equilibrium—The catalytic activity of nNOSr is thought to involve a conformational equilibrium that controls the movement and electron transfer functions of its FMN subdomain (Fig. 1) (4, 22, 26–30). We hypothesized that eNOSr and nNOSr might differ regarding their conformational equilibrium in ways that might help explain their different catalytic activities. We therefore studied and compared FMN shielding in eNOSr and nNOSr in their fully oxidized, 1-electron reduced, and 4-electron reduced states.
Oxidized eNOSr and nNOSr—FMN shielding in oxidized nNOSr (i.e. an nNOSr that contains both flavins in the fully oxidized state) can be investigated by fluorescence spectroscopy (32, 35, 40). In general, the intensity of flavin fluorescence is expected to be inversely proportional to the degree of FMN shielding. Thus, we measured flavin fluorescence of oxidized eNOSr and nNOSr to compare their FMN shielding. Increasing concentrations5 of our CaM-free NOSr samples were excited at 457 nm, and the intensity of their fluorescence emission was monitored at 525 nm. To determine the fluorescence emission that was solely because of the bound FMN cofactor, we subtracted fluorescence emission values obtained for corresponding concentrations of the nNOS FNR subdomain in replica experiments (see “Experimental Procedures”). These values were assumed to give the amount of fluorescence emission that is due only to the bound FAD in nNOSr or eNOSr, and typically accounted for a minor portion (6–15%) of the total fluorescence. The resulting corrected FMN fluorescence emission values for eNOSr and nNOSr are plotted versus protein concentration in Fig. 4A, along with the calculated lines of best fit. The slopes of the two lines are plotted as bar graphs in Fig. 4B. Similar data were obtained with the CaM-bound NOSr enzymes (data not shown). The data show that eNOSr has greater FMN shielding than does nNOSr in the fully oxidized state.
FIGURE 4.

Fluorescence intensities of the FMN subdomain of the NOSr proteins. A, fluorescence intensity of fully oxidized eNOSr and nNOSr proteins were determined at the indicated concentrations by excitation at 457 nm and monitoring the fluorescence emission at 525 nm. A correction factor for the fluorescence intensity of the FNR subdomain of NOSr was subtracted from the total flavin fluorescence intensity to obtain the indicated values. B, fluorescence intensity versus concentration slope values for the eNOSr and nNOSr proteins obtained by linear regression analysis of data in A. Data are representative of at least two experiments.
1-Electron Reduced eNOSr and nNOSr—1-Electron reduced NOSr proteins contain an air-stable FMNsq radical (31, 33, 37). The average amount of shielding of the FMNsq radical, and therefore its average solvent accessibility as well, can be measured by determining the effect of the soluble spin-relaxing agent DyIII-HEDTA on the microwave power saturation characteristics of the FMNsq radical (57). Previously, we used this method to demonstrate that the amount of shielding of the nNOSr FMN cofactor is directly correlated to its rate of electron transfer to external acceptors such as cytochrome c (24). This method also allowed us to characterize the role of Phe-1395 in FMN shielding in nNOSr. In this study, we used this EPR method to determine the amount of FMN shielding in CaM-free and CaM-bound eNOSr for comparison with nNOSr. The CaM-bound and CaM-free eNOSr and nNOSr proteins were first treated with a slight molar excess of NADPH and then allowed to air-oxidize for several minutes until they reached the 1-electron reduced state. Varying amounts of DyIII-HEDTA were added to the samples, and they were immediately frozen and kept in liquid nitrogen for EPR measurements.
The power saturation of the FMNsq radical in eNOSr and nNOSr was affected by DyIII-HEDTA in a concentration-dependent manner. Fitting of the power saturation curves obtained for each sample yielded P½ values (data not shown), which were converted to ΔP½ values as described under “Experimental Procedures.” The ΔP½ values for eNOSr are plotted versus DyIII-HEDTA concentration in Fig. 5A. We performed an identical analysis of nNOSr (Fig. 5B) utilizing the same conditions and EPR instrumentation. Linear regression analysis of the data gave the slope of the lines, m (milliwatts/mm), which is given in Fig. 5. A larger slope indicates an FMNsq radical that is more solvent-accessible and is on average therefore more deshielded. In the absence of CaM, the m value for eNOSr was less than that for nNOSr. Binding CaM caused their m values to increase, although the m value for eNOSr remained less than that for nNOSr. The data show that the FMNsq in 1-electron reduced eNOSr is more shielded than in nNOSr in both the CaM-free and CaM-bound states.
FIGURE 5.

Comparative shielding of the FMNsq radical in eNOSr and nNOSr as measured by EPR spectroscopy. EPR power saturation measurements were recorded at 150 K for either NOSr protein in the presence of the indicated concentrations of DyIII-HEDTA, and the ΔP½ values were calculated as described under “Experimental Procedures.” Values of ΔP½ for eNOSr (A) and nNOSr (B) are plotted versus [DyIII-HEDTA] in the presence (filled circles) or absence (open circles) of bound CaM. The lines of best fit were calculated by linear regression, and the slope of the lines, m (milliwatts/mm), was determined. Data are representative of at least two experiments.
Fully Reduced eNOSr and nNOSr—The degree of FMN shielding in a fully reduced NOSr (i.e. the 4-electron reduced state that contains both FADH2 and FMNH2) can be determined by an established stopped-flow spectroscopic method (24, 27, 28) that measures the rate of electron transfer to cytochrome c by an excess of the reduced NOSr protein. We first confirmed that photoreduction of eNOSr would generate the fully reduced form that contains FMNH2 (supplemental Fig. 1). We then determined FMN shielding in photo-reduced eNOSr under three different conditions, -CaM/-NADPH, -CaM/+NADPH, and +CaM/+NADPH; and ran replica experiments using photo-reduced nNOSr for a direct comparison. In all cases, the reactions contained a 5-fold molar excess of NOSr over cytochrome c. We determined the rate of cytochrome c reduction by monitoring the absorbance increase at 550 nm and fitting the kinetic trace to exponential functions.
