

Abstract
The publication of the crystal structure of the β2-adrenergic receptor (β2-AR) proved that G protein-coupled receptors (GPCRs) share a structurally conserved rhodopsin-like 7TM core. Here, to probe to which extent realistic GPCR structures can be recreated through modeling, carazolol was docked at two rhodopsin-based homology models of the human β2-AR. The first featured a rhodopsin-like second extracellular loop, which interfered with ligand docking and with the orientation of several residues in the binding pocket. The second featured a second extracellular loop built completely de novo, which afforded a more accurate model of the binding pocket and a better docking of the ligand. Furthermore, incorporating available biochemical and computational data to the model by correcting the conformation of a single residue lining the binding pocket – Phe290(6.52) – resulted in significantly improved docking poses. These results support the applicability of GPCR modeling to the design of site-directed mutagenesis experiments and to drug discovery.
Introduction
For many years rhodopsin has been the only G protein-coupled receptor (GPCR) with crystallographic structural information available.1–4 In this context, structural studies of other G protein coupled receptors heavily relied on mutagenesis experiments combined with sequence comparison and homology modeling.5–20 The latter technique is based on the general assumption that evolutionary related proteins – i.e. homologous proteins - conserved more of their three-dimensional structure (3D) than their amino acid sequence. This allows inferring protein models even from significantly distant templates.21 The first rhodopsin-related GPCR models were based on Shertler’s low resolution projection map obtained by electron crystallography and on the associated Baldwin’s rhodopsin three-dimensional (3D) molecular model published in 1993.2, 22 More recently, GPCR modeling has been based on the 3D crystal structure of bovine rhodopsin published for the first time by Palczewski and coworkers in 2000.1
The first evidence of a general structure shared by the family of receptors that were later identified under the label of GPCRs came during the eighties, with the report of a significant amino acid homology between rhodopsin and the β2-adrenergic receptor (β2-AR).23–25 In subsequent years, it became clear that, just like rhodopsin, GPCRs are formed by a single polypeptide chain that crosses seven times the cell membrane with seven α-helical transmembrane domains (7TMs) bundled together in a very similar manner.26 Supporting the idea of a common folding of the 7TMs, sequence comparison revealed specific amino acid patterns characteristic of each TM and highly conserved in the great majority of Class A GPCRs.27–29 These conserved residues constitute the basis for the identification of the 7TMs within GPCR amino acid sequences. They are also the foundation of the GPCR residue indexing system introduced by Ballesteros and Weinstein, which is used throughout this paper to facilitate the comparison among receptors.28 Briefly, the most conserved residue in a given TM is assigned the index X.50, where X is the TM number, and the remaining residues are numbered relatively to this position.
Homology models of GPCRs, especially those supported by experimental data, and molecular docking experiments have been widely used in computational medicinal chemistry to guide site-directed mutagenesis and for drug discovery purposes, pursued also through virtual screenings and through the generation of docking-based quantitative structure-activity relationship (QSAR) models.30–39 Encouraging results led to a general acceptance of these models, which, however, although corroborated by indirect experimental evidence could not be ultimately validated. The recent publication of the crystal structure of the human β2-AR proved conclusively that GPCRs indeed share a very structurally conserved 7TM core, strongly supporting the body of literature and the hypotheses that were built on the basis of homology modeling and molecular docking.40–43
To probe to which extent molecular modeling of GPCRs can recreate realistic structures potentially applicable to computer-aided drug discovery, here I compare molecular docking of carazolol at two different rhodopsin-based homology models of the β2-AR with the corresponding high resolution crystal structure (pdb ID: 2rh1).42, 43 The two homology models differ in the way the second extracellular loop (EL2) was constructed: in the first model (model 1) it was partially built by homology to rhodopsin, while in the second model (model 2) it was built completely de novo.
Results and Discussion
Homology models of the β2-AR
The alignment of the amino acid sequences, which is provided in the supporting information, was performed following a facile and straightforward procedure generally applicable to all Class A GPCRs. The 7TMs have been aligned by identification of the conserved amino acid patterns.27 The termini were not modeled. Loops of comparable lengths have been aligned introducing, when necessary, short gaps in positions where an insertion or deletion is compatible with the 3D structure of rhodopsin. Loops of significantly different lengths were modeled de novo without aligning them with those of rhodopsin. Due to the amount of experimental evidence suggesting a role of EL2 in ligand recognition and receptor activation,8, 31, 44–48 two different models of the β2-AR were built: model 1 was built aligning the conserved Cys in EL2 and the four adjacent residues that in rhodopsin form a β-strand with the corresponding residues in the β2-AR; model 2 was built completely de novo, without any alignment between the sequences of rhodopsin and the β2-AR. The models were built with Modeler,49, 50 using as a template the crystal structure of rhodopsin published by Schertler and coworkers in 2004 (1gzm) that, among the published rhodopsin structures, is thought to provide a better conformation for the cytoplasmic ends of TM5 and TM6.4 The latter Retinal – the inverse agonist covalently bound to rhodopsin - was included in the structural template in the model building process. Four Cys residues are present in the β2-AR EL2. It is well known that two of them (Cys184 and Cys190) form a disulfide bridge internal to EL2, while the other (Cys191) forms a disulfide bridge – virtually conserved in all GPCRs – with a Cys located in the extracellular side of TM3.51 In the construction of the models, the formation of both disulfide bridges was enforced. The models were processed with the protein preparation tool of the Schrödinger package to add hydrogens and optimize the protonation state of the His residues, the orientation of the hydroxyl groups, and that of the Asn and Gln residues.
As expected, in the structurally conserved regions both β2-AR models showed a high degree of similarity with bovine rhodopsin, with a root mean square deviation (RMSD) of the backbone atoms of the TMs of 0.32 Å for model 1 and 0.34 Å for model 2 – in the remainder of the manuscript RMSD values will always refer to backbone atoms, unless specified.
Due to the high structural similarity shared by the β2-AR and rhodopsin, the models effectively approximate the crystal structure in the structurally conserved regions, with an RMSD about the TMs of 2.04 Å for both models, where the RMSD between the TMs of the crystal structures of the β2-AR (2rh1) and rhodopsin (1gzm) is 2.07 Å. A superimposition between the models and the crystal structure of the β2-AR is presented in Figure 1. Also the loops without gaps in the alignment, i.e. IL1 and EL1, are well approximated by the models, while the loops with insertions or deletions were not predicted with accuracy. The RMSD of individual TMs and loops of model 1 and model 2 with respect to the crystal structure are reported in Table 1. No comparison of IL3 was possible due to the fact that, in the crystallized β2-AR, this region is replaced by the T4 lysozyme.
