

Abstract
Ca++ signaling is an important component of signal transduction pathways regulating B and T lymphocyte proliferation, but the functional role of Ca++ signaling in regulating myeloid leukemia cell proliferation has been largely unexplored. We observe that the activated (autophosphorylated) Ca++/calmodulin-dependent protein kinase II (CaMKIIγ) is invariably present in myeloid leukemia cell lines as well as in the majority of primary acute myelogenous leukemia (AML) patient samples. In contrast myeloid leukemia cells induced to terminally differentiate or undergo growth arrest display a marked reduction in this CaMKIIγ autophosphorylation. In cells harboring the bcr-abl oncogene, the activation (autophosphorylation) of CaMKIIγ is regulated by this oncogene. Moreover, inhibition of CaMKIIγ activity with pharmacological agents, dominant negative constructs or shRNAs inhibits the proliferation of myeloid leukemia cells, and this is associated with the inactivation/downregulation of multiple critical signal transduction networks involving the MAP kinase, JAK/Stat and GSK3β/β-catenin pathways. In myeloid leukemia cells CaMKIIγ directly phosphorylates Stat3 and enhances its transcriptional activity. Thus CaMKIIγ is a critical regulator of multiple signaling networks regulating the proliferation of myeloid leukemia cells. Inhibiting CaMKIIγ may represent a novel approach in the targeted therapy of myeloid leukemia.
Keywords: myeloid leukemia, Ca++/calmodulin dependent protein kinase II, bcr-abl oncogene, Stat 3
Introduction
The myeloid leukemias including acute myelogenous leukemia (AML) and chronic myelogenous leukemia (CML) are neoplastic disorders characterized by the aberrant proliferation and differentiation of myeloid precursor cells. AML is a remarkably heterogenous disease and is associated with multiple different types of genetic mutations. These mutations are generally of two functional types including those which (1) interfere with the differentiation of myeloid precursors and (2) dysregulate the proliferation and viability of these myeloid precursors (1). For example AML-specific chromosome translocations and mutations involving the RARα (2), AML-1(3) and C/EBPα (4) genes interfere with the differentiation of myeloid precursors while mutations involving tyrosine kinase receptors such as c-kit (5) and flt-3 (6) dysregulate their proliferation and viability. Other specific signal transduction pathways that are aberrantly activated in AML include those involving ras/raf/MAPKinase (7), Stat3 (8), SRC - related kinases (9) and PI3 kinase/Akt(10). Similarly in CML the oncogenic bcr-abl tyrosine kinase that is generated by the Ph chromosome is associated with the activation of multiple downstream signal transduction pathways including Stat5 (11), MAPK (12), PI3 Kinase (13) and SRC-family kinases (14).
The Ca++/calmodulin-dependent protein kinases (CaMKs) are ubiquitously expressed multifunctional Ser/Thr protein kinases that function through Ca++ signaling to regulate the development and activity of many different cell types (15). CaMKs include CaMKI, II and IV, each of which have multiple isoforms. A critical feature shared by members of the CaMK family involves the regulation of their enzymatic activity by changes in intracellular Ca++ concentration. Ca++ binding to the ubiquitously expressed calmodulin triggers a conformational change that enhances its binding to the different CaMKs. This binding of Ca++/calmodulin in turn triggers a conformational change in the CaMKs leading to activation of these enzymes.
The most widely studied of these Ca++ regulated enzymes is CaMKII, which consists of four homologous but distinct genes (CaMKIIα, β, γ, δ). One of the critical functional characteristics of CaMKII is its distinct holoenzyme structure consisting of 12 identical subunits(16). Activation of individual subunits of this dodecameric holoenzyme by Ca++/calmodulin leads to the phosphorylation of adjacent enzyme subunits at threonine286 (for the α isoform) or at threonine287 (for the β, γ, and δ isoforms) (17). This CaMKII autophosphorylation leads to increased enzymatic activity and markedly diminishes the requirement for Ca++/calmodulin binding to maintain enzymatic activity (18). Thus the autophosphorylated CaMKII is autonomous and relatively independent of Ca++ concentration and Ca++/calmodulin regulation.
CaMKII comprises approximately 1-2% of total brain protein, and previous studies have identified an important role of this enzyme in regulating neuronal cell development and activity (19). Other studies using pharmacological inhibitors of the CaMKs have indicated a role for CaMKII in regulating the cell cycle progression of cultured NIH 3T3 fibroblasts (20, 21). In contrast studies determining the role of CaMKII in regulating hematopoiesis have been relatively limited. In T lymphocytes CaMKII is involved in regulating cytokine expression (22) and TCR-triggered cell proliferation (23) as well as the generation of memory T cells (24). In previous studies we have observed that CaMKIIγ is the CaMKII isoform that is preferentially expressed in myeloid cells, and this enzyme inhibits the retinoic acid induced differentiation of myeloid leukemia cell lines by phosphorylating and inhibiting the transcriptional activity of the retinoic acid receptor (RARα) (25). In these previous efforts we observed that CaMKII inhibitors enhance the differentiation of certain myeloid leukemia cell lines, but this effect was only noted in leukemias that were responsive to retinoic acid (25). In the present study we extend these studies beyond the retinoic acid responsive myeloid leukemias and describe a critical and central role of CaMKIIγ in regulating the proliferation of a wide variety of myeloid leukemia cells. We observe that autophosphorylated CaMKIIγ is commonly present in myeloid leukemias. Inhibitors of CaMKIIγ including pharmacological agents and dominant negative constructs inhibit myeloid leukemia cell proliferation, and this is associated with the inactivation of multiple signal transduction pathways including MAPK p44,42, Stat3/Stat5, and GSK 3β/β catenin. Moreover, we observe that Stat3 is directly phosphorylated by CaMKII at Ser727. These observations highlight the importance of CaMKII in regulating the proliferation of myeloid leukemia cells and suggest that inhibiting CaMKIIγ activity may be of significant value in the targeted therapy of myeloid leukemia.
Materials and Methods
Cell lines
All leukemia cell lines are cultured at 37° in RPMI supplemented with 5% heat-inactivated fetal calf serum.
Antibodies and Western blots
Protein extracts, immunoprecipitations and Western blots were performed as previously detailed (26). Antibodies against CaMKI, CaMKIIγ, CaMKIV, c-myc, Stat3, Stat5, calmodulin, as well as the phosphospecific CaMKII (T286, 287) and phosphospecific CaMKI (T177) antibodies were from Santa Cruz Biotechnology. Antibodies against phospho-Stat1 (Ser727), phospho-Stat3 (Ser727 and Tyr 705), phospho-Stat5 (Tyr 694), phospho-p44/42 MAP kinase (Thr202/Tyr 204), p44/42 MAP kinase, phospho-p38 MAP kinase (Thr 180/Tyr 182), phospho-GSK3β (Ser9) were from Cell Signaling Technology. Antibodies against Stat1, GSK3β, β-catenin, PP2Ac, and cyclin D1 were from BD Biosciences.