The kinetic traces obtained for nNOSr under each of the three reaction conditions fit well to a single exponential equation as reported previously (22, 24, 27, 41). In contrast, the kinetic traces obtained for eNOSr fit best to a double-exponential equation under two of the three experimental conditions, such that those reactions included a fast phase k1 and a slow phase k2. The rate constants and the percentage of absorbance change occurring in each phase over 0.5 s of reaction are reported in Table 3. Among the different reaction conditions for eNOSr, the slow phase contributed most in the -CaM/-NADPH condition, to a lesser extent in the -CaM/+NADPH condition, and to an undetectable extent in the +CaM/+NADPH condition. CaM binding increased the reaction rates k1 and k2 for eNOSr and the single rate k1 in nNOSr. Remarkably, NADPH binding to CaM-free eNOSr did not diminish its reaction rates. This differs from the 4-fold rate decrease that is typically observed when NADPH binds to the fully reduced, CaM-free nNOSr in Table 3 and in Refs. 22, 24, 27, 41. The inability of NADPH to lower the eNOSr reaction rate was not because of eNOS containing residual bound NADP(H), because we pre-treated eNOSr with 2′-AMP to remove any residual NADP(H) (see “Experimental Procedures”), and our eNOSr exhibited spectral changes upon NADP+ addition that indicate it binds (see Fig. 2A). Together, our data suggest fully reduced eNOSr has greater FMN shielding than nNOSr and differs in its regulation by NADPH.
TABLE 3.
Effect of CaM and NADPH on FMN shielding in fully reduced NOSr proteins An excess of each pre-reduced NOSr protein in the presence or absence of NADPH and CaM was mixed with cytochrome c in a stopped-flow instrument under anaerobic conditions at 10 °C. The molar ratio of NOSr enzyme to cytochrome c was 5:1. The observed rates of absorbance increase at 550 nm are reported as the mean ± S.D. of 5–6 single mixing experiments and are representative of at least two different enzyme preparations. The values in parentheses are the relative percentage of absorbance change with respect to the full reduction of 1.5 μm cytochrome c used in the reaction. ND is not detected.
|
Enzyme
|
Observed rate
|
||
|---|---|---|---|
| –CaM/–NADPH | –CaM/+NADPH | +CaM/+NADPH | |
| s–1 | |||
| eNOSr | |||
| k1 | 52.9 ± 1.8 (54%) | 60.9 ± 3.4 (81%) | 72.5 ± 3.9 (100%)a |
| k2 | 7.1 ± 0.2 (46%) | 16.3 ± 1.2 (19%) | ND |
| nNOSr | 23 ± 1.2 | 5.8 ± 0.5 | 41 ± 0.5 |
For eNOSr, the total absorption change measured after 0.5 s corresponded to 85% of the expected calculated absorbance change at 550 nm for cytochrome c
Effect of Varying the eNOSr to Cytochrome c Ratio—The reactions described above contained fully reduced NOSr proteins in 5-fold molar excess to cytochrome c. Under this condition, observing a slow phase implies that a large percentage of the fully reduced NOSr molecules exist in a slow-reacting state. For example, in the -CaM/-NADPH reaction of eNOSr described above, ∼9 of 10 eNOSr molecules would have to have been in a slow-reacting state for the slow phase to represent 50% of the total absorbance change. A similar calculation for the -CaM/+NADPH reaction of eNOSr suggests that ∼5 of 6 protein molecules were present in a slow-reacting state. To examine this further, we performed similar stopped-flow experiments but varied the eNOSr to cytochrome c concentration ratio. Fig. 6A contains representative stopped-flow traces that we obtained at three different ratios (4, 8, and 12 mol of eNOSr per mol of cytochrome c). Table 4 reports the cytochrome c reduction rates and the percentage of absorbance change observed for each phase in the reactions. The contribution of the slow phase diminished as the eNOSr to cytochrome c concentration ratio increased. This effect was linearly related to the concentration ratio (Fig. 6B). These data establish that a majority (∼80–90%) of the fully reduced, CaM-free eNOSr molecules exist in a slow-reacting state, irrespective of bound NADPH.
FIGURE 6.

Rates of cytochrome c reduction by fully reduced eNOSr at different eNOSr to cytochrome c concentration ratios. Different concentrations of fully reduced eNOSr were mixed with cytochrome c (2.5 μm) in the stopped-flow instrument under anaerobic conditions at 10 °C. A, kinetic traces at 550 nm obtained for the indicated eNOSr/cytochrome c ratios. B, relative percentage of the total absorbance change at 550 nm within 0.5 s of reaction represented by the fast phase k1 and slow phase k2 of each reaction as reported in Table 4. Data are representative of at least two experiments.
TABLE 4.
Effect of eNOSr to cytochrome c ratio on the percentage of fast and slow phase in the reaction The ratio of fully reduced eNOSr versus cytochrome c was varied to 4, 8, and 12 in the absence of NADPH and CaM. A known concentration of fully reduced eNOSr was mixed with a fixed concentration of cytochrome c (2.5 μm) in a stopped-flow instrument under anaerobic conditions at 10 °C. The observed rates and percentages of absorbance change of the fast and slow phase at 550 nm are reported as the mean ± S.D. of 5–6 single mixing experiments and are representative of at least two different enzyme preparations. The relative percentages of absorption change with respect to the full reduction of 1.25 μm cytochrome c are given together with the observed rates, k1 and k2 (s–1) in parentheses. The total absorbance change, ΔA, was measured after 0.5 s.
| Ratio eNOSr/cyt c | % A (k1) | % A (k2) | ΔA (mA)a |
|---|---|---|---|
| 4 | 48 ± 3 (38 ± 2) | 52 ± 3 (3.5 ± 0.2) | 19.3 (77%) |
| 8 | 64 ± 5 (55 ± 3) | 36 ± 4 (9.4 ± 0.5) | 23.1 (93%) |
| 12 | 82 ± 2 (57 ± 1) | 18 ± 2 (3.2 ± 0.2) | 24.4 (98%) |
The expected absorbance (A) chance for 1.25 μm cytochrome c is 25 mA (Δε550 = 21 mm–1 cm–1)
Reaction of Fully Reduced NOSr Proteins with Excess Cytochrome c—Another way to examine the relationship between the NOSr conformational state and reactivity of its reduced FMN subdomain is to monitor the reaction of a fully reduced NOSr with excess cytochrome c. In this circumstance, the electron transfer from FMNH2 to cytochrome c will occur very rapidly for the portion of NOSr molecules that are in a fast-reacting state. Thus, the extent to which the first electron transfer to cytochrome c exhibits a fast and a slow phase can directly indicate what proportion of the NOSr molecules is in a fast-reacting versus a slow-reacting conformation state.