Figure 1.
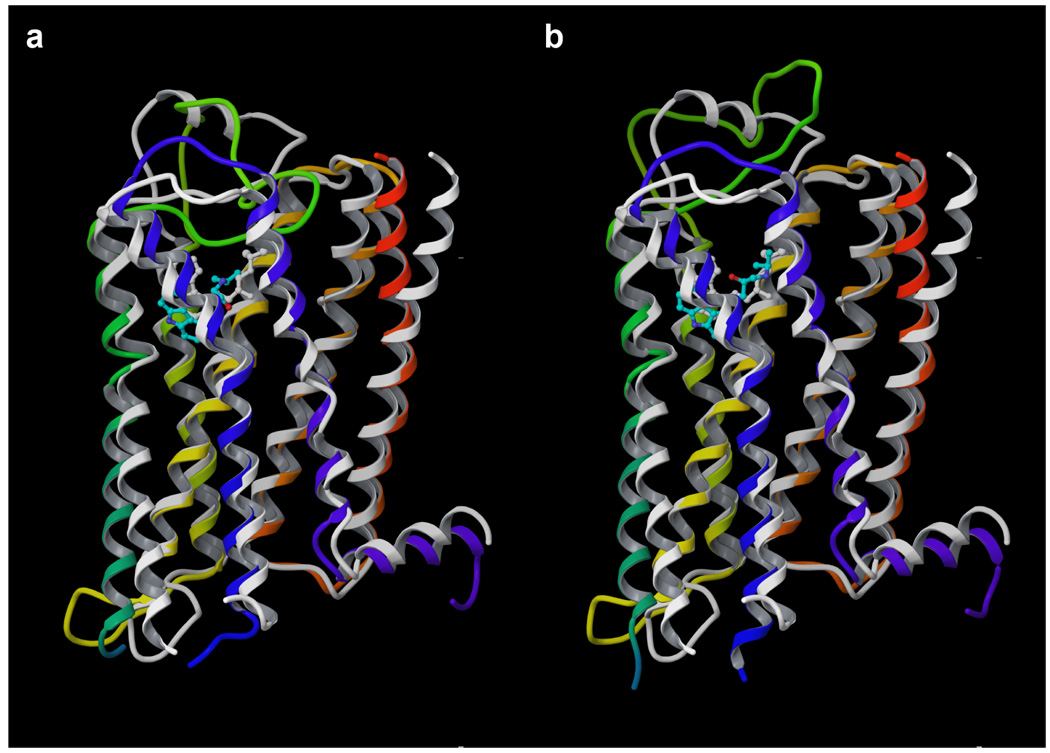
Superimposition of the crystal structure (2rh1) and the two homology models of the β2-AR schematically represented as ribbons. The homology models approximate well the structure of the receptor with an RMSD of the backbone of the TMs comparable to that observed between the crystal structure of the β2-AR and rhodopsin. Model 1 is in panel a, while model 2 is in panel b. The crystal structure is in white, while the models are depicted with the colors of the rainbow going from the N-terminus to the C-terminus (TM1: orange; TM2: yellow; TM3: green; TM4: green/blue; TM5: cyan; TM6: blue; TM7: violet). The docked ligand is colored according to the CPK scheme with cyan carbons.
Table 1.
RMSD of the backbone of individual TMs and loops of the rhodopsin-based β2-AR homology models with respect to the β2-AR crystal structure (2rh1). No comparison of IL3 was possible due to the fact that, in the crystallized β2-AR receptor, this region is replaced by the T4 lysozyme.
| Model 1 | Model 2 | |
|---|---|---|
| TM1 | 2.77 | 2.71 |
| TM2 | 1.89 | 1.87 |
| TM3 | 1.89 | 1.88 |
| TM4 | 2.09 | 2.07 |
| TM5 | 2.08 | 2.14 |
| TM6 | 1.46 | 1.56 |
| TM7 | 1.78 | 1.81 |
| IL1 | 2.20 | 2.41 |
| IL2 | 6.09 | 7.17 |
| EL1 | 1.91 | 1.98 |
| EL2 | 8.82 | 11.13 |
| EL3 | 4.10 | 3.78 |
Model 1, just like most rhodopsin-based GPCR models, features a rhodopsin-like EL2 laying flat over the opening of the receptor cavity. The crystal structure of the β2-AR revealed that the buried β-hairpin conformation of EL2 featured by rhodopsin is not shared by all GPCRs. In the β2-AR, EL2 appears to be more solvent exposed, resulting in a more open configuration of the binding pocket (Figure 2). This characteristic is captured by model 2, which features an EL2 even more solvent exposed than what is revealed by the crystal structure (Figure 2). Although the overall RMSD of the modeled and the experimentally determined EL2 remains high, the RMSD of the residues that face the binding pocket (Cys190-Thr195) is reduced from 5.86 Å to 3.84 Å, going from model 1 to model 2. As I will demonstrate, the conformation of EL2 significantly affects molecular docking.
Figure 2.
