

SUMMARY
Although p120-catenin regulates adherens junction (AJ) stability in cultured cells, genetic studies in lower eukaryotes have not revealed a role for this protein in vivo. Using conditional targeting in mice, we show that p120 null neonatal epidermis exhibits reduced intercellular AJ components but no overt disruption in barrier function or intercellular adhesion. As the mice age, however, they display epidermal hyperplasia and chronic inflammation, typified by hair degeneration and loss of body fat. Using skin engraftments and anti-inflammatory drugs, we show that these features are not attributable to reductions in junctional cadherins and catenins, but rather NFkB activation. Both in vivo and in vitro, p120 null epidermal cells activate nuclear NFkB, triggering a cascade of proinflammatory NFkB targets. Although the underlying mechanism is likely complex, we show that p120 affects NFkB activation and immune homeostasis in part through regulation of Rho GTPases. These findings provide important new insights into p120 function.
INTRODUCTION
Found throughout epithelial tissues, adherens junctions (AJs) are actin-based intercellular junctions whose constituents are often reduced or mutated in human carcinomas (Perez-Moreno et al., 2003). The transmembrane core of epithelial AJs is composed of E-cadherin, whose extracellular domain physically links cells together. The cytoplasmic domain of E-cadherin associates with catenins, which coordinate actin dynamics to assemble and stabilize these junctions.
Two conserved members of the Armadillo repeat family of catenins are β-catenin and p120, which bind to E-cadherin at distinct sites. β-Catenin associates with α-catenin, which in turn recruits proteins that dynamically remodel and polymerize the associated actin cytoskeleton to seal intercellular membranes (reviewed by Kobielak and Fuchs [2004]). By contrast, p120 promotes lateral clustering of cadherins (Thoreson et al., 2000; Yap et al., 1998), and regulates cadherin stability and turnover at the cell membrane (Davis et al., 2003; Ireton et al., 2002; Xiao et al., 2003). p120 may also promote cell surface trafficking of cadherins through its ability to complex with kinesin and move vesicles along microtubules (Chen et al., 2003; Franz and Ridley, 2004; Yanagisawa et al., 2004).
Constituents of the cadherin-catenin complex have also been implicated in physiological processes that extend beyond the governance of intercellular connections. The best characterized is β-catenin, which is an essential mediator of the Wnt transduction pathway (reviewed by Bienz [2005]; Nelson and Nusse, 2004). Crystallographic analyses indicate that the structural interface between Wnt-regulated β-catenin and the Lef1/Tcf family of DNA binding proteins is similar to that between β-catenin and E-cadherin (Graham et al., 2000; Huber and Weis, 2001). Despite considerable sequence identity with β-catenin, p120 does not bind Lef1/Tcf proteins, consistent with their differential binding to E-cadherin. By contrast, p120 binds to another transcription factor, Kaiso (Daniel and Reynolds, 1999; Park et al., 2005). Although the functional significance of these interactions is unknown, the diversity of putative binding partners for p120 and β-catenin is likely to further diversify their functions in biological processes.
Several lines of evidence suggest that p120 can influence the activities of small Rho GTPases, perhaps acting as a molecular switch to orchestrate the balance between cellular adhesion and migration (Anastasiadis et al., 2000; Magie et al., 2002; Noren et al., 2000). The activity of Rho GTPases is finely tuned by GTP binding and hydrolysis. It has been reported that p120 binds directly to RhoA (Magie et al., 2002), and it has been postulated to maintain it in its inactive GDPbound state (Anastasiadis et al., 2000). If p120 regulates Rho GTPases in vivo, it could potentiate a variety of Rho GTPase-mediated cellular processes, including not only cell adhesion and actin organization, but also proliferation, cell polarity, activation of transcription factors, apoptosis/survival, and vesicle trafficking (Etienne-Manneville and Hall, 2002). If true, then loss ofp120 might alter tissue homeostasis andpromote disease in a fashion independent of its role in adhesion.
Despite the tantalizing roles that have emerged for p120 from these in vitro studies, the functional importance of this protein remains unclear. The Drosophila genome has only one p120 gene, and yet null alleles are viable and fertile, with no substantial changes in junction structure or function (Myster et al., 2003). Additionally, while Drosophila p120 seems to modulate intercellular adhesion, the juxtamembrane domain of DE-cadherin that is essential for its interaction with p120 does not appear to be required in vivo (Pacquelet et al., 2003). Similar genetic studies in C. elegans suggest a supporting but nonessential role for p120 in development and maintenance of intercellular adhesion (Pettitt et al., 2003).
Given the relatively modest role for p120 in lower eukaryotes, a critical in vivo role for p120 in higher eukaryotes has seemed unlikely, particularly since vertebrates have multiple p120 family members, including p0071, δ-catenin/NPRAP and ARVCF, as well as more distantly related plakophilins 1–3 (reviewed by Hatzfeld [2005]). This said, morpholinomediated reduction of p120 expression in Xenopus embryos affects convergent extension movements during gastrulation and axial elongation, in a manner that appears to be independent of p120’s ability to stabilize AJs (Fang et al., 2004). Additionally, when p120 morpholino injections are targeted to neuroectodermal tissues, developing eyes and craniofacial cartilage skeleton are perturbed (Ciesiolka et al., 2004).
Elucidating the precise roles of p120 and its relatives in higher eukaryotes is predicated on direct functional studies using null mutations in their genes, which to date have not been generated for p120. To this end, we have now employed conditional gene targeting to ablate p120 expression in the epidermis and its appendages. In this report, we show that quantitative and specific loss of p120 is deleterious to mouse physiology. Unexpectedly, abrogating p120 expression leads to activation of nuclear factor kappa B (NFkB) transcription factor, sparking a surge of proinflammatory changes in gene expression. We show that NFkB activation in p120 null keratinocytes is accompanied by alterations in Rho GTPase activity that appear to reside upstream from NFkB activation and downstream from p120 loss. Our results not only identify a function for p120 in counteracting inflammatory responses but also raise new insights that extend beyond intercellular adhesion in explaining why reductions in p120 have been associated with epithelial hyperproliferation and tumorigenesis, including squamous cell carcinomas of the skin.
RESULTS
Ablation of p120 in Skin Epidermis Results in Decreased AJ Components without Grossly Perturbing Intercellular Adhesion
The generation of p120 (fl/fl) mice harboring loxP sites flanking exons 3–8 will be reported elsewhere (M.A.D. and A.B.R., unpublished data). Conditional targeting of p120 in skin epidermis was achieved by breeding to K14-Cre recombinase transgenic mice, which efficiently excised the floxed exon by embryonic day E15.5 (Vasioukhin et al., 1999; Figure 1A). Newborn (postnatal day zero; P0) mice genotypic for the K14-Cre allele and homozygous for the p120 floxed allele appeared indistinguishable from wild-type (wt) counterparts (Figure 1B). Histologically, no major morphological alterations were detected in the skin epithelium, and as judged by “outside-in” X-gal permeability and “inside-out” biotin penetration assays, the epidermal barrier was intact (Figures 1C and 1D).
Figure 1. Phenotypic and Biochemical Alterations in Newborn Mice Conditionally Null for p120 in Skin Epidermis.

(A) PCR analysis to confirm genotype. All PCR fragments were of the expected sizes.
(B) P0 mice.
(C and D) Outside-in (β-galactosidase, blue) and inside-out (biotin, red) dye penetration assays on E18.5mice to test epidermal integrity. Sections are counterstained with anti-occludin to verify tight junction integrity and barrier exclusion. sc, stratum corneum.