The experiments mixed photo-reduced eNOSr or nNOSr with excess cytochrome c in the stopped-flow spectrometer at 10 °C, and then monitored the absorbance increase at 550 nm during the first few electron transfers to cytochrome c (i.e. the first few turnovers). Representative traces are shown in Fig. 7. In these experiments it is essential that we obtain an accurate initial absorbance reading at time = 0. We determined this value by mixing enzyme-free reaction buffer with cytochrome c in the instrument. Our experimental data were then plotted in terms of the increase in absorbance from that point (a value designated as 0 on the left y axis in Fig. 7, A–D). The right axis of each panel in Fig. 7 indicates the calculated moles of cytochrome c reduced per mol of total NOSr protein that was present in each reaction.
FIGURE 7.

Reaction of fully reduced NOSr proteins with excess cytochrome c. Solutions of pre-reduced, CaM-free, or CaM-bound NOSr (5–20 μm) containing 200 μm NADPH were rapidly mixed with cytochrome c (100 μm) in a stopped-flow instrument under anaerobic conditions at 10 °C. Kinetic traces were recorded at 550 nm during the first few electron transfers to cytochrome c. The absorbance change representing the first turnover is shown by dotted lines according to the right-hand scale in each figure. Data are representative of at least two experiments.
The traces in Fig. 7 show a rate deflection occurred during all four reactions. There was an initial burst phase followed by a slower and nearly constant rate of cytochrome c reduction, as reported previously for pre-reduced nNOSr under similar reaction conditions (28). Notably, in the reactions of fully reduced, CaM-free eNOSr, and nNOSr (Fig. 7, A and C), it is clear that the rate deflection occurred within the time frame of the first turnover. This is particularly apparent in the reaction of CaM-free eNOSr (Fig. 7A), where the slow phase describes a majority of the first turnover. In comparison, the first turnover in the CaM-free nNOSr reaction (Fig. 7C) is mostly described by the fast phase, including a portion that took place within the mixing dead time of the instrument. Together, the results confirm that a subpopulation of fully reduced eNOSr and nNOSr maintains their FMNH2 in a slow-reacting state in the CaM-free condition. On the basis of the traces in Fig. 7, A and C, we estimate that the slow-reacting species represents about 80% of the total eNOSr but about 25–50% of the total nNOSr.
In similar reactions catalyzed by the fully reduced, CaM-bound eNOSr and nNOSr (Fig. 7, B and D), the first turnovers were completed more rapidly and either mostly or completely took place within the mixing dead time of the instrument. This suggests that CaM shifted the conformational equilibrium of the fully reduced eNOSr and nNOSr so that a majority of protein molecules populate a fast-reacting state.
We used the post-burst phase, near-linear portion of each trace in Fig. 7 to calculate rates of cytochrome c reductase activities for eNOSr and nNOSr in the presence and absence of CaM. The calculated activities are given in supplemental Table 1 and match fairly well with the reductase activities we determined for eNOSr and nNOSr in steady-state assays at 10 °C (Table 1).
Reaction of 1-Electron Reduced NOSr Proteins with Excess Cytochrome c—The reactions described above used fully reduced NOSr proteins that had already achieved an equilibrium regarding the slow- versus fast-reacting states before they were mixed with cytochrome c. We also wished to study our NOSr proteins under conditions more akin to steady-state catalysis, where their reaction with cytochrome c also includes steps that form FMNH2. This can be accomplished by mixing the 1-electron reduced NOSr proteins (which contain an air-stable FMNsq) with a solution that contains excess NADPH plus excess cytochrome c in the stopped-flow spectrometer. Under this condition, NADPH reduction of FAD and subsequent electron transfer to FMN must occur before the FMN subdomain (containing FMNH2) can move away from the FNR subdomain and react with cytochrome c (as in Fig. 1). Fig. 8 contains representative absorbance traces from eNOSr and nNOSr reactions run under this condition. The right axis of each panel in Fig. 8 indicates the moles of cytochrome c reduced per mol of NOSr protein. A burst phase was no longer present, consistent with the reactions containing no pre-reduced, fast-reacting NOSr species that contains FMNH2. Instead, we observed an initial slow phase that led to a near-linear rate of cytochrome c reduction in each reaction. The estimated delay time required for each reaction to reach the near-linear phase is shown in the figure. The delay period may reflect build up of FMNH2 within NOSr. The estimated delay times were similar in eNOSr and nNOSr, except for it being a bit faster in the CaM-bound nNOSr reaction (Fig. 8D). Notably, in both eNOSr reactions (-/+ CaM; Fig. 8, A and B) and in the CaM-free nNOSr reaction (Fig. 8C), the linear phase of cytochrome c reduction began well before a single turnover was completed. Only the CaM-bound nNOSr reaction completed its first turnover mostly or completely within the delay period.
FIGURE 8.

Reaction of 1-electron reduced NOSr proteins with excess cytochrome c. 1-Electron reduced CaM-free or CaM-bound NOSr proteins (5–20 μm) were rapidly mixed with a solution containing excess cytochrome c (100 μm) and NADPH (200 μm) in a stopped-flow instrument at 10 °C. Kinetic traces were recorded at 550 nm during the first few electron transfers to cytochrome c. Arrows indicate the estimated delay time before the start of linear phase cytochrome c reduction. Data are representative of at least two experiments.