Chemicals
Different inhibitors of specific signal transduction pathways including AG490, Jak3 inhibitor II, PP1, PD98059, UO 126, SB 203580, wortmannin, LY 294002, JNK inhibitor II, raf kinase inhibitor I, pho kinase inhibitor, RO-31-8220, GÖ 6983,cucurbitacin I, AMP kinase inhibitor compound C, KN62, KN93 and STO609 were all from Calbiochem. All-trans retinoic acid (ATRA), phorbol myristate acetate (TPA), arsenic trioxide and SB216367 were from Sigma.
Patient AML and normal CD34+ samples
Patient AML samples were obtained from the Childrens Oncology Group (COG) bank of AML cells. Ficoll gradient centrifugation was utilized to isolate mononuclear cells from bone marrow aspirates from newly diagnosed patients with AML. These cells consisted of > 80% blasts by morphology and were stored frozen under liquid nitrogen. Normal CD34+ cells were immunopurified from G-CSF mobilized, leukophoresed peripheral blood stem cell donors. Appropriate IRB approval was obtained for utilizing these patient and normal donor samples which are anonymized such that individuals cannot be identified.
GST-fusion proteins
The coding region of the human Stat3α gene was amplified by RT-PCR and subcloned into the GST fusion protein expression vector, pGEX5x.1. The GST-Stat3 fusion protein was purified from bacteria extracts using glutathione-4B Sepharose (Amersham Biosciences).
Expression vectors
A cDNA harboring the coding sequence of amino acids 1-290 of CaMKIIγ was cloned into the LXSN retroviral expression vector (M28248). Utilizing site-directed mutagenesis we mutated Lys43 of this construct to methionine to generate a kinase deficient CaMKII mutant labeled kdCaMKII.
CaMKIIγ cDNA amplification and sequencing
Total cellular RNA from the leukemia cell lines and normal tissue was isolated by Trizol reagent (Invitrogen). First strand cDNA was made by the superscript amplification system (Invitrogen) with the oligo(dT)12-18 primer. To determine the expression of the different CaMKIIγ isoforms we performed RT-PCR utilizing primers flanking the variable region of CaMKIIγ including:
Sense 5′ CGTCAGGAGACTGTGGAGTGT
Antisense 5′ TCACTG CAG CGG TGC GGC AGG
To amplify and sequence the complete CaMKIIγ coding sequence the cDNA from the HL60 and NB4 cell lines was amplified in separate reactions with two different primer pairs flanking the amino terminal and COOH terminal regions of the CaMKIIγ coding sequence. The sequence of these primers as well as the GAPDH control primers are available on request. The PCR products were isolated and subcloned into the TA cloning vector for DNA sequencing.
Transient transfections and luciferase assays
K562 cells were transiently transfected utilizing electroporation. 5×106 cells were electroporated at 500 μF, 250 V, and cultured for 24 h under standard conditions. NIH3T3 cells were transiently transfected utilizing lipofectamine 2000 (Invitrogen). Stat3 transcriptional activity was determined in K562 cells transfected with the pSIE-tk-LUC luciferase reporter which harbors a Stat3 binding site (CCTTAATCCTTCTGGGAATTCTGGCT). The β-gal reporter driven by the β-actin promoter was used as an internal control for transfection efficiency.
CaMKIIγ RNAi constructs
Three LXSN based plasmids harboring three different synthetic human CaMKIIγ oligos designed to generate shRNA hairpins were constructed as previously detailed (25). We synthesized and inserted an additional oligo generating an shRNA corresponding to human CaMIKIIγ (NM_172171) coding region (954-976) into the pLL3.7 lentiviral vector plasmid which harbors a GFP marker (27). These four plasmids were simultaneously electroporated into K562 cells which were selected in G418 (800 ug/ml) for 10 days. GFP+ cells from this culture were then FACS enriched and cell lysates subjected to Western blot analysis.
Construction and utilization of doxycycline inducible expression vectors
A full length CaMKIIγ cDNA was mutated at Lys43 to methionine to render it kinase deficient. It was then FLAG-tagged at the amino terminus and cloned into the pTRE-Tight expression vector which harbors a tet-responsive element (www.clontech.com). This plasmid was electroporated into K562 cells together with the Clontech pTet-On Advanced plasmid, which codes for a doxycycline-regulated transactivator protein. The electroporated cells were selected in G418 (800 ug/ml) for 10 days. Cell lysates for western blot analysis were obtained from these transfected cells as well as from the same cells treated for two days with doxycycline (1 ug/ml).
Results
Activated CaMKIIγ is frequently present in myeloid leukemia cell lines and primary AML cells
We previously observed that CaMKIIγ is the CaMKII isoform that is predominantly expressed in myeloid cells (25). The binding of Ca++/calmodulin to the inactive CaMKIIγ triggers autophosphorylation of this enzyme at threonine 287 leading to autonomous enzyme activity that is relatively independent of further Ca++/calmodulin regulation (17, 28, 29). This autophosphorylated CaMKIIγ can be detected utilizing a CaMKII threonine 287 phospho-specific antibody. Utilizing this antibody on Western blots we observed that the autophosphorylated (activated) CaMKIIγ was invariably present in different human leukemia cell lines (Figure 1A). To further investigate the role of CaMKIIγ in the pathogenesis of human leukemia the present study focuses on the myeloid leukemias, and we observe that the activated, autophosphorylated CaMKIIγ is also present at varying levels in the majority (11/16) of primary patient AML samples (Figure 1B). In contrast Western blots did not reveal any detectable autophosphorylated CaMKIIγ in three different normal peripheral white blood cell samples (not shown).
Figure 1. CaMKIIγ activation in myeloid leukemia cells.