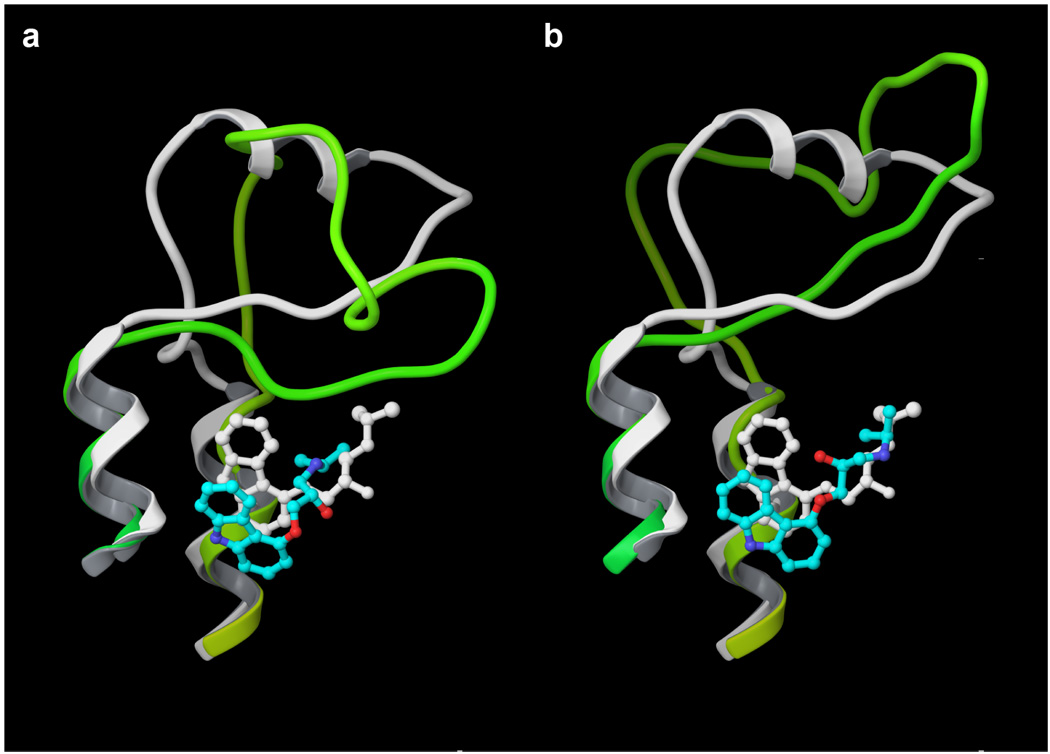
Model 1 (panel a) features a rhodopsin-like EL2 that is buried more deeply than in the crystal structure and interferes with ligand docking, causing an unnaturally deep pose. Model 2 (panel b) features a solvent exposed EL2, which allows a better placement of the positively charged tail of the ligand. The tricyclic moiety still docks deeper than in the crystal structure, due to the incorrect orientation of Phe290(6.52) in the model. The receptor models are represented in yellow /green going from the N-terminus to the C-terminus, with the docked ligand colored according to the CPK scheme with cyan carbons. The crystal structure (2rh1) is represented in white.
Docking carazolol at the β2-AR homology models
After the construction of the two β2-AR homology models, I docked the inverse agonist carazolol at them by means of the InducedFit procedure implemented in the Schrödinger package.52, 53 Briefly, the procedure is intended to explore the flexibility of both ligand and receptor in the course of the docking process and is composed of three sequential steps: 1) docking of the ligand at the receptor; 2) optimization of the side chains of the residues in the binding pocket; 3) final docking of the ligand at the optimized receptor. The docking region was defined as a box with a side of 26 Å and centered on Val114(3.33), a residue which is located in a central position within the 7TM helical bundle. A figure representing the box is available in the supporting information, while further details on the docking procedure are given in the Methodologies section.
With model 1, the docking procedure yielded thirteen poses, eleven of which – ranked 1st – 10th and 12th – were consistent with each other and with the crystal structure. The same procedure, when applied to model 2, which is characterized by a more open configuration of the binding pocket, produced fourteen poses, seven of which – ranked 1st – 5th, 8th, and 9th – were consistent with each other and with the crystal structure. Four of these poses - ranked 2nd, 3rd, 5th, and 9th - feature the tricyclic carbazol moiety rotated about 180° and pointing toward the center of the receptor. In the following paragraphs, I will always describe the top scoring poses.
A comparison of the modeled and experimentally elucidated ligand binding pockets is provided in Figure 3. As in the crystal structures, in the two models the ligand docks in the cavity formed by TM3, TM5, TM6, and TM7, with the aromatic carbazol moiety pointing toward TM5 – specifically toward Ser203(5.42), Ser204(5.43), and Ser207(5.46) – and the positively charged amino group pointing toward TM3 and TM7 – specifically toward Asp113(3.32) and Asn312(7.39).
Figure 3.
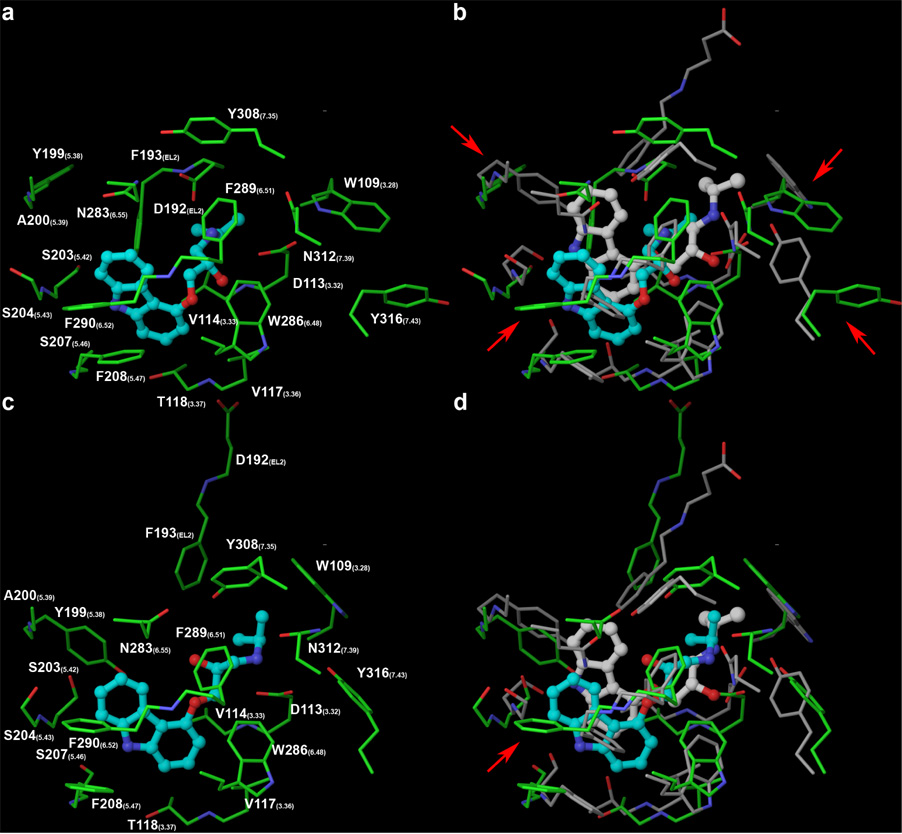
Molecular docking of carazolol at Model 1 (panels a and b) and model 2 (panels c and d). Model 1 features a rhodopsin-like EL2, which pushes carazolol more deeply into the pocket compared to the crystal structure. Besides incorrectly predicting an interaction between the ligand and Asp192(EL2), Model 1 fails in predicting the orientation of four additional residues (indicated with red arrows). Model 2 correctly placed Asp192(EL2) far away from the ligand, thus allowing an accurate reproduction of the binding mode of the positively charged tail of carazolol. The different conformation of EL2 in model 2 also led to the accurate prediction of the orientation of all the side chains of the residues lining the binding pocket, with the notable exception of Phe290(6.52). In model 2, the incorrect orientation of the latter is the only reason for the too deep docking of the tricyclic moiety of carazolol. The carbon atoms are colored in green for the modeled receptor, in gray for the crystallized receptor, in cyan for the modeled ligand, and in white for the crystallized ligand.