For the CaM-free nNOSr reaction in Fig. 8, it is apparent that the linear rate achieved within the first turnover was maintained during subsequent turnovers. This is also true for the CaM-free and CaM-bound eNOSr reactions when they were followed over longer reaction times (data not shown). The rates of cytochrome c reduction that we estimated from the linear portions of each trace in Fig. 8 (supplemental Table 1) matched well with the steady-state activities we measured under the same reaction conditions (Table 1). In addition, the linear phase rates for the reactions of Fig. 8 were close to the slow-phase reaction rates that we observed in reactions that were started with fully reduced eNOSr and nNOSr in Fig. 7 (supplemental Table 1). Together, these results argue that a step occurring after FMNH2 formation limits the cytochrome c reductase activities of eNOSr and nNOSr, particularly in the absence of CaM.
DISCUSSION
Mechanisms that control electron transfer by eNOSr and nNOSr are of current interest in the NOS and flavoprotein fields. Our current study reveals that eNOSr and nNOSr differ in the set point and regulation of a conformational equilibrium that controls the reactivity of their reduced FMN cofactors. Moreover, the different set points appear to define the individual electron transfer activities on eNOSr and nNOSr, as will be discussed below. Although we also found that eNOSr and nNOSr differ in their NADP(H) interaction and thermodynamics of flavin reduction, the differences were comparatively minor and are unlikely to explain the distinct electron transfer activities of the two enzymes.
General Conclusions Regarding FMN Shielding and the Conformational Equilibrium of eNOSr and nNOSr—We measured FMN shielding in eNOSr and nNOSr under conditions where the bound FMN cofactor was poised in its three possible oxidation states, namely oxidized FMN, the 1-electron reduced FMN (FMNsq), and FMN hydroquinone (FMNH2). We found that eNOSr maintains a greater degree of FMN shielding than nNOSr, largely independent of the FMN redox state or whether CaM is bound.
Conceivably, the greater degree of FMN shielding in eNOSr could be due to both “static” and “dynamic” differences in its protein conformation relative to nNOSr. For example, an inherent structural difference in the FMN subdomains of eNOSr and nNOSr could reduce the solvent exposure of bound FMN in eNOSr. This static difference in conformation could cause greater FMN shielding in eNOS. Likewise, a dynamic difference in the conformational equilibrium of the FMN subdomain could cause a greater proportion of eNOSr molecules to exist in the FMN-shielded state, and thus result in greater FMN shielding. The methods we used to measure FMN shielding in the fully oxidized or 1-electron reduced forms of our NOSr enzymes (fluorescence and EPR methods) cannot distinguish the relative contributions of static and dynamic differences in the protein conformation. However, the method we used to measure FMN shielding in the fully reduced NOSr proteins (cytochrome c reactivity) does by nature indicate the sole contribution that dynamic differences make in determining FMN shielding. Our results shown in Fig. 7 indicate there exists an FMN-shielded:FMN-deshielded conformational equilibrium of ∼8:1 versus 1:1 for the fully reduced, CaM-free eNOSr and nNOSr, respectively. Thus, the greater FMN shielding we observed for the fully reduced eNOSr appears to be due to its conformational equilibrium being set to favor the FMN-shielded state more than in nNOSr. CaM binding shifted the conformational equilibrium of both fully reduced enzymes toward the FMN-deshielded state, suggesting a common mechanism for CaM function. In contrast, NADPH did not significantly impact the conformational equilibrium of fully reduced eNOSr, despite it shifting the conformational equilibrium of nNOSr toward the FMN-shielded state (22, 24, 27, 41). We conclude the following. (i) Fully reduced eNOSr and nNOSr maintain different set points for a conformational equilibrium that determines the distribution of their FMN-shielded versus FMN-deshielded forms. (ii) CaM similarly regulates the conformational equilibrium of eNOSr and nNOSr, whereas NADPH does not. (iii) Whether the FMN redox state influences the set point for the conformational equilibrium is still unclear. Our findings are surprising because the basis for greater FMN shielding in eNOSr, or its lack of regulation by NADPH, is not obvious from the protein sequences or from the crystal structure of nNOSr (4, 58). Indeed, eNOSr and nNOSr contain similar protein regulatory elements, and their C-terminal tail elements both contain a conserved Arg residue that is implicated in NADPH regulation of the conformational equilibrium of nNOSr (27). The basis for their differences merits further investigation.
Relationship between Electron Transfer from FMNH2 and the NOSr Conformational Equilibrium—During catalysis by NOSr, the bound FMN cycles between its 1- and 2-electron reduced states (FMNsq and FMNH2). In this context, it is informative to consider the conformational equilibrium of NOSr when it contains FMNH2, because this is the entity that donates an electron to acceptors like cytochrome c or the NOS heme domain. As first noted by Daff and co-workers (13, 28), cytochrome c is particularly useful for studying the reactivity of FMNH2 bound in NOSr proteins for at least three reasons. First, cytochrome c reacts appreciably only with fully reduced FMN in NOSr. This is indicated by the slow reaction of the FMNsq in NOSr (31, 33) and by the slow reductase activity of NOSr constructs that can only generate FADH2 (i.e. FMN-free NOSr mutants or the FNR protein fragment of NOSr) (40, 41, 53, 59). Second, electron transfer from bound FMNH2 to cytochrome c is rapid and practically irreversible. This is indicated by an estimated second-order rate constant of 35.5 ± 1.9 μm-1s-1 for the reaction (28) and a thermodynamic driving force of -12.7 kcal based on the relative midpoint potentials of the reactants (-280 mV for FMNH2/FMNsq and +270 mV for ferrous/ferric cytochrome c) (60). Third, cytochrome c greatly amplifies detection of FMNH2 electron transfer events because of the large extinction coefficient difference between its reduced and oxidized forms (21 mm-1cm-1). Thus, cytochrome c is useful in determining the relative proportion of FMNH2 that can react directly with an electron acceptor, and in measuring the changes in this proportion affected by the presence and absence of bound NADPH or CaM.