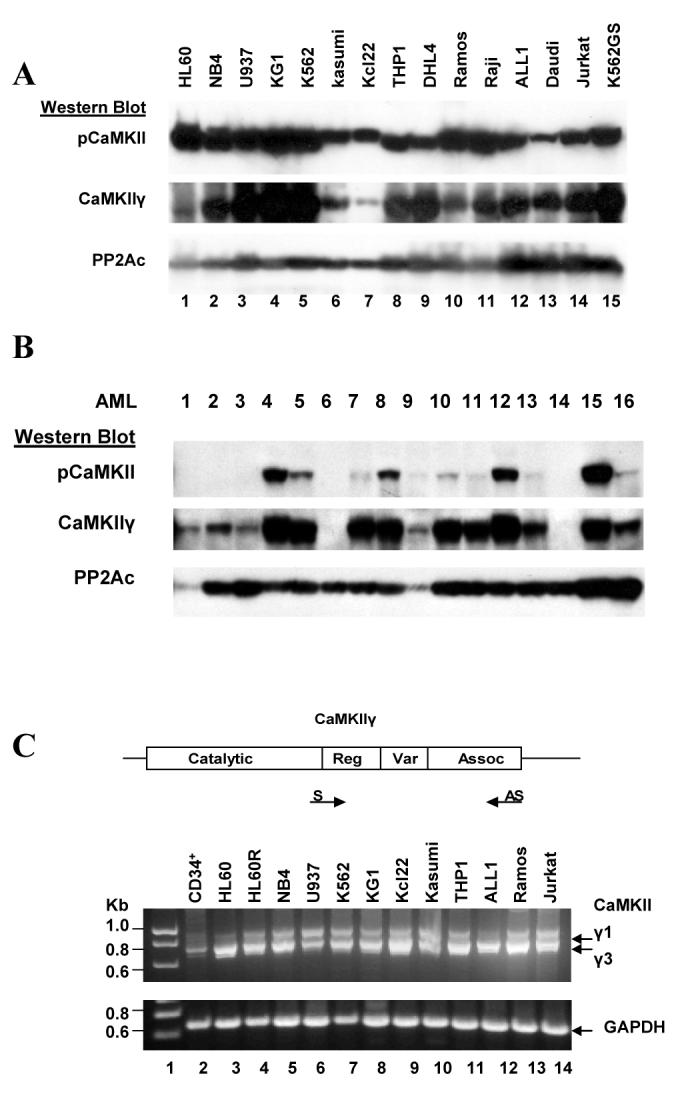
A). Cell lysates from the indicated human leukemia cell lines were subjected to Western blots utilizing the indicated CaMKII antibodies. Antibodies to the catalytic subunit of PP2A (PP2Ac) serve as a loading control.
B). Cell lysates from primary AML samples were subjected to Western blots utilizing the indicated CaMKII antibodies. Antibodies to PP2Ac serve as a loading control.
C) RT-PCR was performed on RNA extracted from the indicated leukemia cell lines and normal CD34+ cells and the products were displayed on an agarose gel. The location of the sense (S) and antisense (AS) primers in relationship to the catalytic, regulatory, variable and association domains of the human CaMKIIγ coding sequence are indicated.
Since alternative splicing within a variable region of CaMKIIγ generates a number of different isoforms (30), we determined whether the myeloid leukemia cells exhibiting CaMKIIγ autophosphorylation expressed any unusual CaMKIIγ isoform(s). An RT-PCR analysis utilizing primers flanking this alternatively spliced CaMKIIγ variable region (Figure 1C) indicated that in normal CD34+ cells as well as in leukemia cells the previously described CaMKIIγ1 and γ3 isoforms were predominantly and commonly expressed (Fig. 1C). In addition we determined whether the activation (autophosphorylation) of CaMKIIγ observed in the myeloid leukemia cell lines was associated with any particular mutations within the CaMKIIγ coding region in these cells. We PCR - amplified and sequenced the entire CaMKIIγ coding region cDNA from the HL60 and NB4 myeloid cell lines. The CaMKIIγ cDNA coding sequence from both these cell lines matched the GenBank CaMKIIγ1 and γ3 sequences. Thus the CaMKIIγ activation exhibited by the myeloid leukemia cells is not associated with the expression of any aberrantly spliced CaMKIIγ isoform(s) nor with any particular CaMKIIγ coding region mutation(s).
Terminal differentiation/apoptosis of myeloid leukemia cell lines is associated with decreased CaMKIIγ activation
All-trans retinoic acid (ATRA) induces the terminal granulocytic differentiation of certain myeloid leukemia cell lines including HL60 (31) and NB4 (32), and ATRA also induces terminal monocytic differentiation of the U937 cell line (33). We utilized Western blots to compare CaMKIIγ expression in these cells at different times after this ATRA-induced terminal differentiation. We observed in all three of these cell lines that ATRA-induced terminal differentiation was associated with a marked decrease in the levels of activated CaMKIIγ, while total CaMKIIγ levels remain essentially unchanged (Fig. 2A). This decrease in activated CaMKIIγ was most prominent 3-5 days post differentiation induction which, as previously observed (31, 32), is the time that these cells undergo terminal differentiation and cease to proliferate. This ATRA-induced decrease in activated CaMKIIγ was not associated with any decrease in cellular calmodulin levels (not shown). This decreased CaMKIIγ activation was ATRA-dose dependent (Fig. 2B, rows 1-3) and appeared to closely correlate with differentiation induction since a similar decrease was not observed in ATRA-treated HL60R cells, which harbor an inactivating point mutation in RARα that renders them unresponsive to ATRA - induced granulocytic differentiation (34) (Figure 2B, row 4). Similarly other myeloid leukemia cell lines that are insensitive to ATRA-induced differentiation including K562, KG1, THP1, and KCL22 did not exhibit any decrease in activated CaMKIIγ expression following ATRA induction (Figure 2B, rows 5-8). A similar marked decrease in activated CaMKIIγ was also observed in HL60 cells treated with phorbol myristate acetate (TPA), which induces terminal macrophage-like differentiation of these cells (35) (Figure 2C). Similarly we observe that the terminal differentiation/apoptosis induced by arsenic trioxide in promyelocytic leukemia cell lines such as NB4 (36) is accompanied by a downregulation in phosphorylated CaMKII (Figure 2D). Thus the terminal differentiation of myeloid leukemia cells is associated with a marked downregulation of the autophosphorylated CaMKIIγ.
Figure 2. Terminal differentiation/apoptosis of myeloid leukemia cells is associated with decreased CaMKIIγ activation.

A). Lysates from the HL60, NB4 and U937 myeloid cell lines treated with ATRA (1μM) for the indicated period of time were subjected to Western blots.
B). Lysates from the indicated myeloid cell lines treated for five days with the indicated concentration of ATRA were subjected to Western blots.
C). Lysates from HL60 cells treated with TPA (0.1 μM) for the indicated time were subjected to Western blots.
D). Lysates from NB4 cells treated with arsenic trioxide (1 uM) for the indicated time were subjected to Western blots.