The complex of carazolol with model 1 shows RMSDs about the heavy atoms of the ligands and the residues lining the pocket of 3.69 Å and 3.28 Å, respectively, while the complex with model 2 shows RMSDs about the heavy atoms of the ligands and the residues lining the pocket of 2.90 Å and 2.84 Å, respectively. The interactions between carazolol and the binding pocket of the β2-AR are exhaustively described by Rosenbaum and coworkers. In the following paragraphs, I want to describe the main differences between the model and the crystal structure. In particular, I want to highlight the major impact of the conformation of EL2 on the docking pose of the ligand and on the orientation of the side chains that line the binding pocket.
Both homology models placed Phe193(EL2) in the vicinity of the carbazol moiety of the ligand, in line with the crystal structure. However, due to the rhodopsin-like conformation of its EL2, model 1 erroneously places also the adjacent Asp192(EL2) in close proximity to the ligand, interacting with its positively charged amino group. As a consequence, the latter lost its interaction with Asn312(7.39), although still conserving the one with Asp113(3.32). Furthermore, due to the buried conformation of EL2, (Figure 3, panels a and b) carazolol is pushed into the binding pocket significantly more deeply than what is indicated by the crystal structure. Conversely, model 2, which features a more solvent exposed EL2, correctly placed Asp192(EL2) far away from the ligand, with an orientation consistent with the crystal structure, thus allowing an accurate reproduction of the binding mode of the positively charged tail of carazolol, coordinated by Asp113(3.32) and Asn312(7.39) (Figure 3, panels c and d). The biological relevance of Asp113(3.32) is well established.54 In analogy with carazolol binding, Asp113(3.32) is thought to establish interactions with the positively charged amino group of isoproterenol, epinephrine, and norepinephrine.55–57
Besides the orientation of Asp192(EL2), there are four other notable differences between the binding pockets of the crystal structure and model 1, namely the orientations of Trp109(3.28), Tyr199(5.38), Phe290(6.52), and Tyr316(7.43). Remarkably, the different conformation of EL2 in model 2 not only caused the correct placement of Asp192(EL2), but also led to accurate predictions of all the sidechains lining the binding pocket, with the notable exception of Phe290(6.52). The latter has been proposed to form with Trp286(6.48) a rotamer toggle switch for the activation of the β2-AR. In particular, its trans conformation is maintained to be associated with the active state of the receptor, while its gauche+ conformation is thought to be associated with the inactive state.58, 59 Furthermore, the corresponding residue of the dopamine D2 receptor has been proposed to be accessible in the binding site crevice and to establish contact with antagonists on the basis of substitute-cysteine accessibility studies.60 Consistently, in the crystal structure with the inverse agonist carazolol (2rh1), Phe290(6.52) adopts the gauche+ conformation and points toward the center of the pocket, making it relatively shallow. This feature is not captured by the two rhodopsin-based homology models, in which Phe290(6.52) adopts the trans conformation and points away from the pocket, thus making it deeper.
The gauche+ of Phe290(6.52) conformation is the only cause of the excessively deep docking of the carbazol moiety of carazolol in model 2. Homology modeling cannot contribute to the prediction of its Ch1 angle, since rhodopsin features an Ala at the 6.52 position. However, incorporating the available biochemical and computational data to the homology model by changing the conformation of the side chain of Phe290(6.52) from trans to gauche+ in model 2 prior to docking remarkably resulted in significantly improved docking poses. In particular, the top scoring pose showed RMSDs about the heavy atoms of the ligands and the residues lining the pocket of 1.70 Å and 2.67 Å, respectively (Figure 4). The carbazol moiety of the ligand docked as shallowly as revealed by the crystal structure, allowing the formation of a hydrogen bond between the aromatic amine and Ser203(5.42). Conversely, this interaction, which is evident in the crystal structure, could not be detected with the docking experiments performed with Phe290(6.52) in the trans conformation. The importance of this particular Ser, together with two other Ser located in TM5 at positions 5.43 and 5.46, in ligand recognition and activation is well established.61 Relatively to agonist binding, it has been hypothesized that the three residues are in close proximity with the hydroxyl groups of the catechol ring of norepinephrine.55
Figure 4.
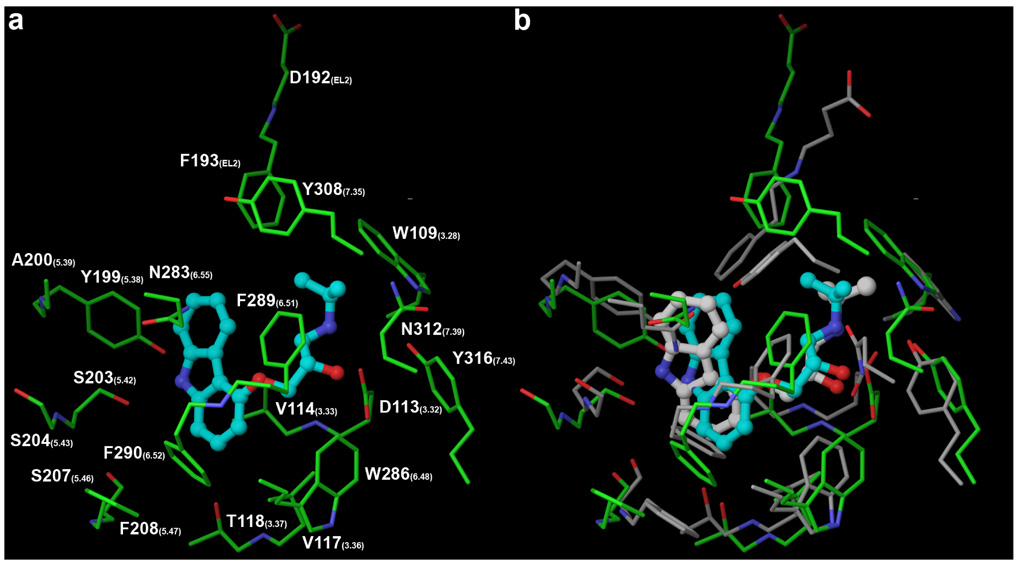
Molecular docking of carazolol at a modified Model 2 featuring Phe290(6.52) in the gauche+ conformation resulted in significantly improved docking poses (panels a and b). The carbon atoms are colored in green for the modeled receptor, in gray for the crystallized receptor, in cyan for the modeled ligand, and in white for the crystallized ligand.