(E) Ultrastructural analysis of basal epidermal layer. Insets are magnified views of boxed areas. Intercellularmembrane ultrastructure was indistinguishable in wt and KO epidermis (apparent variations in desmosome sizes are due to angling of sections). BL, basal lamina; AJ, putative adherens junctions; De, desmosomes; HD, hemidesmosomes; der, dermis; bl, basal layer. Scale bars, 500 nm. Quantification of the amount of intercellular membranes sealed (including putative AJs) or occupied by desmosomes.
(F) Immunofluorescence of frozen backskin sections (10 μm) labeled with the Abs indicated. Color coding is according to secondary Abs; nuclei were counterstained with DAPI (blue). *Nonspecific secondary Ab staining of cornified layer. Dotted lines mark the epidermal-dermal border.
(G) Immunoblot analysis of total epidermal lysates.
Ultrastructural analyses revealed that intercellular borders were sealed and desmosomes were intact (Figure 1E). This was true for both P0 and adult cKO skin epidermis, in both basal and suprabasal compartments. Shown are data from adult epidermal cells of the basal (innermost) layer, where the ratio of sealed membranes/AJs to desmosomes is the greatest and junctional defects would be expected to be most noticeable. Quantification verified that the degree of intercellular membrane sealing/AJs and desmosomes per unit of membrane was comparable in wt and cKO skin. Other ultrastructural features of the epidermis also appeared to be normal.
Immunofluorescence and immunoblot analysis confirmed the absence of p120 in cKO epidermis (Figures 1F and 1G). Based upon our ultrastructural analyses and also prior genetic studies conducted in lower eukaryotes, we did not anticipate that the localization or levels of AJ components would be affected by loss of p120 (Myster et al., 2003; Pettitt et al., 2003). However, there were markedly reduced levels of the two major classical cadherins in skin epithelium, as well as the two core components of AJs, i.e., β-catenin and α-catenin (Figures 1F and 1G). These perturbations were not observed in tissues where p120 was not targeted for ablation (see Figure S1 in the Supplemental Data available with this article online).
ARVCF is one of p120’s most closely related family members, and like p120, it binds the juxtamembrane domain of E-cadherin (Mariner et al., 2000; Paulson et al., 2000). We therefore expected that if ARVCF had have been expressed in p120 null epidermis, it should have stabilized junctional AJ proteins as it seems to do in Xenopus embryogenesis treated with p120 mopholinos (Fang et al., 2004). Surprisingly, however, not only was ARVCF expressed throughout wt epidermis, but its expression was also sustained in the absence of p120 (Figures 1F and 1G).
Taken together, these data revealed three important points regarding intercellular junctions in the p120 null epidermis in vivo: (1) it is accompanied by reduced levels of cadherins and catenins at sites of intercellular contacts; (2) it does not affect ARVCF at these sites; and (3) it does not disrupt intercellular membrane sealing. A final note is that desmosomal and tight junction proteins appeared to be largely unaffected by the loss of p120 in vivo, as were classical markers of terminal differentiation (Figures 1F, 1G, and S2).
In Vitro, p120 null Keratinocytes Are Inefficient at Assembling into Epithelial Sheets
Although our findings on p120 null mouse epidermis differed from other in vivo studies on p120-deficient Drosophila and C. elegans embryos, they were consistent with in vitro dominant- negative and RNAi knockdown studies in cultured p120+/+ mammalian transformed cell lines (Davis et al., 2003; Xiao et al., 2003). To place p120 null epidermal cells in the context of past in vitro studies, we cultured primary wt and KO epidermal keratinocytes and analyzed their kinetics of AJ assembly, induced by elevating calcium levels to 1.8 mM. In vitro, the cells behaved similarly to their in vivo counterparts in expression and levels of AJ proteins, including ARVCF (data not shown).
At high density, wt keratinocytes began forming AJs within 3 hr after the calcium switch (Figure 2A). By 6 hr, AJ proteins localized to most cell-cell borders, revealing a typical honeycombed anti-E-cadherin staining pattern. By 24 hr, an intact epithelial sheet had formed and some stratification was present. In contrast, KO keratinocytes plated at similar density displayed a relative paucity of cell-cell border anti-E-cadherin staining even after 24 hr exposure to high calcium.
Figure 2. Alterations in Intercellular Junction Assembly in p120 null Keratinocytes.

Primary keratinocytes cultured from wt and cKO neonatal epidermis were induced to form cell-cell junctions by switching from low-calcium (0.06 mM) to high-calcium (1.8 mM) medium for the times indicated. Cells were washed in PBS, fixed and stained with E-cadherin Ab (green), DAPI (blue), phalloidin (red), which binds filamentous actin or anti-desmoplakin (green), a marker for desmosomes. Arrowheads denote presence of E-cadherin in cytoplasmic vesicles.
Primary wt keratinocytes go through an intermediate step in epithelial sheet formation that involves the formation of an adhesion zipper composed of discrete puncta associated with radial cables of actin filaments. Such zippers were prevalent by 6 hr after the calcium switch (Figure 2B). To a limited extent, p120 null keratinocytes formed puncta and organized actin cables. In this regard, the loss of p120 was not as deleterious as the loss of α-catenin (Vasioukhin et al., 2000). This said, zipper formation was inefficient, and numerous small vesicles containing AJ proteins were seen throughout the cytoplasm of p120 null keratinocytes (arrowheads). Overall, these findings provide important genetic support for the hypothesis that AJ stability is governed by p120.
Eventually, confluent cultured p120 null keratinocytes did seal their membranes, as judged by antidesmoplakin staining (Figure 2C). Even so, the process was inefficient, as re- flected by the fact that stratification in vitro, which also involves actin-AJ dynamics (Vaezi et al., 2002), was impaired. Although culture-related deviations in gene expression could contribute to the differences between our in vitro and in vivo results, it is more likely that physiological and mechanistic differences are responsible. We revisit this issue in the Discussion.
Epidermal Hyperproliferation Occurs in the Absence of p120
As p120 null animals aged, they developed overt phenotypic abnormalities that distinguished them from their wt counter-parts. Normal epidermis is a self-renewing tissue that maintains homeostasis by precisely balancing cellular proliferation in the innermost basal layer with the outward flux of terminally differentiating cells moving outward and being sloughed from the skin surface (Fuchs and Raghavan, 2002). Neonatal p120 cKO mice exhibited mild flakiness at their skin surface, which was particularly severe around the severed umbilical cord (Figure 3A). The flakiness was suggestive of epidermal hyperplasia, and this was confirmed at the histological level (Figure 3B).
Figure 3. Signs of Epidermal Hyperplasia in Neonatal p120 null Epidermis.

A) wt and cKO animals. Arrows denote backskin scaliness in P3 cKO mice and blistered, crusty skin in the umbilical cord region of P10 cKO mice.
(B) P3 backskin sections stained with hematoxylin and eosin. Note hyperthickening in cKO epidermis.
(C) Anti-BrdU immunohistochemistry, FACS cell cycle profiles, and quantification of BrdU-labeled skins from wt and cKO mice. Error bars denote the standard error of the mean (SEM).
(D) Immunofluorescence microscopy with Abs indicated. Nuclei are counterstained with DAPI (blue).
(E) Immunohistochemistry and immunoblot analyses with Abs to phosphorylated (activated) Erk1/2 (pMAPK).
(F) Immunoblot analysis of epidermal lysates probed with Abs against total Erk1/2 (MAPK) and active (phospho) Erk1/2 (pMAPK). Epi, epidermis; der, dermis; hf, hair follicle. Dotted lines represent the epidermal-dermal borders.