Oncogenic bcr-abl regulates CaMKIIγ activation
In contrast to its lack of response to ATRA, the K562 cell line, derived from a patient with CML, undergoes proliferative arrest following exposure to imatinib (gleevec), which is a potent inhibitor of the tyrosine kinase activity of the bcr-abl oncogene. We observed that this imatinib -induced proliferation arrest is accompanied by a rapid, marked decrease in autophosphorylated CaMKIIγ, while there was no significant change in the total levels of CaMKIIγ following this imatinib exposure (Figure 3A).
Figure 3. Inhibiting bcr-abl activity/expression inhibits CaMKIIγ activation.

A). Lysates from K562 cells undergoing growth arrest following treatment with gleevec (5uM) for the indicated times were subjected to Western blots.
B). Lysates from TonB210.1 cells depleted of doxycycline for the indicated period of time were subjected to Western blots.
C). IL-3 dependent TonB210.1 cells were deprived of IL-3 for 24 hours (lane 1). The cells were then treated with the indicated compounds simultaneously with the addition of doxycycline (lanes 2-8). After 24 hours cell lysates were harvested and subjected to Western blots with the indicated CaMKII antibodies.
D). K562 cells were cultured for 48 hours with the indicated chemical inhibitors of specific signal transduction pathways. Cell lysates were harvested and subjected to Western blot analysis.
The marked downregulation of CaMKII activation (autophosphorylation) by the bcr-abl inhibitor, imatinib observed in K562 cells suggests that the activation of CaMKIIγ in these CML cells requires bcr-abl activity. To confirm this we utilized the TonB210.1 cell line, which is a Baf3 - derived line whose proliferation/viability is dependent upon either IL-3 alone or the tet-regulated expression of bcr-abl (tet-on) (37). We observe in the TonB210.1 cells that the decreased bcr-abl expression associated with tet withdrawal is also accompanied by a marked and relatively rapid (within 4 hours) reduction in the levels of activated (autophosphorylated) CaMKII, while total CaMKIIγ levels remain unchanged (Figure 3B). To determine if enhanced bcr-abl expression can initiate CaMKII activation, we first deprived the IL-3 dependent TonB210.1 cells of IL-3 for 24 hours, which induced cell proliferative arrest associated with decreased autophosphorylated CaMKIIγ (Fig. 3C, lane 1). Treatment of these cells with tet restores CaMKII autophosphorylation (Fig. 3C, compare lanes 1 and 2), and this phosphorylation was inhibited in a dose dependent manner by the CaMK chemical inhibitors KN62 and KN93 (Fig. 3C, lanes 3-6), which interfere with the binding of the Ca++/calmodulin complex to the CaMKs (38). In contrast this bcr-abl mediated enhancement of CaMKIIγ phosphorylation was not inhibited by the CaM kinase kinase (CaMKK) chemical inhibitor STO-609 (Fig. 3C, lanes 7-8). Taken together these observations with the K562 and TonB210.1 cells indicate that oncogenic bcr-abl directly or indirectly activates CaMKIIγ and that this bcr-abl induced CaMKIIγ activation is dependent on Ca++/calmodulin binding to CaMKIIγ.
Effect of inhibitors of different signal transduction pathways on CaMKIIγ activation
The above observations indicate that CaMKII activation is downstream of bcr-abl, but we don’t know whether this oncogene directly or indirectly (through one or more of its downstream effectors) mediates this activation. The molecular basis for bcr-abl mediated cell transformation is complex and involves a number of specific downstream signal transduction pathways activated by bcr-abl including Stat5 (11), Ras/Raf1/MAPK (12), PI3 Kinase (13) and SRC-family kinases (14). Indeed the cooperative activation of these pathways may be necessary for full bcr-abl mediated transformation (39). We utilized specific chemical inhibitors to determine whether any of these signal transduction pathways downstream of bcr-abl might be involved in the bcr-abl mediated activation of CaMKIIγ in K562 cells. In contrast with the gleevec induced downregulation of CaMKIIγ activation (Figure 3D, lane 17), none of these chemical inhibitors reduced CaMKIIγ activation including those which target Jak/Stat (Figure 3D, lanes 3,4,16), PI3 kinase (lanes 9,10), p38 (lane 8), JNK, (lane 11), Ras/Raf/MEK/ERK (lanes 6,7,12), PKC (lanes 14,15), AMP kinase (lane 18) and SRC-family kinases (lane 5). Thus the bcr-abl mediated activation of CaMKIIγ does not appear to involve any of the signal transduction pathways that are known downstream effectors of bcr-abl.
Inhibitors of CaMKII inhibit the proliferation of myeloid leukemia cell lines
To determine whether inhibiting CaMKIIγ activity might have any effect on myeloid leukemia cell proliferation/viability, we treated K562 cells with the CaMK inhibitor KN93. We found that KN93 inhibited the proliferation of K562 cells in a dose dependent manner (Fig. 4A, i). In contrast STO609, a potent small molecule inhibitor of CaMKinase Kinase (CaMKK)(40) did not significantly inhibit K562 proliferation nor did KN92, an inactive structural analog of KN62 (Fig. 4A, ii). The KN93 induced inhibition of K562 cell proliferation was accompanied by a reduction in the levels of the activated (autophosphorylated) CaMKIIγ (Fig. 5A, row 1). KN93 did not affect K562 viability, and no effect on the morphologic differentiation of these cells was observed. A similar KN93-mediated inhibition of cell proliferation without any significant effect on cell viability or morphologic differentiation was also noted in other cultured myeloid leukemia cell lines including KG-1, KCL22, THP1, and Kasumi (Fig. 4B).
Figure 4. CaMKII inhibitors inhibit myeloid leukemia cell proliferation.

A). K562 cells were seeded in liquid suspension culture at 5 × 104 cells/ml in the presence or absence of the indicated concentration of compounds, and cell counts were obtained at the indicated time.
B). The indicated myeloid leukemia cell lines were seeded in liquid suspension culture at 5 × 104 cells/ml in the presence or absence of KN93, and cell counts obtained at the indicated time.
C). K562 cells were electroporated with the LXSN expression vector harboring the kinase dead, Lys43 mutated, truncated CaMKII construct (kdCaMKII) as well as with the control (empty) LXSN vector. The electroporated cells were diluted into 96 well plates in liquid suspension culture in the presence of G418 (1 mg/ml). Following eight days of culture the total number of discrete, actively proliferating G418 resistant colonies within individual wells was determined.
Figure 5. CaMKII regulates multiple signal transduction pathways in myeloid cells.