The other component of the rotamer toggle switch, i.e. Trp286(6.48), corresponding to Trp265(6.48) in rhodopsin,58, 59 adopts the gauche+ conformation in the crystal structure and in both rhodopsin-based homology models. This result is consistent with the fact that the β2-AR and rhodopsin were both crystallized in complex with inverse agonists.
To confirm the robustness of the InducedFit docking procedure as implemented in the Schrödinger software package, Carazolol was extracted from the crystal structure and docked back following the same procedure utilized when docking at the homology models. Also in this case the conformation of carazolol was generated by means of a Monte Carlo conformational search prior to docking. The docked complex approximates the crystal structure remarkably well, with RMSDs about the heavy atoms of the ligands and the residues lining the pocket of 0.67 Å and 0.41 Å, respectively (Figure 5).
Figure 5.
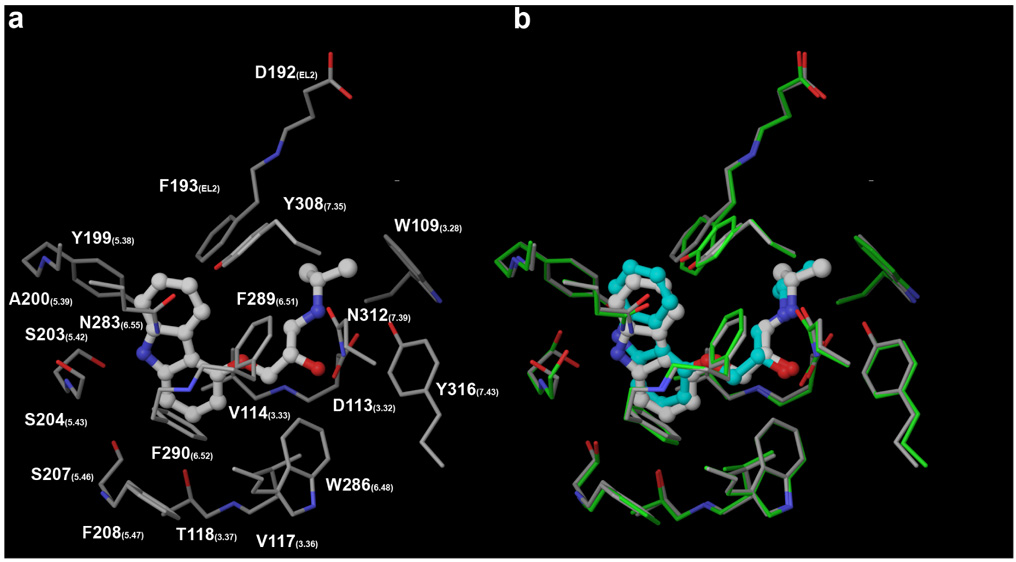
Redocking of carazolol at the crystal structure confirms the robustness of the InducedFit docking procedure. The carbon atoms are colored in green for the modeled receptor, in gray for the crystallized receptor, in cyan for the modeled ligand, and in white for the crystallized ligand.
Conclusions
The results reported in this paper suggest that, at least in some instances, molecular docking at GPCR homology models can produce 3D structures that approximate the real picture well, amply supporting its wide-spread application. This is confirmed at least in the case of the complex of the inverse agonist carazolol and the β2-AR. The resolution of the models is not high enough to detect all receptor-ligand interactions. However, it is sufficient to: a) design site-directed mutagenesis experiments intended to experimentally probe the interactions, which in turn can be incorporated into the models in an iterative manner; b) predict conformation and orientation of the ligands, which can be applied to the construction of docking-based 3D-QSAR models; c) design fuzzy receptor-based pharmacophores to be applied in virtual screening experiments. It is worth noting that retinal and carazolol are significantly similar in size and shape, and that the binding pockets of rhodopsin and of the β2-AR largely overlap. Less accurate results may be obtained by modeling receptors with ligands significantly different from those bound to the templates or characterized by a high level of flexibility. It is also worth noting that the results obtained through docking at homology models based on templates in the inactive conformation may be more accurate in the case of inverse agonists – such as carazolol – and antagonists than in that of agonists.
From the computational medicinal chemistry perspective, modeling EL2 seems a crucial point to be addressed. In most of the current rhodopsin-based homology models, EL2 is buried in the 7TM pocket. The crystal structure of the β2-AR revealed that this is not a general feature shared by all GPCRs. In the case of the β2-AR homology models presented here, the rhodopsin-like EL2 of model 1 not only interfered with the docking of the ligand, suggesting wrong interactions and pushing the ligand excessively toward the core of the receptor, but also caused the incorrect modeling of many of the side chains of the residues that line the binding pocket. Most of those problems were solved when the model was generated without aligning the residues of EL2 of the β2-AR and rhodopsin (model 2). We have noticed interferences of EL2 in ligand docking in the past. In the case of FFAR1 we addressed them by removing the loop prior to docking and reinserting it only afterwards.18 Due to the inherent difficulties of modeling and validating the structures of the loops, the development and testing of GPCR models built without EL2 may prove a viable alternative.