To further characterize these defects, we administered a 2h pulse of bromodeoxyuridine (BrdU) to P4 mice and then analyzed incorporation into S phase, α6 integrin-positive basal keratinocytes by immunohistochemistry and flow cytometry of dispase-isolated epidermis (Figure 3C). Similar to wt, p120 null epidermis displayed only a single layer of dividing basal cells. However, a nearly 2-fold increase was observed in the number of BrdU-positive cells (11% ± 1.4% versus 6% ± 1%). This was accompanied by an increase in the number of cells expressing the proliferation marker Ki67 (data not shown), the hyperproliferation-associated suprabasal keratin K6 (Figure 3D) and the phosphorylated (activated) form of the MAPK proteins Erk1/2 (Figure 3E). The increased Erk1/2 activity was confirmed by immunoblot analyses (Figure 3F). Together, these differences are hallmarks of hyperproliferative skin epidermis. Moreover, as is often typical of epidermal hyperproliferation involving only a single layer of mitotically active cells, the terminal differentiation program still progressed to completion morphologically and biochemically (Figures 3B and S2). These findings were consistent with the intact epidermal barrier.
Searching for a Mechanistic Link between Loss of p120 and Epidermal Hyperplasia: Clues in Hair Loss and Inflammation
A priori, several possible mechanisms might account for how a reduction and/or imbalance in AJ components at the skin surface might lead to a hyperproliferative state. If β-catenin was free to interact with members of the Lef1/Tcf family of DNA binding proteins, its ability to function as a bipartite transcription factor might have activated cell cycle regulatory genes (Clevers, 2004; Nelson and Nusse, 2004). If α-catenin was missing, association of E-cadherin/β-catenin complexes with downstream effectors might have led to activation of tyrosine kinase receptor pathways (Vasioukhin et al., 2001). We found no evidence to support these circuits in p120 null epidermal cells. Rather, AJ components that were not localized at sites of cell-cell contact still appeared to be in cytoplasmic complexes involving E-cadherin (Figure 2B and data not shown). Although more rigorous investigation would be required to unequivocally rule out these pathways, we began to search for other clues that might explain the hyperproliferative response.
Adult cKO mice were markedly smaller than their wt counterparts and displayed large patches of hair loss over their dorsal skin surface (Figure 4A). The hair coat was also sparser than normal, and gentle tugging released more hairs. Scaly plaques affected their ears and tails. Toluidine blue-stained semithin sections revealed that the hyperplasia first observed in neonatal mice had persisted as the animals aged (Figure 4B). Normal telogen-phase hair follicles were absent, and follicles often exhibited large orifices packed with keratinized material. Dermal blood vessels (bv) were more prevalent, while subcutaneous fat (sf) was markedly reduced. The hair disintegration, wasting, hyperplasia, and enhanced vasculature are classical features of chronic subcutaneous inflammation.
Figure 4. p120 null Mice Develop Inflammatory Skin Lesions with Age.

(A) Visible abnormalities in P60 cKO animals. As cKO mice aged, they diverged in weight from their wt counterparts and displayed hair loss over their skin surface. The P60 cKO hair coat is sparser and scaly plaques affect their ears and tails.
(B) Toluidine blue-stained semithin sections (1 μm) P60 backskins. Insets are magnified views of boxed epidermal areas. cKO skin displayed abnormally thickened epidermis, hyperkeratosis and enlarged blood vessels (bv), as well as degenerated remnants of hair follicles. sf, subcutaneous fat.
(C) Ultrastructure of cKO P60 dermis. Note marked inflammatory infiltration of lymphocytes, macrophages, granulocytes, and mast cells. These cells were rare in the wt P60 counterpart. Scale bar, 2 microns.
Ultrastructural analyses revealed that the cKO underlying dermis was infiltrated with immune cells, including lymphocytes, mast cells, granulocytes (eosinophils and neutrophils), and macrophages (Figure 4C), when compared to controls (data not shown). Although immune cell infiltration was less pronounced in younger cKO mice, it was detectable within the first few days after birth. This was best evidenced by immunofluorescence microscopy with antibodies against T lymphocyte cell surface markers such as CD3 or CD4, the granulocyte marker Gr1, the macrophage marker F4/80, and neutrophils (Figure 5 and data not shown).
Figure 5. Characterization of the Inflammatory Infiltrate Present in p120 cKO Skin.

Immunofluorescence of backskin sections from P3 (A) and P60 (B) wt and cKO mice. Paraffinembedded and cut sections were stained with markers of immune cells (green): CD3, T cells; Gr1, granulocytes; and F4/80, macrophages. Sections were counterstained with DAPI (blue). Epi, epidermis; der, dermis. *Nonspecific secondary Ab staining of cornified layer.
Irrespective of whether animals were neonates or adults, immune cells were always more abundant in cKO versus wt skin (Figure 5). Although the immune infiltration was more pronounced in P60 dermis, inflammatory cells were even observed in the p120 cKO skins of P3 mice. This was quite remarkable, given that the inflammatory response is largely a process that develops postnatally. These findings extended our ultrastructural observations, indicating that a skin inflammatory reaction began early in p120 null mice.
The Epidermal Hyperplasia Can Be Alleviated by Immunosuppressive Drugs, but the Defects in Cadherin and Catenin Distribution Cannot
The proinflammatory response raised a concern that the altered distribution of cadherin/catenins might have subtly compromised the epidermal barrier in a fashion that escaped our detection. To explore this possibility further, we first grafted P0 cKO and wt skins onto the backs of immunocompromised Nude mice, defective in forkhead transcription factor FoxN1, which is essential for T lymphocyte and hair cell differentiation. In control skin epithelial grafts from wt mice, hair growth was restored (first panel of Figure 6A). In marked contrast, cKO grafts still displayed hair defects, indicative of an intrinsic defect to the donor cKO epithelium (second panel of Figure 6A). The altered distribution of cadherins and catenins and epidermal hyperproliferation were also maintained in the p120 null grafts, indicating that these differences were also intrinsic to the p120 null epidermis (Figures 6B and 6C).
Figure 6. Epidermal Hyperplasia, Hair Loss, and Inflammation in p120 cKO Skin Are Rescued by Dexamethasone but Border Localization of Cadherins and Catenins Is Not.

P0 wt and cKO skins were grafted onto the backs of Nude mice, defective in T lymphocyte and hair production. Five days postengraftments, mice were either untreated or treated with dexamethasone (DEX), an inhibitor inflammatory responses. Following an additional 15 days, mice were sacrificed and the engrafted skins were analyzed for hair loss (A), cadherin/catenin localization at intercellular borders (B), epidermal hyperproliferation ([C] and Figure S3) and the presence of inflammatory cells in the dermis ([D] and Figure S3).