(A,B). Lysates from K562 cells cultured for the indicated time in KN93 were subjected to Western blots. (C) The K562 cells transduced as described in Materials and Methods with either (i) the doxycycline (DOX) inducible expression vector/transactivator harboring the full length, Lys43 mutated, kinase dead CaMKIIγ (kdCaMKIIγ (Tet-on)) or (ii) the CaMKII shRNA generating plasmids, were cultured in liquid suspension in the presence or absence of DOX as indicated, and cell counts obtained after 48 hours. (D) Lysates from K562 cells transduced with the indicated vectors were subjected to Western blots utilizing the indicated antibodies. For lanes 2 and 4 doxycycline (DOX) treatment was for two days.
To further assess the role of CaMKIIγ in regulating myeloid leukemia cell proliferation, we transiently transfected K562 cells with an expression vector harboring a truncated CaMKIIγ construct mutated at Lys43, a conserved residue near the ATP-binding site of this enzyme. This mutation results in a ‘kinase dead’ enzyme (41), and expression of this mutant inhibits endogenous CaMKII activity in a dominant negative manner (42). We observed a marked reduction in K562 colony formation in cells transfected with this mutated CaMKIIγ compared with cells transfected with vector alone (Fig. 4C). Thus both pharmacological and dominant negative construct inhibition of CaMKII results in inhibition of myeloid leukemia cell proliferation.
Pharmacologic, dominant negative or shRNA-mediated inhibition of CaMKIIγ is associated with the downregulation of multiple signal transduction pathways
We utilized phosphorylation specific antibodies to determine whether the KN93-induced inhibition of K562 cell proliferation (Fig. 4A, i) was associated with any changes in specific phosphoprotein signal transduction pathways. As noted above KN93 treatment of K562 cells is associated with a time dependent reduction in CaMKIIγ phosphorylation (Figure 5A, row 1). This reduction was associated with a decrease in the activation (phosphorylation) of a number of different critical components of signal transduction pathways including pMAPK p44/42 (Figure 5A, row 3), p38 (row 5), pStat3 (Y705, S727) (rows 10,11) and pStat5 (Y694) (row 13). These changes induced by KN93 were not accompanied by any change in the total levels of these phosphorylated proteins (Figs. 5A). These observations indicate that in K562 myeloid cells activated CaMKIIγ is involved in regulating the activation (phosphorylation) of MAPK p44/42, Stat3/Stat5, and p38.
We also observe that the KN93 induced downregulation of CaMKII activation is associated with a marked reduction in GSK3β Ser9 phosphorylation (Fig. 5A, row 6). This is of particular interest since GSK3β, a serine/threonine kinase, phosphorylates and promotes the degradation of β-catenin, a key component of the Wnt signaling pathway, which is frequently activated in myeloid leukemia (43, 44). Since phosphorylation of GSK3β at Ser9 inhibits the activity of this enzyme (45), we would predict that the KN93 induced reduction in GSK3β Ser9 phosphorylation would lead to enhanced GSK3β enzyme activity associated with enhanced degradation of β-catenin. Indeed a marked reduction in the expression of β-catenin is observed in the KN93 treated K562 cells (Fig. 5B, row 3). Consistent with this, in the KN93 induced cells we also observe a reduction of c-myc and cyclin D1 protein levels, both of whose expression is normally upregulated by β catenin (46, 47) (Fig. 5B, rows 4,5).
KN93 inhibits CaMKII activity by blocking its interaction with the Ca++/calmodulin complex. However, this compound can potentially inhibit other enzymes activated by Ca++/calmodulin including CaMKI, CaMKIV, CKLiK as well as the CaMK-kinases (CaMKKs). Nevertheless, several lines of evidence indicate that the effects of KN93 are mediated through CaMKII inhibition rather than through inhibiting any of these other CaMkinases. First, CaMKIV is not expressed in K562 cells (Supplemental Fig. 1A), and the CaMKI expressed by K562 is primarily in the inactive (unphosphorylated) form (Supplemental Fig. 1B). Second, there is little if any CKLiK (CaMKIδ) expression in K562 cells compared with other cell types (Supplemental Fig. 1C). Moreover, STO609, a potent small molecule inhibitor of the CaMKKs, exhibits minimal effects on K562 proliferation (Fig. 4A, ii) indicating that neither the CaMKKs nor their downstream substrate, CaMKI are the targets of KN93. In addition we transduced a FLAG-tagged Lys43 mutated, dominant negative CaMKIIγ under control of a doxycycline inducible promoter (designated kdCaMKIIγ (Tet-on)) into K562 cells. Treatment of these cells with doxycycline upregulated expression of this construct (Fig. 5D, row 1, lane 4), and this induced expression was temporally associated with inhibition of K562 proliferation (Fig. 5C, i) together with downregulation of the activation (phosphorylation) of the MAPkinase, Stat3/Stat5 and β-catenin pathways (Fig. 5D, compare lanes 3 vs.4). Finally we utilized an siRNA approach to knockdown the expression of CaMKIIγ in K562 cells. The shRNA transduced K562 cells with reduced expression of CaMKIIγ (Figure 5D, row 2, compare lanes 5 vs. 6) exhibited reduced cell proliferation (Fig.5C, ii), and this is associated with downregulation of the MAPkinase, Jak/Stat and GSK3β/β -catenin pathways (Figure 5D, compare lanes 5 and 6). Thus the effects of KN93 on K562 cells are likely mediated through its inhibitory activity on CaMKIIγ rather than through any of the other CaMkinases.
CaMKIIγ directly phosphorylates and activates Stat3 in myeloid leukemia cells
In addition to tyrosine phosphorylation of Stat3 at Y705, Stat3 Ser727 phosphorylation is required for full Stat3 transcriptional activity (48). The activated Stat3 has oncogenic activity (49), and Stat 3 activation is frequently observed in myeloid leukemia cell lines (Fig. 6A, i) as well as myeloid leukemia patient samples (8) (Fig. 6A, ii). Our observation that the CaMKII inhibitor KN93 induces a decrease in Stat3 Ser727 phosphorylation (Fig. 5A, row 11) suggests that Stat3 may be a direct substrate of CaMKIIγ in myeloid leukemia cells. Consistent with this we observe that 9/11 of the primary AML samples that exhibit CaMKIIγ activation (Fig. 1B) also exhibit Stat3 Ser727 phosphorylation, while none of the five primary AML samples that were negative for CaMKIIγ activation exhibit Stat3 Ser 727 phosphorylation (Fig. 6A, ii). In addition we observe in the TonB210 cells that the tet-induced bcr-abl induction is associated with a marked increase in Stat3 Ser727 phosphorylation without any change in total Stat3 levels (Fig. 6B, compare lanes 1 vs 2). Moreover, this enhanced Stat3 Ser727 phosphorylation is blocked by KN62 or KN93 (Fig. 6B, lanes 3, 4) indicating that a CaM kinase is involved in this bcr-abl induced Stat3 Ser727 phosphorylation. We also observe that cucurbitacin I, a small molecule Stat3 inhibitor, will, as expected, inhibit Stat3 responsive reporter activity, and this reporter activity is also inhibited by KN93 in a dose dependent manner (Fig. 6C). These latter observations indicate a functional interaction between Stat3 and CaMKII in myeloid leukemia cells.