The orientation of the side chains that line the binding pocket is also of critical importance to obtain accurate docking poses. The data shown here demonstrate that even a single residue with the side chain in an incorrect orientation – in this case Phe290(6.52) – can affect the pose of the ligand and cause the loss of important interactions – in this case between the ligand and the crucial Ser203(5.42). Consequently, incorporating available data on the conformation of a residue or on a specific receptor-ligand interaction, gathered, for example, through biochemical or biophysical experiments, significantly enhances the quality of the models.
In any case, the newly published structure of the β2-AR will afford the modeling community the opportunity to perfect techniques and strategies, towards more reproducible and reliable in silico predictions. First priority items on the agenda of computational medicinal chemists will be the identification of protocols to generate accurate GPCR models, to optimize loops and side chain conformations, and to perform molecular docking. The availability of the structure of a second GPCR, in addition to validating and encouraging modeling studies, will provide the long awaited benchmark to perform the necessary optimization and calibration procedures.
Experimental Section
Homology Modeling
On the basis of two sequence alignments differing in the EL2 region, two alternative homology models of the human β2-AR were built with the program Modeler.49, 50 In both cases, the two disulfide bridges in the second extracellular loop were manually defined. For each alignment, three models were built, and each of them was subjected to five loop refinements setting the optimization level to high. The quality of the models was examined in terms of residue-based probability density function and residue-based energy, as calculated by Modeler. For each of the two alignments, the soundest models were chosen for the subsequent docking experiments.
Protein and Ligand Preparation
Carazolol was sketched in Maestro and subjected to a Monte Carlo Multiple Minimum conformational search using the OPLS_2005 force field and water as implicit solvent (Surface Generalized Born (SGB) model).62, 63 The lowest energy conformation of the ligand was used as starting point for the docking experiments. Model 1 and Model 2 were imported into the Maestro interface of the Schrödinger software and subjected to the Protein Preparation Workflow to: 1) add hydrogens; 2) add N-acetyl and N-methyl amide capping groups to the N-terminus and C-terminus, respectively; 3) optimize the orientation of hydroxyl groups, Asn, and Gln, and the protonation state of His; 4) perform a constrained refinement with the impref utility, setting the max RMSD of 0.30. The impref utility consists a cycle of energy minimizations based on the impact molecular mechanics engine and on the OPLS_2001 force field. The first minimization is performed constraining the heavy atoms with the hydrogen torsion parameters turned off, to allow free rotation of the hydrogens. Subsequently up to five minimizations are performed, gradually decreasing the constraints on the positions of the heavy atoms. If, at the end of any minimization cycle, the RMSD of the heavy atoms is greater than the max RMSD from the original structure, the calculation terminates and returns the structure resulting from the previous cycle.
The model with Phe290(6.52) in the gauche+ conformation was built from model 2 after impref refinement by rotating the χ1 angle of the residue of 90°.
Induced Fit Docking
Molecular docking was performed with the Induced Fit Docking procedure based on Glide 4.5 and Prime 1.6, as implemented in the Schrödinger package.52, 53 The procedure is composed by a Glide SP docking, followed by a Prime refinement of the side chains of the residues in the binding pocket, and then by a final Glide XP docking of the ligand into the receptor in the refined conformations.
The docking box was centered on Val114(3.33) and featured a side of 26 Å (see figure in supporting information). In the initial Glide SP docking (Glide 4.5), the vdW scaling was set to 0.5 for non polar atoms of receptor and ligand, defined as the atoms with partial charge lower than 0.25 for the receptor and lower than 0.15 for the ligand. For each obtained docking pose, a Prime refinement (Prime 1.6) was performed on all the residues located within 5 Å from the ligand. Brifely, the Prime refinement starts with the optimization of the side chains of the selected residues performed through randomization and subsequent exploration of various combinations of rotamers; the optimization is followed by a truncated-Newton minimization of the selected residues and the ligand using the OPLS_2000 all-atom force field for the receptor and OPLS_2001 for the ligand, treating solvation with the SGB continuum solvation model. All the obtained complexes within an energy range of 30 kcal/mol from the best were passed onto the final step, in which the ligand was extracted and redocked with Glide XP (Glide 4.5), with a vdW scaling factor of 0.8 for the non polar atoms of the ligand only. The InducedFit docking procedure was always preceded by an IMPREF refinement of the receptor with a max RMSD of 0.18 Å.
Supplementary Material
Acknowledgment
The author thanks Dr. Kenneth A. Jacobson and Dr. Marvin Gershengorn for the helpful discussions, and Joel Karpiak for critical reading of the manuscript. This research was supported by the Intramural Research Program of the NIH, NIDDK.