Further analyses revealed that mature T lymphocytes were now absent in the p120 null grafts, but they were heavily in- filtrated with host-derived granulocytes, mast cells, and macrophages (Figure S3). To determine the extent to which this partial immune infiltration might still be impacting on the p120 null phenotype, we treated engrafted Nude mice with dexamethasone, a potent and general immune suppressant. As shown in the right panels of Figure 6A, dexamethasone treatment restored the ability of the p120 null graft to produce a hair coat. In addition, treatment caused the epidermal hyperproliferation in cKO skin to vanish along with the vestiges of the proinflammatory response (Figures 6C, 6D, and S3). Despite the return to normal epidermal behavior, however, the cKO-grafted epidermis continued to show altered cadherin and catenin distribution. These findings dissected the p120 null phenotype into two parts: one that affected classical intercellular adhesion components of AJs and the other that recruited a robust inflammatory response.
p120 null Epidermal Cells Activate Transcription Factor NFkB, Unleashing Expression of Proinflammatory Cytokines
Our grafting studies revealed that alterations in cadherins/catenins cannot on their own cause epidermal hyperproliferation. We therefore focused on elucidating a dexamethasone- suppressible mechanism that could account for this phenomenon. Since dexamethasone treatment exerts its anti-inflammatory effects in part by inactivating NFkB signaling (Auphan et al., 1995; Scheinman et al., 1995), we began by using immunohistochemistry to evaluate the status of NFkB in cKO skin grafts. In the absence of dexamethasone, only the cKO and not the wt epidermis showed intense nuclear staining for the active (phosphorylated) form of the p65 subunit of the NFkB complex (Figure 7A). Consistent with the immune infiltration in the cKO dermis, NFkB-positive cells were also detected in below the dermo-epidermal junction (Figure 7A, arrowheads). Staining was quantitatively eliminated in both dermis and epidermis by dexamethasone treatment of the cKO graft (shown, Figure 7A).
Figure 7. p120 null Epidermal Keratinocytes In Vivo and In Vitro Intrinsically Activate NFkB and Express a Variety of Proinflammatory NFkB Target Genes.

(A) Left four frames: grafted skins ± dexamethasone were processed for immunohistochemistry with Abs against the phosphorylated (active) p65 subunit of NFkB. Dexamethasone treatment eliminated nuclear NFkB throughout grafted skin. Right four frames: E18.5 and P60 skin sections from wt and cKO mice stained for active NFkB. Note nuclear NFkB in embryonic p120 cKO skin. Note more intense nuclear staining at P60 and in grafted cKO skins, where it is also found not only in epidermis but also in dermal cells, reflective of inflammatory cells (arrowheads).
(B) Immunofluorescence of cultured keratinocytes labeled with Abs against NFkB. The nuclear localization of NFkB is intrinsic to the loss of p120.
(C) Immunoblot analysis of total epidermal lysates. p120 null epidermis displays increased phosphorylation of the NFkB inhibitor IκBα, a target of IKK activity.
(D) In vitro IKKβ assays were performed on IKKβ immunoprecipitates of cKO and control epidermal lysates, using a recombinant GST-IκBα as substrate. Relative to total IKKβ, the IKKβ kinase activity was higher in p120 null cells.
(E) Primary wt and KO keratinocytes ± TNFα were electroporated with an NFkB reporter gene and Renilla luciferase control to measure the relative NFkB activities. *t test statistical analysis showed a p ≤ 0.005.
(F) Real-time PCRs to measure the relative mRNA levels of the NFkB target genes indicated. The data depicted in the histogram are from ≥5 independent sets of experiments (±SEM).
To evaluate the extent to which the NFkB staining pattern in the cKO epidermis was dependent upon a robust inflammatory response in the dermis, we examined E18.5 skin. Although not as strong, antiphospho NFkB still exhibited prominent nuclear staining in embryonic cKO epidermis (Figure 7A). More intense nuclear staining was observed in P60 epidermis, but in this case phospho NFkB-positive nuclei were also prevalent below the dermo-epidermal border, reflective of the abundant inflammatory cells (Figure 7A).
The presence of an intensified nuclear distribution of NFkB was also observed in p120 null keratinocytes in culture (Figure 7B). Moreover, the enhanced nuclear localization of active NFkB in KO cells was observed irrespective of calcium conditions. Taken together, these data indicated that the initial step in activation of NFkB was an intrinsic response due to the loss of p120 rather than an extrinsic consequence of inflammation. Moreover, it could not be attributable to perturbations in intercellular junction formation.
As judged by immunoblot analyses, overall NFkB levels were similar in wt and p120 null epidermis (Figure 7C). Further analysis revealed that although IκBα was comparably expressed in both cKO and wt epidermis, phosphorylated IκBα was markedly elevated in the absence of p120. IκBα kinase (IKKβ) activity was indeed elevated in the absence of p120, as judged by in vitro kinase assays using recombinant GST-IκBα protein as substrate and epidermal protein extracts as the source of enzyme (Figure 7D). Overall levels of IKKβ were unchanged. These data suggested that increased IKK activity in p120 null epidermis led to enhanced phosphorylation and inactivation of the IκBα inhibitor, which in turn led to the nuclear localization and activation of NFkB. Luciferase reporter assays further verified this difference in NFkB activity, with KO keratinocytes consistently displaying an ~2-fold increase in NFkB activity (Figure 7E).
Well-established target genes for the NFkB transcription factor include TNFα and interleukin 1β, whose activity has been broadly implicated in inflammation, cell proliferation, and apoptosis (reviewed by Karin and Greten [2005]; Viatour et al., 2005). To test whether these and other putative NFkB target genes were upregulated in p120 null epidermis, we selectively removed epidermis from P0 and P3 skins and conducted real-time PCR on their mRNAs. Reflective of NFkB’s activity in p120 null epidermis, many NFkB target genes were upregulated by as much as 10-fold (Gilmore, 2005; Figure 7F). These included TNFα, the interleukins IL1β, IL6, IL13, and IL15, ICAM-1, neutrophil-activating peptide-78 (NAP-78), macrophage inflammatory protein 3 α (MIP3α), macrophage chemotactic protein (MCP1), regulated upon activation normal T lymphocyte expressed and secreted (RANTES), cutaneous T cell attracting chemokine (CCL27), Cox2, and Gro-1. In addition, epidermal cells have receptors for some of these cytokines, e.g., TNFα (shown), and notably, the p120-mediated difference in NFkB reporter activity was bolstered considerably by treatment with TNFα (Figure 7E). This finding revealed the existence of a possible feedback loop to account for how the inflammatory response and epidermal proliferation could escalate over time.
RhoA GTPase Levels Are Elevated in p120 Null Keratinocytes
The engraftment experiments in vivo and the calcium experiments in vitro suggested that p120’s role in NFkB regulation and inflammation is not dependent upon its function in maintaining cadherin-mediated intercellular adhesion. In considering other possible roles for p120, we noted that p120 null keratinocytes displayed more robust stress fibers than their wt counterparts. Given p120’s previously postulated role in the activation/suppression of Rho GTPases, we began by using pull-down assays (see Experimental Procedures) to investigate whether Rho activity might be elevated in the absence of p120. No obvious differences were detected in the Rac1 and Cdc42 GTP pull-down assays (data not shown). However, relative to wt controls, p120 null epidermis displayed a marked increase in GTP bound, i.e., active, RhoA even though total RhoA levels were unchanged (Figure 8A).
Figure 8. Sustained NFkB Activity in p120 null Keratinocytes Is Elicited through RhoA Activation.