Figure 6. Stat3 is directly phosphorylated and activated by CaMKIIγ at Ser727.

A, i). Cell lysates from the indicated human leukemia cell lines were subjected to Western blots utilizing the indicated Stat3 antibodies. ii). Cell lysates from the same primary AML samples depicted in Fig. 1B were subjected to Western blots utilizing the indicated Stat3 antibodies.
B). The IL-3 dependent TonB210.1 cells were deprived of IL-3 for 24 hours. The cells were then treated with the indicated compounds immediately prior to the addition of doxycycline. After an additional 24 hours cell lysates were harvested and subjected to Western blots with the indicated Stat3 antibodies.
C). K562 cells were electroporated with a luciferase reporter driven by a Stat3 response element. The indicated concentrations of KN93 and the Stat 3 inhibitor, cucurbitacin I, were added and relative luciferase activity determined on cell lysates following 48 hours of culture.
D, i) K562 cell lysates were immunoprecipitated with control IgG or a Stat3 antibody. The immunoprecipitates were then subjected to Western blot analysis with the indicated antibodies.
ii, ) A bacterial - expressed GST-Stat3 fusion protein was incubated in vitro with CaMKIIγ that had been immunoprecipitated from HL60 cells. Western blots were then performed on the reaction mixtures utilizing the indicated Stat3 antibodies.
iii,). NIH3T3 cells were transfected with the empty LXSN expression vector (lane 1), the same vector harboring the coding sequences of a constitutively active (ca) CaMKIIγ (lane 2), or vector harboring the kinase dead (kd) CaMKIIγ (lane 3). After 48 hours Western blots were performed on cell lysates utilizing the indicated Stat3 antibodies.
To further assess the CaMKIIγ/Stat3 interaction we observed that in K562 cells Stat3 co-immunoprecipitates with CaMKIIγ (Fig. 6D, i). Moreover, CaMKIIγ phosphorylates Stat3 at Ser727 both in vitro (Fig. 6D, ii) and in vivo (Fig. 6D, iii). Together these observations suggest that Stat3 is likely a direct substrate of CaMKIIγ in myeloid leukemia cells and that the KN93 induced inhibition of K562 cell proliferation (Fig. 4A) may be mediated, at least in part, through its inhibition of Stat3 Ser727 phosphorylation.
Discussion
The CaM kinases (CaMKs) are major targets of Ca++ regulated physiological events, and the role of a particular CaMK, (i.e. CaMKII), in regulating both neuronal and cardiac muscle cell development and activity has been well established. Our present study defines a novel and critical role for the activated (autophosphorylated) CaMKII (CaMKIIγ) in regulating the proliferation of myeloid leukemia cells. Indeed we observe that relatively high amounts of the autophosphorylated CaMKIIγ are invariably present in myeloid leukemia cell lines, and this activation is also observed in the majority of primary patient AML samples. In different in vitro models of myeloid leukemia cell terminal differentiation/growth arrest including those triggered by ATRA, TPA, arsenic trioxide or gleevec, the loss of proliferation of the induced leukemia cells is accompanied by a marked reduction in CaMKIIγ activation. This marked downregulation of CaMKIIγ activation likely directly contributes to the loss of leukemia cell proliferative capacity since we observe that both pharmacologic, dominant negative and shRNA-mediated inhibition of CaMKII expression/activity inhibits myeloid leukemia cell proliferation. Moreover, we observe that CaMKIIγ directly or indirectly regulates multiple signaling pathways previously implicated in leukemia cell proliferation including the MAPKinase, Jak/Stat and GSK3β/β-catenin pathways.
What are the molecular events that trigger and maintain CaMKIIγ constitutive activation (autophosphorylation) in the myeloid leukemia cell lines and primary patient AML samples? The myeloid leukemias are remarkably heterogenous, and diverse molecular events may trigger CaMKIIγ activation in these leukemias. Our observations in cell lines driven by oncogenic bcr-abl indicate that CaMKII activation is downstream of bcr-abl but is not regulated by other bcr-abl activated signal transduction events involving the MAP kinase, Jak/Stat, SRC - related kinase or PI3 kinase pathways (Fig. 3D). The primary trigger of CaMKII activation is its binding to Ca++/calmodulin, whose levels are regulated by intracellular Ca++ concentration. Our observations in TonB210.1 cells suggest that the bcr-abl mediated CaMKII activation is dependent, at least in part, upon Ca++/calmodulin binding to CaMKII since this bcr-abl induced activation is inhibited by KN62 or KN93, which block Ca++/calmodulin binding to CaMKII (Fig. 3C). This suggests that bcr-abl might regulate intracellular Ca++/calmodulin levels, and this would likely involve the regulation by this oncogene of intracellular Ca++ concentration. Nevertheless how bcr-abl might regulate intracellular Ca++ levels and Ca++/calmodulin activity is currently unknown.
Closely related to the question of how CaMKIIγ is activated in myeloid leukemia cells is how this enzyme becomes inactivated (dephosphorylated) during terminal myeloid differentiation/growth arrest (Fig. 2). Indeed we observe a prominent decrease in CaMKIIγ activation during ATRA-induced terminal granulocytic differentiation of myeloid leukemia cells and also note that normal granulocytes harbor relatively low levels of the activated CaMKIIγ. This terminal myeloid differentiation is not associated with any decrease in total cell calmodulin concentration to account for the reduced CaMKIIγ activity. One possibility to explain this downregulation of CaMKIIγ autophosphorylation is that myeloid differentiation is accompanied by changes in Ca++ channel activity with an associated decrease in intracellular Ca++ concentration that reduces the Ca++/calmodulin activation of CaMKII. Another possibility is that an increase in phosphatase activity that targets the activated (autophosphorylated) CaMKIIγ accompanies myeloid differentiation. Consistent with this latter possibility we have observed that PP2A, a phosphatase known to dephosphorylate CaMKII in neuronal cells (50), coimmunoprecipitates with CaMKIIγ in myeloid cells (not shown). However the levels of PP2A do not appear to change during myeloid leukemia cell differentiation (not shown), and whether the phosphatase activity that specifically targets the activated (autophosphorylated) CaMKIIγ is developmentally regulated during myeloid differentiation is presently unknown.