A simple model that can describe the relationship between the NOSr conformational equilibrium and the reactivity of bound FMNH2 is shown in Scheme 1. The main assumption of the model is that bound FMNH2 can only react with cytochrome c after the FMN subdomain has moved away or “dissociated” from the FNR subdomain to exist in the FMN-deshielded conformation. The considerations outlined above regarding cytochrome c help simplify analysis of this model, because they minimize or negate the importance of the reverse reactions (i.e. k-1 and k-2) in Scheme 1. Thus, if the dissociation of the reduced FMN subdomain from FNR (k1 in Scheme 1) is slow relative to the reaction of bound FMNH2 with cytochrome c (k2), then (i) the relative proportion of fast- and slow-reacting enzyme that is observed in the first catalytic turnover will be a direct indication of the NOSr conformational equilibrium Keq, and (ii) the “slow phase” rate of cytochrome c reduction, if observed during the first catalytic turnover, will be an estimate of the macroscopic dissociation rate of the reduced FMN subdomain from FNR (k1). These experimental conditions were met for the reactions reported in Fig. 7. Specifically, cytochrome c was present at a sufficiently high concentration so that its reaction with the deshielded FMNH2 of NOSr was so fast (estimated rate is 3550 s-1) that it took place within the mixing dead time of the instrument.
SCHEME 1.

Relationship between the NOSr conformational equilibrium and the reactivity of the bound FMNH2.
The estimated 8:1 ratio for the eNOSr conformational equilibrium that we derive from data in Fig. 7 is reasonably close to the 9:1 ratio we estimated independently using data from reactions that contained fully reduced eNOSr in molar excess to cytochrome c (Table 3). The slope of the slow-phase component recorded during the first catalytic turnovers in Fig. 7, A and C, can provide an estimate of the macroscopic dissociation rate k1 for the reduced FMN subdomain in either CaM-free enzyme. These values are 0.5 s-1 for eNOSr and ∼8 s-1 for nNOSr at 10 °C (supplemental Table 1). By using the ratios noted above to derive estimated Keq values for the conformational equilibrium, we can also estimate the macroscopic association rates (k-1 in Scheme 1) for the FMN subdomain in either fully reduced, CaM-free NOSr enzyme, according to Keq = (k1/k-1). The k-1 estimates are 4.0 s-1 for eNOSr and 8 s-1 for nNOSr at 10 °C. These rate estimates are derived from ensemble measures and therefore are not equivalent to the microscopic rates of FMN subdomain association/dissociation in NOSr. However, they can advance our understanding because they link the kinetics of a conformational step to catalysis of electron transfer by eNOSr and nNOSr. Unfortunately, we cannot derive similar rate estimates from the reactions of CaM-bound eNOSr and nNOSr (Fig. 7, B and C), because their first catalytic turnover occurred mostly within the mixing dead time as a consequence of CaM shifting their conformational equilibrium toward a predominantly FMN-deshielded state.
The data we obtained from reactions of the 1-electron reduced forms of eNOSr and nNOSr (Fig. 8) is also informative in this regard. CaM-free eNOSr and nNOSr began a linear phase of cytochrome c reduction well before their first turnovers were completed. The linear-phase rates observed within the first turnover matched fairly well with the estimated dissociation rates for the reduced FMN subdomain (k1) that we derived from the data in Fig. 7 as noted above. This equivalence further argues that a common conformational step is rate-limiting for the reductase activities of CaM-free eNOSr and nNOSr.
The Conformational Equilibrium and the Reductase Activities of eNOSr and nNOSr—Our findings suggest that the steady-state reductase activities of CaM-free eNOSr and nNOSr are limited by a conformational motion of their FMN subdomains. Specifically, dissociation of the fully reduced FMN subdomain away from the FNR (k1 in Scheme 1) appears to be rate-limiting for catalysis by the CaM-free enzymes. Other potentially rate-limiting steps like inter-flavin electron transfer or NADP+ release need not be invoked. These results that highlight the importance of a conformational step fully support early work by Daff and co-workers (13, 28) who first uncovered the existence of a CaM-sensitive “conformational lock” in nNOSr, and related work (32, 35) that suggested the CaM-induced conformational changes may be independent of flavin redox state. Most importantly, this work provides new and specific mechanistic information; we can now state that the approximate 10-fold difference in reductase activities of CaM-free eNOSr and nNOSr is caused by an approximate 10-fold difference in the macroscopic dissociation rate (k1 in Scheme 1) of their reduced FMN subdomains. Likewise, CaM must increase the reductase activities of eNOSr and nNOSr, at least in part, by increasing the dissociation rate (k1) of their reduced FMN subdomains. However, for the CaM-bound enzymes our data cannot exclude the possibility that dissociation of the reduced FMN subdomains may become so fast that other reaction steps (for example, inter-flavin electron transfer (36, 38)) could begin to limit the steady-state reductase activities of CaM-bound eNOSr and nNOSr. Further work is needed to clarify this issue. Another question is whether the FMN redox state may impact the conformational equilibrium of an NOSr. In CPR, dissociation of the FMN subdomain from the FNR subdomain may depend on its reduction to FMNH2 (61, 62). Our current data are consistent with a similar behavior for eNOSr and nNOSr but also point out that dissociation of the FMN subdomain is not complete upon FMNH2 formation, as evidenced by the sizeable fractions of fully reduced eNOSr and nNOSr that remain in the FMN-shielded conformation under the CaM-free condition. Perhaps CaM binding enables FMN reduction to trigger a more complete shift in the NOSr conformational equilibrium toward the FMN-deshielded state. This possibility can now be explored.