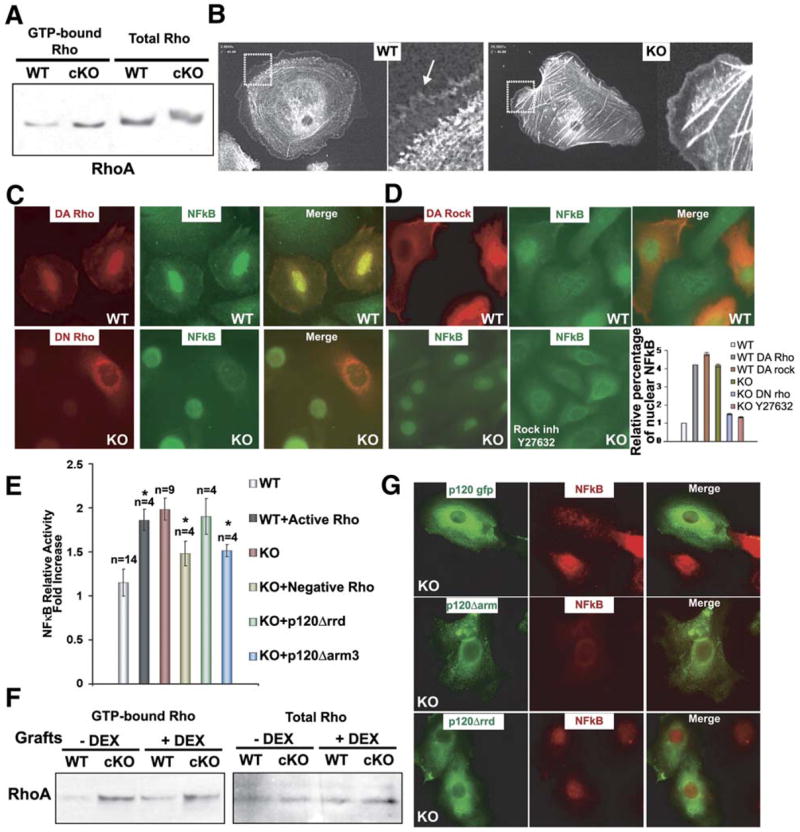
(A) RhoA activity assays. Total protein extracts from P0 wt and cKO epidermis were either used directly or first treated with GST-Rhotekin binding domain bound to glutathione-coupled Sepharose beads to selectively pull down active GTP-RhoA.
(B) Primary keratinocytes were plated on fibronectin and transduced with adenoviral vectors expressing GPF-actin. Live videomicroscopy was conducted to visualize actin dynamics. Movies were focused on representative cells from each of the two cultures. Shown are frames from Movies S1 and S2. Magnified views of boxed areas are to the right. Actin-rich filopodial and lamellipodial extensions, abundant in wt keratinocytes (arrow), are reduced in the KO cells. Actin stress fibers, reflective of active RhoA, are markedly more abundant in KO cells.
(C) Primary keratinocytes were electroporated with expression vectors encoding myc-tagged forms of either DA (L63) or DN (N19) RhoA. Forty-eight hours postelectroporation, cells were fixed and stained with Abs against Myc (red) and NFkB (green). Shown are representative frames. wt cells expressing DA-RhoA- GFP display atypical nuclear NFkB, while KO keratinocytes expressing DN-RhoA-GFP are not longer positive for nuclear NFkB.
(D) Primary KO keratinocytes were either untreated or treated with Y-27632, an inhibitor of the ROCK kinase that is activated by GTP-RhoA. Cells were fixed and stained for NFkB. Quantification of the data for (C) and (D) are presented at right.
(E) Primary wt and KO keratinocytes were coelectroporated with expression vectors encoding either DA or DN RhoA and an NFkB reporter gene and a Renilla luciferase control. Forty-eight hours later, cells were processed for NFkB activities. Although relative differences were small due to the inefficiency of electroporation (~2%–3%), DA Rho consistently had a positive effect and DN Rho a negative effect on NFkB reporter gene activity. *t test statistical analysis shows a p ≤ 0.05. Although subtle, each effect was statistically significant and highly reproducible.
(F) RhoA activity assays of grafted wt and cKO epidermis following dexamethasone treatment. Assays were performed as in (A). Note that despite dexamethasone rescue of nuclear NFkB, RhoA-GTP levels were still higher in treated cKO skins relative to wt.
(G) Transfected keratinocytes expressing GFP fusions proteins of p120, p120 cateninΔarm 3 (lacking E-cadherin binding domain), or p120Δrrd (lacking Rho regulatory binding domain). Note that p120Δrrd is the only one that does not alleviate nuclear localization of NFkB (red). Relative number of transfected cells with nuclear NFkB cells: p120 (1 ± 0.08), p120 cateninΔarm 3 (0.96 ± 0.015), p120Δrrd (1.94±.06).
We next used live-imaging microscopy to explore Rho GTPase-mediated alterations in actin cytoskeletal dynamics caused by p120 deficiency. To perform the imaging analyses, we infected primary wt and p120 null epidermal cells with an adenoviral vector expressing GFP-actin, which in keratinocytes functions normally in all aspects of actin dynamics (Vaezi et al., 2002). We then plated the cells on fibronectin and in low-calcium medium to prevent intercellular adhesion, identified representative cells within each of the two cultures, and imaged them by videomicroscopy. Figure 8B shows frames of Movies S1 and S2.
wt keratinocytes spread quickly and extended filopodia, characteristic of Cdc42 activity, lamellipodia and membrane ruffles, typical of activated Rac1, and actin bundles or stress fibers, a diagnostic feature of activated RhoA (Etienne-Manneville and Hall, 2002). Fluorescing actin filaments concentrated at the leading edges of the membrane ruffles, while a continuous flux of concentric rings of actin filaments moved centripetally from the cytoplasmic periphery toward the nucleus. By contrast, p120 null keratinocytes displayed robust actin stress fibers and a decrease in overall actin dynamics. p120 null cells also displayed significantly fewer filopodial extensions and more restricted areas of lamellipodia, suggesting reduced Cdc42 and Rac1 activities in these cellular compartments.
Dependency of NFkB and GTPase Activities
To test whether the altered levels of small GTPases might be contributing to NFkB activation, we first transfected primary keratinocytes with expression vectors encoding myctagged dominant-active (DA) and dominant-negative (DN) forms of RhoA and the Rho kinase (ROCK), which is downstream of activated RhoA. At 48 hr postelectroporation, cells were fixed and stained with antibodies against either Myc (red) or NFkB (green). As shown in Figure 8C, wt cells receiving the DA Rho construct displayed intense NFkB staining which included prominent nuclear labeling, more typical of p120 null cells. By contrast, p120 null cells expressing DN Rho displayed reduced nuclear NFkB staining, more typical of wt cells. Finally, expression of DA ROCK, downstream of activated Rho, resulted in nuclear staining of NFkB in wt cells, while conversely, treatment of KO keratinocytes with the ROCK inhibitor Y-27632, resulted in a quantitative loss of nuclear NFkB staining. Quantification of these data are presented in the accompanying graph (Figure 8D).
To verify that the differences in NFkB localization reflected the status of activated NFkB, we used a luciferase reporter gene driven by an NFkB-responsive element. The activity measurements were expectedly modest, given that multiple plasmids needed to be taken up by the primary keratinocytes, which only display an optimal transfection efficiency of 2%–3%. This said, the differences were reproducible and statistically significant. Indeed, the levels of NFkB activity were elevated by DA RhoA and suppressed by DN RhoA (Figure 8E). Thus, although immunofluorescence enabled preferential focus on transfected cells which reporter gene assays did not, both sets of data were consistent and suggested that RhoA activity is upstream of nuclear activated NFkB and downstream of p120 targeting. Further documentation that RhoA activity is independent of NFkB activity but dependent upon the status of p120 came from measuring RhoA activities in wt and cKO skin grafts ± dexamethasone treatment (Figure 8F).