ABBREVIATIONS
- 3D
three-dimensional
- β2-AR
β2-adrenergic receptor
- EL2
second extracellular loop
- GPCR
G protein-coupled receptor
- OPLS
optimized potential for liquid simulations
- QSAR
quantitative structure-activity relationships
- RMSD
root mean square deviation
- SGB
surface generalized born
- SP
standard precision
- TM
transmembrane domain
- XP
extra precision
Footnotes
Supporting Information Available. The alignments of the β2-AR and rhodopsin sequences utilized for the construction of the homology models and a figure representing the docking box are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le TI, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 2.Schertler GF, Villa C, Henderson R. Projection structure of rhodopsin. Nature. 1993;362:770–772. doi: 10.1038/362770a0. [DOI] [PubMed] [Google Scholar]
- 3.Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GF. Electron crystallography reveals the structure of metarhodopsin I. EMBO J. 2004;23:3609–3620. doi: 10.1038/sj.emboj.7600374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J, Edwards PC, Burghammer M, Villa C, Schertler GFX. Structure of bovine rhodopsin in a trigonal crystal form. Journal of Molecular Biology. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 5.Kim J, Wess J, van Rhee AM, Schoneberg T, Jacobson KA. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2A adenosine receptor. J. Biol. Chem. 1995;270:13987–13997. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobson KA, Gao ZG, Chen AS, Barak D, Kim SA, Lee K, Link A, Van Rompaey P, Van Calenbergh S, Liang BT. Neoceptor concept based on molecular complementarity in GPCRs: A mutant adenosine A(3) receptor with selectively enhanced affinity for amine-modified nucleosides. Journal of Medicinal Chemistry. 2001;44:4125–4136. doi: 10.1021/jm010232o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gershengorn MC, Osman R. Minireview: Insights into G protein-coupled receptor function using molecular models. Endocrinology. 2001;142:2–10. doi: 10.1210/endo.142.1.7919. [DOI] [PubMed] [Google Scholar]
- 8.Costanzi S, Mamedova L, Gao ZG, Jacobson KA. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J. Med. Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu XP, Huang W, Worthington S, Drabik P, Osman R, Gershengorn MC. A model of inverse agonist action at thyrotropin-releasing hormone receptor type 1: Role of a conserved tryptophan in helix 6. Molecular Pharmacology. 2004;66:1192–1200. doi: 10.1124/mol.104.000349. [DOI] [PubMed] [Google Scholar]
- 10.Jacobson KA, Kim SK, Costanzi S, Gao ZG. Purine receptors: GPCR structure and agonist design. Molecular Interventions. 2004;4:337–347. doi: 10.1124/mi.4.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bissantz C, Logean A, Rognan D. High-throughput modeling of human G-protein coupled receptors: Amino acid sequence alignment, three-dimensional model building, and receptor library screening. Journal of Chemical Information and Computer Sciences. 2004;44:1162–1176. doi: 10.1021/ci034181a. [DOI] [PubMed] [Google Scholar]
- 12.Moro S, Spalluto G, Jacobson KA. Techniques: Recent developments in computer-aided engineering of GPCR ligands using the human adenosine A3 receptor as an example. Trends Pharmacol. Sci. 2005;26:44–51. doi: 10.1016/j.tips.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Huang W, Osman R, Gershengorn MC. Agonist-induced corformational changes in thyrotropin-releasing hormone receptor type I: Disulfide cross-linking and molecular modeling approaches. Biochemistry. 2005;44:2419–2431. doi: 10.1021/bi048808+. [DOI] [PubMed] [Google Scholar]
- 14.Ivanov AA, Costanzi S, Jacobson KA. Defining the nucleotide binding sites of P2Y receptors using rhodopsin-based homology modeling. Journal of Computer-Aided Molecular Design. 2006;20:417–426. doi: 10.1007/s10822-006-9054-2. [DOI] [PubMed] [Google Scholar]
- 15.Moro S, Deflorian F, Bacilieri M, Spalluto G. Ligand-based homology modeling as attractive tool to inspect GPCR structural plasticity. Curr. Pharm. Des. 2006;12:2175–2185. doi: 10.2174/138161206777585265. [DOI] [PubMed] [Google Scholar]
- 16.Patny A, Desai PV, Avery MA. Homology modeling of G-protein-coupled receptors and implications in drug design. Current Medicinal Chemistry. 2006;13:1667–1691. doi: 10.2174/092986706777442002. [DOI] [PubMed] [Google Scholar]
- 17.Kleinau G, Krause G. Implications for understanding molecular function and dysfunction of glycoprotein hormone receptors by a new sequence-structure-function analysis resource. Experimental and Clinical Endocrinology & Diabetes. 2007;115:S34. doi: 10.1210/me.2006-0309. [DOI] [PubMed] [Google Scholar]
- 18.Tikhonova IG, Sum CS, Neumann S, Thomas CJ, Raaka BM, Costanzi S, Gershengorn MC. Bidirectional, iterative approach to the structural delineation of the functional "Chemoprint" in GPR40 for agonist recognition. Journal of Medicinal Chemistry. 2007;50:2981–2989. doi: 10.1021/jm0614782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deupi X, Dolker N, Lopez-Rodriguez ML, Campillo M, Ballesteros JA, Pardo L. Structural models of class A G protein-coupled receptors as a tool for drug design: Insights on transmembrane bundle plasticity. Current Topics in Medicinal Chemistry. 2007;7:991–998. doi: 10.2174/156802607780906799. [DOI] [PubMed] [Google Scholar]
- 20.Costanzi S, Ivanov AA, Tikhonova IG, Jacobson KA. Structure and function of G protein-coupled receptors studied using sequence analysis, molecular modeling, and receptor engineering: Adenosine receptors. In: Caldwell GE, Rahman AU, Player MR, Chouday MI, editors. Frontiers in Drug Design and Discovery. Bentham: 2007. pp. 63–79. [Google Scholar]
- 21.Costanzi S, Vincenzetti S, Cristalli G, Vita A. Human cytidine deaminase: A three-dimensional homology model of a tetrameric metallo-enzyme inferred from the crystal structure of a distantly related dimeric homologue. Journal of Molecular Graphics & Modelling. 2006;25:10–16. doi: 10.1016/j.jmgm.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 22.Baldwin JM. The probable arrangement of the helices in G protein-coupled receptors. EMBO J. 1993;12:1693–1703. doi: 10.1002/j.1460-2075.1993.tb05814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobilka BK, Dohlman HG, Frielle T, Bolanowski MA, Dixon RAF, Sigal I, Keller P, Caron MG, Lefkowitz RJ. Homology Between the Human and Hamster Beta-2-Adrenergic Receptors and Rhodopsin Revealed by Molecular-Cloning. Circulation. 1986;74:155. [Google Scholar]
- 24.Dixon RAF, Kobilka BK, Strader DJ, Benovic JL, Dohlman HG, Frielle T, Bolanowski MA, Bennett CD, Rands E, Diehl RE, Mumford RA, Slater EE, Sigal IS, Caron MG, Lefkowitz RJ, Strader CD. Cloning of the Gene and Cdna for Mammalian Beta-Adrenergic-Receptor and Homology with Rhodopsin. Nature. 1986;321:75–79. doi: 10.1038/321075a0. [DOI] [PubMed] [Google Scholar]