Relationship between the NOSr Conformational Equilibrium and NO Synthesis—The conformational equilibrium of NOSr is also an essential component of NO synthesis because the reduced FMN subdomain must dissociate from FNR to transfer an electron to the NOS heme (see Fig. 1). The lowest possible rate for this conformational step (k1) in the CaM-bound enzymes is indicated by the linear phase slopes in Figs. 7 and 8, which are about 1 and 20 s-1 for eNOSr and nNOSr, respectively. These rates still are 5–10 times faster than the measured rates of heme reduction in CaM-bound eNOS or nNOS at 10 °C (0.1–4 s-1) (14, 15). This indicates that electron transfer from the reduced FMN subdomain to the NOS heme is far less efficient than is its electron transfer to cytochrome c. One major difference between the two reactions is entropic; only NOS heme reduction requires specific motions that create a relatively specific interaction of the FMN subdomain with NOSoxy. Another factor is thermodynamic; the driving force is about 55 times less for NOS heme reduction because of the heme having a 540-mV more negative midpoint potential in NOS than in cytochrome c. Although shifting the conformational equilibrium of NOSr toward an FMN-deshielded state must increase the probability of NOS heme reduction, a variety of evidence suggests that this change is insufficient on its own (13–15, 22, 26, 29, 30, 32), and implies that heme reduction must also involve factors that may restrict the motion of the FMN subdomain and/or remove barriers for its productive docking with NOSoxy.
Future Perspectives—This study provides a blueprint to examine how various post-translational modifications (63, 64), NOS protein structural elements (3, 4, 9), and NOS protein-protein interactions (64, 65) control the NOSr conformational equilibrium, and whether this is a way they influence NOS catalytic activities. These studies could also extend to other members of the dual-flavin reductase family to understand the set points and regulation of their conformational equilibria and what role they have in determining their respective electron transfer activities.
Supplementary Material
Acknowledgments
We thank Drs. Kulwant Aulak, Jesus Tejero, and Mohammad Mahfuzul Haque for helpful advice and discussions and Claire Kenney for assistance with protein purification and characterization.
This work was supported, in whole or in part, by National Institutes of Health Grants CA53914, HL76491 (to D. J. S.), ES012658, and GM075036 (to R. H.). This work was also supported by American Heart Association Fellowship 0725341B (to R. P. I.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1 and Table 1.
Footnotes
The abbreviations used are: NO, nitric oxide; NOS, nitric-oxide synthase; NOSr, NOS reductase domain; eNOSr, endothelial NOSr; nNOSr, neuronal NOSr; NOSoxy, NOS oxygenase domain; CaM, calmodulin; DyIII-HEDTA, dysprosium-hydroxyethylethylenediaminetriacetic acid complex; S-NADP+, thio-NADP+; FNR, ferredoxin NADP+ reductase; FMNH2, FMN hydroquinone; FMNsq, FMN semiquinone; EPPS, 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid; CPR, cytochrome P450 reductase.
thio-NADP+ (S-NADP+) is a structural analog of NADP+ that contains a carbothioamide substituent at the 3-position of the pyridine ring. This single atom substitution relative to NADP+ can result in altered binding characteristics in nucleotide-binding proteins (51, 52, 66), and it may also cause larger absorbance shifts near 510 nm (50, 67).
We encountered some difficulty with excessive equilibration/stabilization times for the FMN ox/sq couple during oxidative titrations of eNOSr. Because of this, the oxidative titrations were not complete, but the potential versus absorbance values that we did obtain in the oxidative titrations correlated well with those we collected in the reductive titration.
Each NOSr protein sample used for fluorescence measurement was subsequently denatured by boiling (3–5 min) in a sealed tube, and the insoluble protein was removed by centrifugation. The fluorescence of the released flavins in each supernatant was determined to calculate an exact flavin concentration in each sample and to normalize the eNOSr and nNOSr sample concentrations.
References
- 1.Thomas, D. D., Miranda, K. M., Colton, C. A., Citrin, D., Espey, M. G., and Wink, D. A. (2003) Antioxid. Redox. Signal. 5 307-317 [DOI] [PubMed] [Google Scholar]
- 2.Stuehr, D. J. (1999) Biochim. Biophys. Acta 1411 217-230 [DOI] [PubMed] [Google Scholar]
- 3.Li, H., and Poulos, T. L. (2005) J. Inorg. Biochem. 99 293-305 [DOI] [PubMed] [Google Scholar]
- 4.Garcin, E. D., Bruns, C. M., Lloyd, S. J., Hosfield, D. J., Tiso, M., Gachhui, R., Stuehr, D. J., Tainer, J. A., and Getzoff, E. D. (2004) J. Biol. Chem. 279 37918-37927 [DOI] [PubMed] [Google Scholar]
- 5.Zhang, J., Martasek, P., Paschke, R., Shea, T., Siler Masters, B. S., and Kim, J. J. (2001) J. Biol. Chem. 276 37506-37513 [DOI] [PubMed] [Google Scholar]