Finally, we conducted transient transfections to test the relevance of p120’s Rho GTPase regulatory domains and E-cadherin binding domains to this mechanism. On a p120 null background, expression of p120-GFP restored the wt localization of nuclear NFkB (Figure 8G). Similarly, a p120-GFP mutant lacking the E-cadherin-interacting domain appeared to repress NFkB activity. By contrast, expression of a p120-GFP mutant lacking the putative Rho GTPase regulatory domain (p120Δ622–628 referred as p120Δrrd) still showed nuclear localization of NFkB. Together, these results are suggestive of a role for the Rhoregulatory domain in exerting p120’s effects on NFkB signaling in keratinocytes.
DISCUSSION
Epidermal p120: Distinctions between the Stabilization of AJ Components and Intercellular Adhesion
Prior studies in lower eukaryotes had suggested that p120 is dispensable for intercellular adhesion (Myster et al., 2003; Pettitt et al., 2003). Hence, we were not initially surprised to find that mice targeted for p120 loss in keratinocytes displayed an intact epidermis with a functional barrier and normal architecture. However, in contrast to the developing surface of the Drosophila embryo, p120 null skin epidermis displayed a marked reduction in core AJ components at intercellular borders. Remarkably, at least under conditions of normal stress, these effects did not appear to grossly alter the desmosomes, tight junctions, or the sealing of intercellular membranes within the epidermis.
The ability of p120 null epidermis to sustain intercellular contacts and epidermal differentiation contrasted with epidermis lacking α- or β-catenin (Huelsken et al., 2001; Vasioukhin et al., 2001). Although the milder features of p120 deficiency might be attributable in part to compensatory action provided by ARVCF, the presence of this ARVCF did not completely rescue the cadherin and catenin organization at cell-cell junctions.
Although the mammalian p120 null epidermis behaved differently than p120-defective flies and worms, the p120 null cultured keratinocytes behaved similarly to other cultured cells in which p120 function was compromised by knockdown or dominant-negative approaches (Davis et al., 2003; Ireton et al., 2002; Xiao et al., 2003). In this regard, it was intriguing that reductions in cadherins and catenins were observed irrespective of whether p120 null keratinocytes were cultured or in a tissue, whereas defects in effectively sealing cell-cell junctions were only observed in vitro.
In contemplating the underlying basis for this difference, it is noteworthy that in vitro, epidermal cells are less densely packed and must use filopodial and lamellipodial extensions to physically draw themselves together and seal their intercellular contacts (Vasioukhin et al., 2000). Moreover, in vitro, keratinocytes are flat, and the surface available for cell-cell interactions is significantly reduced. Additionally, this confronts the intercellular junction assembly process with considerable competition from factors promoting cell-substratum adhesion. By contrast, in vivo, epidermal sheet formation and stratification are less affected by actin dynamics, since cells are more columnar and closely packed, providing a greater surface area for cell-cell interactions in the absence of competition from cell-substratum actin dynamics. In the future, it will be interesting to see whether the reductions in AJ components imposed by loss of p120 will manifest adhesive defects under conditions of stress and/or wound healing in vivo.
p120 and Epidermal Homeostasis: Intrinsic versus Extrinsic Effects Leading to NFkB Activation and Inflammation
Skin homeostasis is dependent upon a tightly regulated microenvironment of cytokines and growth factors produced by keratinocytes, dermal cells, and immune cells. Abnormal epidermal proliferation and inflammatory conditions are characteristic of a number of skin disorders. One way in which intrinsic effects can generate an inflammatory response is through disruption of an epidermal barrier, leading to infections that elicit an inflammatory response. Our results show that if intrinsic loss of p120 does cause a defect in barrier function; it is subtle, as neither inside-out nor outside-in barrier assays revealed signs of barrier disruption. In contrast, loss of p120 resulted in NFkB activation by an intrinsic mechanism that does not appear to rely upon bacterial infection. Rather, this response occurs in the embryo, before the epidermis is exposed to the external environment as well as in a sterile environment in vitro.
The effects on epidermal proliferation and immune cell recruitment were rooted in p120 null-mediated activation of NFkB, as dexamethasone treatment of p120 null engraftments not only erased NFkB activation but also abated the epidermal hyperproliferation and inflammatory infiltrate in the graft. Notably, the treatment did not alleviate the paucity of AJ components at intercellular borders, indicating that these perturbations on their own are not sufficient to activate a proliferative response. In this regard, the underlying mechanism operating on hyperproliferation in p120 null epidermis differs from those ascribed to the α-catenin null state, which were traced at least in part to alterations in tyrosine kinase receptor signaling pathways (Vasioukhin et al., 2001).
Although NFkB’s role in skin physiology is still not fully understood, our results were intriguing given that NFkB can act as an antiproliferative safeguard under some circumstances (Dajee et al., 2003; Zhang et al., 2005). At present, we cannot rule out the possibility that NFkB’s action might be due partially to an extension in cell survival. However, in p120 null epidermis, the number of cycling cells in the basal layer was enhanced, suggesting a positive role for NFkB in this situation.
The repertoire of activated NFkB target genes was suffi- cient to explain the inflammatory response in cKO mice. Indeed, many of these cytokines and adhesion molecules had already been implicated in inflammatory responses in skin (Cheng et al., 1992; Pahl, 1999; Robert and Kupper, 1999). Another interesting aspect was that the epidermal hyperplasia and inflammation became progressively more severe as p120 null mice aged. Since the effects were suppressed by dexamethasone treatment, we surmise that the enhanced proliferation may be mediated by the increasingly greater number of dermal inflammatory cells in older cKO mice. Notably, activated inflammatory cells also express activated NFkB and produce and secrete cytokines that epidermal keratinocytes respond to. Thus, the sustained activation of NFkB by p120 null keratinocytes is likely to trigger paracrine NFkB signaling loops between epidermal keratinocytes and various inflammatory cell types that amplifies with age (reviewed by Werner and Smola [2001]; Karin and Greten, 2005). Since an elevated response was also seen in the engrafted Nude mice, the nonlymphocytic immune cells of the animal appear able to carry out this crosstalk with the epidermal cells.
Epidermally expressed TNFα deserves particular note as a pivotal modulator of inflammation which can also enter the bloodstream, leading to loss of body fat and wasting (Cheng et al., 1992). The upregulated expression of TNFα in p120 cKO mice provided a possible explanation for their small size and frailty as the mice aged.
An NFkB-Rho GTPase Connection?
As evidence mounted that p120 loss was affecting normal skin physiology through NFkB activation, our attention shifted to how loss of p120 is involved. There are multiple ways to activate NFkB, some of which involve IKKs to suppress IκB’s inhibitory powers and others which are IKK independent (Makris et al., 2002; Pasparakis et al., 2002; reviewed by Viatour et al. [2005]). Although IκB phosphorylation was enhanced in p120 null epidermis, this inflammatory response differs from that involving loss of IKK’s catalytic subunit (Makris et al., 2002; Pasparakis et al., 2002). Although loss of p120 may elicit multiple contributing factors, one mechanistic distinction is the elevated levels of RhoA.
Several previous studies have led to the view that activation of small Rho GTPases may result in NFkB activation and cytokine production (Perona et al., 1997) and that p120 might be a negative regulator of Rho GTPase and a positive regulator of Rac1/Cdc42 activity (Anastasiadis et al., 2000; Noren et al., 2000; Grosheva et al., 2001; Magie et al., 2002). Our work now shows when p120 is absent RhoA activity is enhanced, and this profoundly alters cytoskeletal- membrane dynamics. Since this happens in lowcalcium medium in vitro, it diminishes a possible role for AJ instability in this response. That the response can be at least partially rescued by a p120 mutant lacking the E-cadherin binding domain provides another clue that p120’s action goes beyond its ability to associate with and regulate AJ stability. While AJ stability and more subtle alterations in transport properties or cell physiology could contribute, both actin-membrane dynamics and NFkB activation also appear to involve the activation of RhoA.