What are the critical substrates of CaMKIIγ that are involved in regulating myeloid leukemia cell proliferation? In HeLa cells CaMKII phosphorylation of the cdc25 phosphatase is associated with cdc2 activation and enhanced G2/M transition (51). Moreover, the localization of CaMKII to the mammalian midbody also suggests a role for this enzyme in regulating cytokinesis (52). We have previously observed that CaMKIIγ directly phosphorylates and inhibits the transcriptional activity of RARα, which is an important regulator of the terminal differentiation of certain myeloid leukemia cells (25). Our present study indicates that Stat3, which exhibits oncogenic activity (49), is directly phosphorylated by CaMKIIγ, and this phosphorylation enhances Stat3 transcriptional activity (Fig. 6). We also observe that CaMKIIγ directly or indirectly promotes the inhibitory phosphorylation of GSK3β, an enzyme which normally downregulates the expression of cell growth/proliferation factors including β-catenin, c-myc and cyclinD1 (Fig. 5B). Thus CaMKII activity appears to orchestrate a complex, interacting network of molecular events that are involved in regulating the proliferation of myeloid leukemia cells.
Our observation that CaMKIIγ is an important regulator of multiple phosphoprotein signaling networks regulating myeloid leukemia cell proliferation suggests that targeting this enzyme might be of therapeutic benefit in human myeloid leukemia. Indeed we reproducibly observe an inhibition of myeloid leukemia cell proliferation triggered by KN93, a small molecule inhibitor of CaMKII. However, the CaMKII pharmacological inhibitors have significant limitations. These compounds inhibit CaMKII activity by interfering with their binding to the Ca++/calmodulin complex. However, once CaMKII has become activated (autophosphorylated) its dependence upon further Ca++/calmodulin binding is markedly diminished (18). Thus these compounds exert considerably less inhibition on CaMKII once it has become activated (autophosphorylated). An “ideal” CaMKIIγ inhibitor would be a small molecule which would directly inhibit the activated CaMKIIγ by directly targeting its ATP binding pocket. Such a CaMKIIγ inhibitor has not been identified, but we would predict that it would exert much greater activity in inhibiting leukemia cell proliferation than KN93 and might be of significant value in the targeted therapy of myeloid leukemia.
Supplementary Material
Acknowledgments
We thank LeMoyne Mueller for excellent technical assistance. J.S. designed, performed and interpreted the experiments and helped write the manuscript. S.C. designed and interpreted the experiments and helped write the manuscript. The authors have no conflicting financial interests.
REFERENCES
- 1.Gilliland DG. Molecular genetics of human leukemias: new insights into therapy. Semin Hematol. 2002;39:6–11. doi: 10.1053/shem.2002.36921. [DOI] [PubMed] [Google Scholar]
- 2.Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999;93:3167–215. [PubMed] [Google Scholar]
- 3.Meyers S, Lenny N, Hiebert SW. The t(8;21) fusion protein interferes with AML-1B-dependent transcriptional activation. Mol Cell Biol. 1995;15:1974–82. doi: 10.1128/mcb.15.4.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pabst T, Mueller BU, Zhang P, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–70. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- 5.Beghini A, Ripamonti CB, Cairoli R, et al. KIT activating mutations: incidence in adult and pediatric acute myeloid leukemia, and identification of an internal tandem duplication. Haematologica. 2004;89:920–5. [PubMed] [Google Scholar]
- 6.Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3:650–65. doi: 10.1038/nrc1169. [DOI] [PubMed] [Google Scholar]
- 7.Radich JP, Kopecky KJ, Willman CL, et al. N-ras mutations in adult de novo acute myelogenous leukemia: prevalence and clinical significance. Blood. 1990;76:801–7. [PubMed] [Google Scholar]
- 8.Steensma DP, McClure RF, Karp JE, et al. JAK2 V617F is a rare finding in de novo acute myeloid leukemia, but STAT3 activation is common and remains unexplained. Leukemia. 2006;20:971–8. doi: 10.1038/sj.leu.2404206. [DOI] [PubMed] [Google Scholar]
- 9.Robinson LJ, Xue J, Corey SJ. Src family tyrosine kinases are activated by Flt3 and are involved in the proliferative effects of leukemia-associated Flt3 mutations. Exp Hematol. 2005;33:469–79. doi: 10.1016/j.exphem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102:972–80. doi: 10.1182/blood-2002-11-3429. [DOI] [PubMed] [Google Scholar]
- 11.de Groot RP, Raaijmakers JA, Lammers JW, Jove R, Koenderman L. STAT5 activation by BCR-Abl contributes to transformation of K562 leukemia cells. Blood. 1999;94:1108–12. [PubMed] [Google Scholar]
- 12.Cortez D, Stoica G, Pierce JH, Pendergast AM. The BCR-ABL tyrosine kinase inhibits apoptosis by activating a Ras-dependent signaling pathway. Oncogene. 1996;13:2589–94. [PubMed] [Google Scholar]
- 13.Skorski T, Bellacosa A, Nieborowska-Skorska M, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. Embo J. 1997;16:6151–61. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Y, Liu Y, Pelletier S, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36:453–61. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- 15.Hook SS, Means AR. Ca(2+)/CaM-dependent kinases: from activation to function. Annu Rev Pharmacol Toxicol. 2001;41:471–505. doi: 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- 16.Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell. 2003;11:1241–51. doi: 10.1016/s1097-2765(03)00171-0. [DOI] [PubMed] [Google Scholar]
- 17.Fong YL, Taylor WL, Means AR, Soderling TR. Studies of the regulatory mechanism of Ca2+/calmodulin-dependent protein kinase II. Mutation of threonine 286 to alanine and aspartate. J Biol Chem. 1989;264:16759–63. [PubMed] [Google Scholar]
- 18.Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 19.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–90. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 20.Tombes RM, Grant S, Westin EH, Krystal G. G1 cell cycle arrest and apoptosis are induced in NIH 3T3 cells by KN-93, an inhibitor of CaMK-II (the multifunctional Ca2+/CaM kinase) Cell Growth Differ. 1995;6:1063–70. [PubMed] [Google Scholar]