- 6.Wolthers, K. R., and Scrutton, N. S. (2007) Biochemistry 46 6696-6709 [DOI] [PubMed] [Google Scholar]
- 7.Finn, R. D., Basran, J., Roitel, O., Wolf, C. R., Munro, A. W., Paine, M. J., and Scrutton, N. S. (2003) Eur. J. Biochem. 270 1164-1175 [DOI] [PubMed] [Google Scholar]
- 8.Munro, A. W., Leys, D. G., McLean, K. J., Marshall, K. R., Ost, T. W., Daff, S., Miles, C. S., Chapman, S. K., Lysek, D. A., Moser, C. C., Page, C. C., and Dutton, P. L. (2002) Trends Biochem. Sci. 27 250-257 [DOI] [PubMed] [Google Scholar]
- 9.Roman, L. J., Martasek, P., and Masters, B. S. (2002) Chem. Rev. 102 1179-1190 [DOI] [PubMed] [Google Scholar]
- 10.Gachhui, R., Abu-Soud, H. M., Ghosha, D. K., Presta, A., Blazing, M. A., Mayer, B., George, S. E., and Stuehr, D. J. (1998) J. Biol. Chem. 273 5451-5454 [DOI] [PubMed] [Google Scholar]
- 11.Abu-Soud, H. M., and Stuehr, D. J. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 10769-10772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siddhanta, U., Presta, A., Fan, B., Wolan, D., Rousseau, D. L., and Stuehr, D. J. (1998) J. Biol. Chem. 273 18950-18958 [DOI] [PubMed] [Google Scholar]
- 13.Daff, S. (2003) Biochem. Soc. Trans. 31 502-505 [DOI] [PubMed] [Google Scholar]
- 14.Stuehr, D. J., Santolini, J., Wang, Z. Q., Wei, C. C., and Adak, S. (2004) J. Biol. Chem. 279 36167-36170 [DOI] [PubMed] [Google Scholar]
- 15.Haque, M. M., Panda, K., Tejero, J., Aulak, K. S., Fadlalla, M. A., Mustovich, A. T., and Stuehr, D. J. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 9254-9259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roman, L. J., Martasek, P., Miller, R. T., Harris, D. E., de la Garza, M. A., Shea, T. M., Kim, J. J., and Masters, B. S. (2000) J. Biol. Chem. 275 29225-29232 [DOI] [PubMed] [Google Scholar]
- 17.Daff, S., Sagami, I., and Shimizu, T. (1999) J. Biol. Chem. 274 30589-30595 [DOI] [PubMed] [Google Scholar]
- 18.Nishida, C. R., and Ortiz de Montellano, P. R. (1999) J. Biol. Chem. 274 14692-14698 [DOI] [PubMed] [Google Scholar]
- 19.Salerno, J. C., Harris, D. E., Irizarry, K., Patel, B., Morales, A. J., Smith, S. M., Martasek, P., Roman, L. J., Masters, B. S., Jones, C. L., Weissman, B. A., Lane, P., Liu, Q., and Gross, S. S. (1997) J. Biol. Chem. 272 29769-29777 [DOI] [PubMed] [Google Scholar]
- 20.Knudsen, G. M., Nishida, C. R., Mooney, S. D., and Ortiz de Montellano, P. R. (2003) J. Biol. Chem. 278 31814-31824 [DOI] [PubMed] [Google Scholar]
- 21.Lane, P., and Gross, S. S. (2002) J. Biol. Chem. 277 19087-19094 [DOI] [PubMed] [Google Scholar]
- 22.Tiso, M., Tejero, J., Panda, K., Aulak, K. S., and Stuehr, D. J. (2007) Biochemistry 46 14418-14428 [DOI] [PubMed] [Google Scholar]
- 23.Adak, S., Santolini, J., Tikunova, S., Wang, Q., Johnson, J. D., and Stuehr, D. J. (2001) J. Biol. Chem. 276 1244-1252 [DOI] [PubMed] [Google Scholar]
- 24.Konas, D. W., Zhu, K., Sharma, M., Aulak, K. S., Brudvig, G. W., and Stuehr, D. J. (2004) J. Biol. Chem. 279 35412-35425 [DOI] [PubMed] [Google Scholar]
- 25.McCabe, T. J., Fulton, D., Roman, L. J., and Sessa, W. C. (2000) J. Biol. Chem. 275 6123-6128 [DOI] [PubMed] [Google Scholar]
- 26.Panda, K., Haque, M. M., Garcin-Hosfield, E. D., Durra, D., Getzoff, E. D., and Stuehr, D. J. (2006) J. Biol. Chem. 281 36819-36827 [DOI] [PubMed] [Google Scholar]
- 27.Tiso, M., Konas, D. W., Panda, K., Garcin, E. D., Sharma, M., Getzoff, E. D., and Stuehr, D. J. (2005) J. Biol. Chem. 280 39208-39219 [DOI] [PubMed] [Google Scholar]
- 28.Craig, D. H., Chapman, S. K., and Daff, S. (2002) J. Biol. Chem. 277 33987-33994 [DOI] [PubMed] [Google Scholar]
- 29.Feng, C. J., Tollin, G., Hazzard, J. T., Nahm, N. J., Guillemette, J. G., Salerno, J. C., and Ghosh, D. K. (2007) J. Am. Chem. Soc. 129 5621-5629 [DOI] [PubMed] [Google Scholar]
- 30.Roman, L. J., and Masters, B. S. (2006) J. Biol. Chem. 281 23111-23118 [DOI] [PubMed] [Google Scholar]
- 31.Guan, Z. W., and Iyanagi, T. (2003) Arch. Biochem. Biophys. 412 65-76 [DOI] [PubMed] [Google Scholar]
- 32.Dunford, A. J., Rigby, S. E. J., Hay, S., Munro, A. W., and Scrutton, N. S. (2007) Biochemistry 46 5018-5029 [DOI] [PubMed] [Google Scholar]
- 33.Matsuda, H., and Iyanagi, T. (1999) Biochim. Biophys. Acta 1473 345-355 [DOI] [PubMed] [Google Scholar]
- 34.Noble, M. A., Munro, A. W., Rivers, S. L., Robledo, L., Daff, S. N., Yellowlees, L. J., Shimizu, T., Sagami, I., Guillemette, J. G., and Chapman, S. K. (1999) Biochemistry 38 16413-16418 [DOI] [PubMed] [Google Scholar]
- 35.Gachhui, R., Presta, A., Bentley, D. F., Abu-Soud, H. M., McArthur, R., Brudvig, G., Ghosh, D. K., and Stuehr, D. J. (1996) J. Biol. Chem. 271 20594-20602 [DOI] [PubMed] [Google Scholar]
- 36.Guan, Z. W., Kamatani, D., Kimura, S., and Iyanagi, T. (2003) J. Biol. Chem. 278 30859-30868 [DOI] [PubMed] [Google Scholar]