The Rho family of GTPases has been implicated in a plethora of cellular processes depending upon context (Etienne- Manneville and Hall, 2002). Exactly how RhoA activation in- fluences NFkB activity remains to be determined. However, in the presence of Rho inhibitory drugs or dominant-negative- acting RhoA, nuclear NFkB was no longer detected by the phosphospecific antibody that detects active NFkB. Collectively, these results place p120 upstream of RhoA activation, which is upstream of NFkB activation, which in turn is upstream from epidermal expression of a host of inflammatory cytokines. The fact that the process occurs in vitro as well as in vivo emphasizes the intrinsic nature of this initial cascade. Figure S4 provides a model which summarizes these results.
In closing, we have uncovered a player, p120, underlying an inflammatory and hyperproliferative response in the skin. Our results are likely to be relevant in understanding why a loss or reduced activity of p120 has been correlated with inflammatory bowel disease, particularly ulcerative colitis and active Crohn’s disease, known to predispose the tissue to cancer (reviewed by Reynolds and Roczniak-Ferguson, 2004). These findings now open new avenues for exploring the relationship between intercellular adhesion molecules, small GTPases, and inflammatory events, a hallmark of several human skin disorders and disease.
EXPERIMENTAL PROCEDURES
Generation of p120 Conditional Null Mice in Skin
Sv/129 mice carrying floxed p120 alleles were crossed to CD-1 transgenic mice expressing Cre-recombinase under the control of the human keratin 14 promoter (Vasioukhin et al., 1999) with p120 mice harboring loxP sites flanking exons 3–8, to obtain p120 (fl/fl) K14-Cre (cKO) mice. p120 fl/+ K14-Cre and p120+/+ K14-Cre mice are referred to as wt, as they were indistinguishable by the assays performed.
Immunofluorescence, Immunohistochemistry, and Antibodies
Unless indicated, dilutions were according to the manufacturer’s instructions. Primary antibodies (Abs) used were: p120 (Zymed), E-cadherin (Ecad; ECCD-1, M. Takeichi), β- and α-catenin (Sigma), P-cadherin (Pcad, Zymed), Desmoglein 1,2 (Dg1,2; W. Franke, German Cancer Research, Heidelberg), desmoplakin (Dp, Research Diagnostics), β4-integrin (BD-Pharmigen), keratin 5 (K5, 1:100; E.F.), K6 (1:100; P. Coulombe, John Hopkins School of Medicine), Ki67 (Novocastra), plakoglobin (P. Cowin, New York University), ARVCF (1:100; Ilse Hofmann, Division of Cell Biology, DKFZ, Heidelberg), Occludin (Zymed), p65 subunit of NFkB (Santa Cruz), phospho NFkB p65 (Cell Signaling), IκBα (Imgenex), pIκBα (Santa Cruz), β-actin (Sigma), Vinculin (Sigma), CD3 (Chemicon), Gr1 (RD Systems), F4/80 (Serotec), CD49f (α6 integrin, Pharmigen); BrdU (Abcam), MAPK (Cell Signaling), and active MAPK (Sigma). Secondary Abs used were conjugated to FITC or Texas red (The Jackson Labs) or biotin (Vector Labs). Skins were frozen and embedded in OCT compound or fixed overnight with buffered 4%PFA at 4°C and embedded in paraffin. Skin sections (10 μM) were stained with hematoxylin and eosin or processed for immunoreactions. Slides were blocked with PBS, 0.3% Triton X-100, 1% BSA, 5% normal goat serum, 5% normal donkey serum, or MOM Basic kit (Vector Labs).
NFkB Luciferase Reporter Assays
Neutralized cell suspensions from neonatal epidermises were washed, counted. 300 × 103 cells per T24-well were electroporated (Amaxa electroporation system) with 500 ng of NFkB-luc reporter construct (PathDetect, Calbiochem) and 100 ng of Renilla luciferase plasmid (Promega) and then plated on mitomicin C-treated fibroblast feeders. Where indicated, electroporations also included 25 ng of activated MEKK (positive control), 3, 6.25, and 12.5 ng of L63RhoA, N19RhoA, L61Rac1, p120Δarm3, and p120Δrrd (the data presented correspond to those experiments in where 12.5 ng were used). Twenty-four hours after transfection, cells were lysed, and firefly and renilla luciferase values were measured using the Dual-Luciferase reporter assay system (Promega). Normalized firefly luciferase values are reported.
Supplementary Material
Supplemental Data include four figures, two movies, and Supplemental Experimental Procedures and can be found with this article online at http://www.cell.com/cgi/content/full/124/3/631/DC1/.
Acknowledgments
We cite our various colleagues in the text for their generous contribution of Abs. We thank M. Karin (UCSD) for TNFα, IL1β, and IL6 primer sequences and A. Hall (MRC, University College London) for dominant-negative and dominant-active forms of Rho. We thank M. Schober, W. Lowry, and C. Blanpain for discussions and suggestions, P. Romero for performing the skin grafts, and L. Polak and N. Stokes for assistance in the LARC animal facility. E.F. is an Investigator of the Howard Hughes Medical Institute, and M.P.-M. was a postdoctoral fellow supported by the Department of Defense Breast Cancer Research Program, U.S. Army. This work was supported by the National Institutes of Health (grant AR27883 to E.F. and grants CA55724 and P50 CA95103 to A.B.R.). Request p120 mice from al.reynolds@vanderbilt.edu.
References
- Anastasiadis PZ, Moon SY, Thoreson MA, Mariner DJ, Crawford HC, Zheng Y, Reynolds AB. Inhibition of RhoA by p120 catenin. Nat Cell Biol. 2000;2:637–644. doi: 10.1038/35023588. [DOI] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286– 290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- Bienz M. β-Catenin: A pivot between cell adhesion and Wnt signaling. Curr Biol. 2005;15:R64–R67. doi: 10.1016/j.cub.2004.12.058. [DOI] [PubMed] [Google Scholar]
- Chen X, Kojima S, Borisy GG, Green KJ. p120 catenin associates with kinesin and facilitates the transport of cadherin-catenin complexes to intercellular junctions. J Cell Biol. 2003;163:547–557. doi: 10.1083/jcb.200305137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Turksen K, Yu QC, Schreiber H, Teng M, Fuchs E. Cachexia and graft-vs. -host-disease-type skin changes in keratin promoter-driven TNF alpha transgenic mice. Genes Dev. 1992;6:1444–1456. doi: 10.1101/gad.6.8.1444. [DOI] [PubMed] [Google Scholar]
- Ciesiolka M, Delvaeye M, Van Imschoot G, Verschuere V, McCrea P, van Roy F, Vleminckx K. p120 catenin is required for morphogenetic movements involved in the formation of the eyes and the craniofacial skeleton in Xenopus. J Cell Sci. 2004;117:4325–4339. doi: 10.1242/jcs.01298. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt breakers in colon cancer. Cancer Cell. 2004;5:5–6. doi: 10.1016/s1535-6108(03)00339-8. [DOI] [PubMed] [Google Scholar]
- Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, Khavari PA. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- Daniel JM, Reynolds AB. The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol Cell Biol. 1999;19:3614–3623. doi: 10.1128/mcb.19.5.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163:525–534. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Fang X, Ji H, Kim SW, Park JI, Vaught TG, Anastasiadis PZ, Ciesiolka M, McCrea PD. Vertebrate development requires ARVCF and p120 catenins and their interplay with RhoA and Rac. J Cell Biol. 2004;165:87–98. doi: 10.1083/jcb.200307109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz CM, Ridley AJ. p120 catenin associates with microtubules: inverse relationship between microtubule binding and Rho GTPase regulation. J Biol Chem. 2004;279:6588–6594. doi: 10.1074/jbc.M312812200. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Raghavan S. Getting under the skin of epidermal morphogenesis. Nat Rev Genet. 2002;3:199–209. doi: 10.1038/nrg758. [DOI] [PubMed] [Google Scholar]
- Gilmore T. NF-kB. Org website. 2005 http://people.bu.edu/gilmore/nf-kb/diseases.