- 21.Morris TA, DeLorenzo RJ, Tombes RM. CaMK-II inhibition reduces cyclin D1 levels and enhances the association of p27kip1 with Cdk2 to cause G1 arrest in NIH 3T3 cells. Exp Cell Res. 1998;240:218–27. doi: 10.1006/excr.1997.3925. [DOI] [PubMed] [Google Scholar]
- 22.Nghiem P, Ollick T, Gardner P, Schulman H. Interleukin-2 transcriptional block by multifunctional Ca2+/calmodulin kinase. Nature. 1994;371:347–50. doi: 10.1038/371347a0. [DOI] [PubMed] [Google Scholar]
- 23.Lin MY, Zal T, Ch’en IL, Gascoigne NR, Hedrick SM. A pivotal role for the multifunctional calcium/calmodulin-dependent protein kinase II in T cells: from activation to unresponsiveness. J Immunol. 2005;174:5583–92. doi: 10.4049/jimmunol.174.9.5583. [DOI] [PubMed] [Google Scholar]
- 24.Bui JD, Calbo S, Hayden-Martinez K, Kane LP, Gardner P, Hedrick SM. A role for CaMKII in T cell memory. Cell. 2000;100:457–67. doi: 10.1016/s0092-8674(00)80681-9. [DOI] [PubMed] [Google Scholar]
- 25.Si J, Mueller L, Collins SJ. CaMKII regulates retinoic acid receptor transcriptional activity and the differentiation of myeloid leukemia cells. J Clin Invest. 2007;117:1412–21. doi: 10.1172/JCI30779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Si J, Collins SJ. IL-3-induced enhancement of retinoic acid receptor activity is mediated through Stat5, which physically associates with retinoic acid receptors in an IL-3-dependent manner. Blood. 2002;100:4401–9. doi: 10.1182/blood-2001-12-0374. [DOI] [PubMed] [Google Scholar]
- 27.Rubinson DA, Dillon CP, Kwiatkowski AV, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–6. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 28.Miller SG, Patton BL, Kennedy MB. Sequences of autophosphorylation sites in neuronal type II CaM kinase that control Ca2(+)-independent activity. Neuron. 1988;1:593–604. doi: 10.1016/0896-6273(88)90109-2. [DOI] [PubMed] [Google Scholar]
- 29.Lou LL, Schulman H. Distinct autophosphorylation sites sequentially produce autonomy and inhibition of the multifunctional Ca2+/calmodulin-dependent protein kinase. J Neurosci. 1989;9:2020–32. doi: 10.1523/JNEUROSCI.09-06-02020.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tombes RM, Faison MO, Turbeville JM. Organization and evolution of multifunctional Ca(2+)/CaM-dependent protein kinase genes. Gene. 2003;322:17–31. doi: 10.1016/j.gene.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 31.Breitman T, Selonick S, Collins S. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci USA. 1980;77:2936–2940. doi: 10.1073/pnas.77.5.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lanotte M, Martin-Thouvenin V, Najman S, Balerini P, Valensi F, Berger R. NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3) Blood. 1991;77:1080–6. [PubMed] [Google Scholar]
- 33.Olsson IL, Breitman TR. Induction of differentiation of the human histiocytic lymphoma cell line U-937 by retinoic acid and cyclic adenosine 3′:5′-monophosphate-inducing agents. Cancer Research. 1982;42:3924–3927. [PubMed] [Google Scholar]
- 34.Robertson K, Emami B, Collins S. Retinoic acid-resistant HL-60R cells harbor a point mutation in the RA receptor ligand binding domain that confers dominant negative activity. Blood. 1992;80:1885–1889. [PubMed] [Google Scholar]
- 35.Rovera G, Santoli D, Damsky C. Human promyelocytic leukemia cells in culture differentiate into macrophage-like cells when treated with a phorbol diester. Proc Natl Acad Sci U S A. 1979;76:2779–83. doi: 10.1073/pnas.76.6.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen GQ, Zhu J, Shi XG, et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood. 1996;88:1052–61. [PubMed] [Google Scholar]
- 37.Klucher KM, Lopez DV, Daley GQ. Secondary mutation maintains the transformed state in BaF3 cells with inducible BCR/ABL expression. Blood. 1998;91:3927–34. [PubMed] [Google Scholar]
- 38.Sumi M, Kiuchi K, Ishikawa T, et al. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. 1991;181:968–75. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- 39.Sonoyama J, Matsumura I, Ezoe S, et al. Functional cooperation among Ras, STAT5, and phosphatidylinositol 3-kinase is required for full oncogenic activities of BCR/ABL in K562 cells. J Biol Chem. 2002;277:8076–82. doi: 10.1074/jbc.M111501200. [DOI] [PubMed] [Google Scholar]
- 40.Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem. 2002;277:15813–8. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]
- 41.Hanson PI, Meyer T, Stryer L, Schulman H. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron. 1994;12:943–56. doi: 10.1016/0896-6273(94)90306-9. [DOI] [PubMed] [Google Scholar]
- 42.Kuhl M, Sheldahl LC, Malbon CC, Moon RT. Ca(2+)/calmodulin-dependent protein kinase II is stimulated by Wnt and Frizzled homologs and promotes ventral cell fates in Xenopus. J Biol Chem. 2000;275:12701–11. doi: 10.1074/jbc.275.17.12701. [DOI] [PubMed] [Google Scholar]
- 43.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–76. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 44.Simon M, Grandage VL, Linch DC, Khwaja A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene. 2005;24:2410–20. doi: 10.1038/sj.onc.1208431. [DOI] [PubMed] [Google Scholar]
- 45.Eldar-Finkelman H, Argast GM, Foord O, Fischer EH, Krebs EG. Expression and characterization of glycogen synthase kinase-3 mutants and their effect on glycogen synthase activity in intact cells. Proc Natl Acad Sci U S A. 1996;93:10228–33. doi: 10.1073/pnas.93.19.10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 47.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 48.Shen Y, Schlessinger K, Zhu X, et al. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol. 2004;24:407–19. doi: 10.1128/MCB.24.1.407-419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 50.Strack S, Barban MA, Wadzinski BE, Colbran RJ. Differential inactivation of postsynaptic density-associated and soluble Ca2+/calmodulin-dependent protein kinase II by protein phosphatases 1 and 2A. J Neurochem. 1997;68:2119–28. doi: 10.1046/j.1471-4159.1997.68052119.x. [DOI] [PubMed] [Google Scholar]
- 51.Patel R, Holt M, Philipova R, et al. Calcium/calmodulin-dependent phosphorylation and activation of human Cdc25-C at the G2/M phase transition in HeLa cells. J Biol Chem. 1999;274:7958–68. doi: 10.1074/jbc.274.12.7958. [DOI] [PubMed] [Google Scholar]
- 52.Skop AR, Liu H, Yates J, 3rd, Meyer BJ, Heald R. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science. 2004;305:61–6. doi: 10.1126/science.1097931. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.