- 37.Knight, K., and Scrutton, N. S. (2002) Biochem. J. 367 19-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishino, Y., Yamamoto, K., Kimura, S., Kikuchi, A., Shiro, Y., and Iyanagi, T. (2007) Arch. Biochem. Biophys. 465 254-265 [DOI] [PubMed] [Google Scholar]
- 39.Chen, P. F., Tsai, A. L., Berka, V., and Wu, K. K. (1996) J. Biol. Chem. 271 14631-14635 [PubMed] [Google Scholar]
- 40.Adak, S., Ghosh, S., Abu-Soud, H. M., and Stuehr, D. J. (1999) J. Biol. Chem. 274 22313-22320 [DOI] [PubMed] [Google Scholar]
- 41.Konas, D. W., Takaya, N., Sharma, M., and Stuehr, D. J. (2006) Biochemistry 45 12596-12609 [DOI] [PubMed] [Google Scholar]
- 42.Grunau, A., Paine, M. J., Ladbury, J. E., and Gutierrez, A. (2006) Biochemistry 45 1421-1434 [DOI] [PubMed] [Google Scholar]
- 43.Gutierrez, A., Paine, M., Wolf, C. R., Scrutton, N. S., and Roberts, G. C. (2002) Biochemistry 41 4626-4637 [DOI] [PubMed] [Google Scholar]
- 44.Gutierrez, A., Munro, A. W., Grunau, A., Wolf, C. R., Scrutton, N. S., and Roberts, G. C. (2003) Eur. J. Biochem. 270 2612-2621 [DOI] [PubMed] [Google Scholar]
- 45.Higashimoto, Y., Sakamoto, H., Hayashi, S., Sugishima, M., Fukuyama, K., Palmer, G., and Noguchi, M. (2005) J. Biol. Chem. 280 729-737 [DOI] [PubMed] [Google Scholar]
- 46.Adak, S., Sharma, M., Meade, A. L., and Stuehr, D. J. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 13516-13521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hubbard, P. A., Shen, A. L., Paschke, R., Kasper, C. B., and Kim, J. J. (2001) J. Biol. Chem. 276 29163-29170 [DOI] [PubMed] [Google Scholar]
- 48.Deng, Z., Aliverti, A., Zanetti, G., Arakaki, A. K., Ottado, J., Orellano, E. G., Calcaterra, N. B., Ceccarelli, E. A., Carrillo, N., and Karplus, P. A. (1999) Nat. Struct. Biol. 6 847-853 [DOI] [PubMed] [Google Scholar]
- 49.Panda, S. P., Gao, Y. T., Roman, L. J., Martasek, P., Salerno, J. C., and Masters, B. S. (2006) J. Biol. Chem. 281 34246-34257 [DOI] [PubMed] [Google Scholar]
- 50.Piubelli, L., Aliverti, A., Arakaki, A. K., Carrillo, N., Ceccarelli, E., Karplus, P. A., and Zanetti, G. (2000) J. Biol. Chem. 275 10472-10476 [DOI] [PubMed] [Google Scholar]
- 51.McTigue, M. A., Davies, J. F., Kaufman, B. T., and Kraut, J. (1993) Biochemistry 32 6855-6862 [DOI] [PubMed] [Google Scholar]
- 52.Singh, A., Venning, J. D., Quirk, P. G., van Boxel, G. I., Rodrigues, D. J., White, S. A., and Jackson, J. B. (2003) J. Biol. Chem. 278 33208-33216 [DOI] [PubMed] [Google Scholar]
- 53.Garnaud, P. E., Koetsier, M., Ost, T. W., and Daff, S. (2004) Biochemistry 43 11035-11044 [DOI] [PubMed] [Google Scholar]
- 54.Daff, S. N., Chapman, S. K., Turner, K. L., Holt, R. A., Govindaraj, S., Poulos, T. L., and Munro, A. W. (1997) Biochemistry 36 13816-13823 [DOI] [PubMed] [Google Scholar]
- 55.Munro, A. W., Noble, M. A., Robledo, L., Daff, S. N., and Chapman, S. K. (2001) Biochemistry 40 1956-1963 [DOI] [PubMed] [Google Scholar]
- 56.Wolthers, K. R., Basran, J., Munro, A. W., and Scrutton, N. S. (2003) Biochemistry 42 3911-3920 [DOI] [PubMed] [Google Scholar]
- 57.Innes, J. B., and Brudvig, G. W. (1989) Biochemistry 28 1116-1125 [DOI] [PubMed] [Google Scholar]
- 58.Ray, S. S., Sengupta, R., Tiso, M., Haque, M. M., Sahoo, R., Konas, D. W., Aulak, K., Regulski, M., Tully, T., Stuehr, D. J., and Ghosh, S. (2007) Biochemistry 46 11865-11873 [DOI] [PubMed] [Google Scholar]
- 59.Miyajima, M., Sagami, I., Daff, S., Taiko, M. C., and Shimizu, T. (2000) Biochem. Biophys. Res. Commun. 275 752-758 [DOI] [PubMed] [Google Scholar]
- 60.Dutton, P. L., Wilson, D. F., and Lee, C. P. (1970) Biochemistry 9 5077-5082 [DOI] [PubMed] [Google Scholar]
- 61.Gutierrez, A., Grunau, A., Paine, M., Munro, A. W., Wolf, C. R., Roberts, G. C., and Scrutton, N. S. (2003) Biochem. Soc. Trans. 31 497-501 [DOI] [PubMed] [Google Scholar]
- 62.Zhao, Q., Modi, S., Smith, G., Paine, M., McDonagh, P. D., Wolf, C. R., Tew, D., Lian, L. Y., Roberts, G. C., and Driessen, H. P. (1999) Protein Sci. 8 298-306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iwakiri, Y., Satoh, A., Chatterjee, S., Toomre, D. K., Chalouni, C. M., Fulton, D., Groszmann, R. J., Shah, V. H., and Sessa, W. C. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 19777-19782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fulton, D., Gratton, J. P., and Sessa, W. C. (2001) J. Pharmacol. Exp. Ther. 299 818-824 [PubMed] [Google Scholar]
- 65.Xu, H., Shi, Y., Wang, J., Jones, D., Weilrauch, D., Ying, R., Wakim, B., and Pritchard, K. A., Jr. (2007) J. Biol. Chem. 282 37567-37574 [DOI] [PubMed] [Google Scholar]
- 66.Dolphin, D., Avramovic, O., and Poulson, R. (1987) Coenzymes and Cofactors, Pyridine Nucleotide Coenzymes: Chemical, Biochemical, and Medical Aspects, p. 776, John Wiley & Sons, New York
- 67.Aliverti, A., Deng, Z., Ravasi, D., Piubelli, L., Karplus, P. A., and Zanetti, G. (1998) J. Biol. Chem. 273 34008-34015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.