- Graham TA, Weaver C, Mao F, Kimelman D, Xu W. Crystal structure of a β-catenin/Tcf complex. Cell. 2000;103:885–896. doi: 10.1016/s0092-8674(00)00192-6. [DOI] [PubMed] [Google Scholar]
- Grosheva I, Shtutman M, Elbaum M, Bershadsky AD. p120 catenin affects cell motility via modulation of activity of Rho-family GTPases: a link between cell-cell contact formation and regulation of cell locomotion. J Cell Sci. 2001;114:695–707. doi: 10.1242/jcs.114.4.695. [DOI] [PubMed] [Google Scholar]
- Hatzfeld M. The p120 family of cell adhesion molecules. Eur J Cell Biol. 2005;84:205–214. doi: 10.1016/j.ejcb.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Huber AH, Weis WI. The structure of the β-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by βcatenin. Cell. 2001;105:391–402. doi: 10.1016/s0092-8674(01)00330-0. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- Ireton RC, Davis MA, van Hengel J, Mariner DJ, Barnes K, Thoreson MA, Anastasiadis PZ, Matrisian L, Bundy LM, Sealy L, et al. A novel role for p120 catenin in E-cadherin function. J Cell Biol. 2002;159:465–476. doi: 10.1083/jcb.200205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Kobielak A, Fuchs E. Alpha-catenin: at the junction of intercellular adhesion and actin dynamics. Nat Rev Mol Cell Biol. 2004;5:614– 625. doi: 10.1038/nrm1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magie CR, Pinto-Santini D, Parkhurst SM. Rho1 interacts with p120ctn and alpha-catenin, and regulates cadherin-based adherens junction components in Drosophila. Development. 2002;129:3771–3782. doi: 10.1242/dev.129.16.3771. [DOI] [PubMed] [Google Scholar]
- Mariner DJ, Wang J, Reynolds AB. ARVCF localizes to the nucleus and adherens junction and is mutually exclusive with p120(ctn) in E-cadherin complexes. J Cell Sci. 2000;113:1481–1490. doi: 10.1242/jcs.113.8.1481. [DOI] [PubMed] [Google Scholar]
- Makris C, Roberts JL, Karin M. The carboxyl-terminal region of IκB kinase γ is required for full IKK activation. Mol Cell Biol. 2002;22:6573–6581. doi: 10.1128/MCB.22.18.6573-6581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myster SH, Cavallo R, Anderson CT, Fox DT, Peifer M. Drosophila p120catenin plays a supporting role in cell adhesion but is not an essential adherens junction component. J Cell Biol. 2003;160:433–449. doi: 10.1083/jcb.200211083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren NK, Liu BP, Burridge K, Kreft B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol. 2000;150:567–580. doi: 10.1083/jcb.150.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacquelet A, Lin L, Rorth P. Binding site for p120/deltacatenin is not required for Drosophila E-cadherin function in vivo. J Cell Biol. 2003;160:313–319. doi: 10.1083/jcb.200207160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Park JI, Kim SW, Lyons JP, Ji H, Nguyen TT, Cho K, Barton MC, Deroo T, Vleminckx K, Moon RT, McCrea PD. Kaiso/p120-catenin and TCF/β-catenin complexes coordinately regulate canonical Wnt gene targets. Dev Cell. 2005;8:843–854. doi: 10.1016/j.devcel.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, Krampert M, Goebeler M, Gillitzer R, Israel A, et al. TNF-mediated inflammatory skin disease in mice with epidermis- specific deletion of IKK2. Nature. 2002;417:861–866. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- Paulson AF, Mooney E, Fang X, Ji H, McCrea PD. Xarvcf, Xenopus member of the p120 catenin subfamily associating with cadherin juxtamembrane region. J Biol Chem. 2000;275:30124–30131. doi: 10.1074/jbc.M003048200. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M, Jamora C, Fuchs E. Sticky business: Orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–548. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- Perona R, Montaner S, Saniger L, Sanchez-Perez I, Bravo R, Lacal JC. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- Pettitt J, Cox EA, Broadbent ID, Flett A, Hardin J. The Caenorhabditis elegans p120 catenin homologue, JAC-1, modulates cadherin-catenin function during epidermal morphogenesis. J Cell Biol. 2003;162:15–22. doi: 10.1083/jcb.200212136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AB, Roczniak-Ferguson A. Emerging roles for p120-catenin in cell adhesion and cancer. Oncogene. 2004;23:7947–7956. doi: 10.1038/sj.onc.1208161. [DOI] [PubMed] [Google Scholar]
- Robert C, Kupper TS. Inflammatory skin diseases, T cells, and immune surveillance. N Engl J Med. 1999;341:1817–1828. doi: 10.1056/NEJM199912093412407. [DOI] [PubMed] [Google Scholar]
- Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS., Jr Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- Thoreson MA, Anastasiadis PZ, Daniel JM, Ireton RC, Wheelock MJ, Johnson KR, Hummingbird DK, Reynolds AB. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J Cell Biol. 2000;148:189–202. doi: 10.1083/jcb.148.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaezi A, Bauer C, Vasioukhin V, Fuchs E. Actin cable dynamics and Rho/Rock orchestrate a polarized cytoskeletal architecture in the early steps of assembling a stratified epithelium. Dev Cell. 2002;3:367–381. doi: 10.1016/s1534-5807(02)00259-9. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Degenstein L, Wise B, Fuchs E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci USA. 1999;96:8551–8556. doi: 10.1073/pnas.96.15.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100:209–219. doi: 10.1016/s0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Degenstein L, Wise B, Fuchs E. Hyperproliferation and defects in epithelial polarity upon conditional ablation of α-catenin in skin. Cell. 2001;104:605–617. doi: 10.1016/s0092-8674(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and in- flammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Werner S, Smola H. Paracrine regulation of keratinocyte proliferation and differentiation. Trends Cell Biol. 2001;11:143–146. doi: 10.1016/s0962-8924(01)01955-9. [DOI] [PubMed] [Google Scholar]
- Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, Kowalczyk AP. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol. 2003;163:535–545. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M, Kaverina IN, Wang A, Fujita Y, Reynolds AB, Anastasiadis PZ. A novel interaction between kinesin and p120 modulates p120 localization and function. J Biol Chem. 2004;279:9512–9521. doi: 10.1074/jbc.M310895200. [DOI] [PubMed] [Google Scholar]
- Yap AS, Niessen CM, Gumbiner BM. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J Cell Biol. 1998;141:779–789. doi: 10.1083/jcb.141.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Tao S, Kimmel R, Khavari PA. CDK4 regulation by TNFR1 and JNK is required for NF-kappaB-mediated epidermal growth control. J Cell Biol. 2005;168:561–566. doi: 10.1083/jcb.200411060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include four figures, two movies, and Supplemental Experimental Procedures and can be found with this article online at http://www.cell.com/cgi/content/full/124/3/631/DC1/.