- 25.Dohlman HG, Bouvier M, Benovic JL, Caron MG, Lefkowitz RJ. The Multiple Membrane Spanning Topography of the Beta-2-Adrenergic Receptor - Localization of the Sites of Binding, Glycosylation, and Regulatory Phosphorylation by Limited Proteolysis. Journal of Biological Chemistry. 1987;262:14282–14288. [PubMed] [Google Scholar]
- 26.Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: Implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Molecular Pharmacology. 2001;60:1–19. [PubMed] [Google Scholar]
- 27.Oliveira L, Paiva ACM, Vriend G. A Common Motif in G-Protein-Coupled 7 Transmembrane Helix Receptors. Journal of Computer-Aided Molecular Design. 1993;7:649–658. [Google Scholar]
- 28.Ballesteros JA, Weinstein H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G-Protein Coupled Receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 29.vanRhee AM, Jacobson KA. Molecular architecture of G protein-coupled receptors. Drug Development Research. 1996;37:1–38. doi: 10.1002/(SICI)1098-2299(199601)37:1<1::AID-DDR1>3.0.CO;2-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim HS, Barak D, Harden TK, Boyer JL, Jacobson KA. Acyclic and cyclopropyl analogues of adenosine bisphosphate antagonists of the P2Y(1) receptor: Structure-activity relationships and receptor docking. Journal of Medicinal Chemistry. 2001;44:3092–3108. doi: 10.1021/jm010082h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA. Human P2Y(6) receptor: Molecular modeling leads to the rational design of a novel agonist based on a unique conformational preference. Journal of Medicinal Chemistry. 2005;48:8108–8111. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krause G, Neumann S, Kleinau G, Moore S, Jaeschke H, Paschke R, Thomas CJ, Gershengorn MC. Design of low molecular weight ligands for the TSH receptor utilizing its sequence homology to the LH receptor. Experimental and Clinical Endocrinology & Diabetes. 2007;115:S7. [Google Scholar]
- 33.Moro S, Braiuca P, Deflorian F, Ferrari C, Pastorin G, Cacciari B, Baraldi PG, Varani K, Borea PA, Spalluto G. Combined target-based and ligand-based drug design approach as a tool to define a novel 3D-pharmacophore model of human A(3) adenosine receptor antagonists: Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as a key study. Journal of Medicinal Chemistry. 2005;48:152–162. doi: 10.1021/jm049662f. [DOI] [PubMed] [Google Scholar]
- 34.Costanzi S, Tikhonova IG, Ohno M, Roh EJ, Joshi BV, Colson AO, Houston D, Maddileti S, Harden TK, Jacobson KA. P2Y(1) antagonists: Combining receptor-based modeling and QSAR for a quantitative prediction of the biological activity based on consensus scoring. Journal of Medicinal Chemistry. 2007;50:3229–3241. doi: 10.1021/jm0700971. [DOI] [PubMed] [Google Scholar]
- 35.Klabunde T, Hessler G. Drug design strategies for targeting G-protein-coupled receptors. Chembiochem. 2002;3:929–944. doi: 10.1002/1439-7633(20021004)3:10<928::AID-CBIC928>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 36.Evers A, Klebe G. Ligand-supported homology modeling of G-protein-coupled receptor sites: Models sufficient for successful virtual screening. Angewandte Chemie-International Edition. 2004;43:248–251. doi: 10.1002/anie.200352776. [DOI] [PubMed] [Google Scholar]
- 37.Bissantz C, Schalon C, Guba W, Stahl M. Focused library design in GPCR projects on the example of 5-HT2c agonists: Comparison of structure-based virtual screening with ligand-based search methods. Proteins-Structure Function and Bioinformatics. 2005;61:938–952. doi: 10.1002/prot.20651. [DOI] [PubMed] [Google Scholar]
- 38.Evers A, Klabunde T. Structure-based drug discovery using GPCR homology modeling: Successful virtual screening for antagonists of the Alpha1A adrenergic receptor. Journal of Medicinal Chemistry. 2005;48:1088–1097. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- 39.Kellenberger E, Springael JY, Parmentier M, Hachet-Haas M, Galzi JL, Rognan D. Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. Journal of Medicinal Chemistry. 2007;50:1294–1303. doi: 10.1021/jm061389p. [DOI] [PubMed] [Google Scholar]
- 40.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta(2) adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 41.Day PW, Rasmussen SG, Parnot C, Fung JJ, Masood A, Kobilka TS, Yao XJ, Choi HJ, Weis WI, Rohrer DK, Kobilka BK. A monoclonal antibody for G protein-coupled receptor crystallography. Nat. Methods. 2007;4:927–929. doi: 10.1038/nmeth1112. [DOI] [PubMed] [Google Scholar]
- 42.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR Engineering Yields High-Resolution Structural Insights into {beta}2 Adrenergic Receptor Function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 43.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-Resolution Crystal Structure of an Engineered Human {beta}2-Adrenergic G Protein Coupled Receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olah ME, Jacobson KA, Stiles GL. Role of the 2Nd Extracellular Loop of Adenosine Receptors in Agonist and Antagonist Binding - Analysis of Chimeric A(1)/A(3)-Adenosine Receptors. Journal of Biological Chemistry. 1994;269:24692–24698. [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JH, Jiang QL, Glashofer M, Yehle S, Wess J, Jacobson KA. Glutamate residues in the second extracellular loop of the human A(2a) adenosine receptor are required for ligand recognition. Molecular Pharmacology. 1996;49:683–691. [PMC free article] [PubMed] [Google Scholar]
- 46.Moro S, Hoffmann C, Jacobson KA. Role of the extracellular loops of G protein-coupled receptors in ligand recognition: A molecular modeling study of the human P2Y(1) receptor. Biochemistry. 1999;38:3498–3507. doi: 10.1021/bi982369v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kleinau G, Claus M, Jaschke H, Muller S, Neumann S, Paschke R, Krause G. Molecular mechanism at the TSH receptor: Modulation of signalling activity via extracellular loop 2. Experimental and Clinical Endocrinology & Diabetes. 2007;115:S75–S76. [Google Scholar]
- 48.Kleinau G, Claus M, Jaeschke H, Mueller S, Neumann S, Paschke R, Krause G. Contacts between extracellular loop two and transmembrane helix six determine basal activity of the thyroid-stimulating hormone receptor. Journal of Biological Chemistry. 2007;282:518–525. doi: 10.1074/jbc.M606176200. [DOI] [PubMed] [Google Scholar]
- 49.Sali A, Blundell TL. Comparative Protein Modeling by Satisfaction of Spatial Restraints. Journal of Molecular Biology. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 50.Sali A, Overington JP. Derivation of Rules for Comparative Protein Modeling from A Database of Protein-Structure Alignments. Protein Science. 1994;3:1582–1596. doi: 10.1002/pro.5560030923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noda K, Saad Y, Graham RM, Karnik SS. The High-Affinity State of the Beta-2-Adrenergic Receptor Requires Unique Interaction Between Conserved and Nonconserved Extracellular Loop Cysteines. Journal of Biological Chemistry. 1994;269:6743–6752. [PubMed] [Google Scholar]
- 52.Glide. [4.5] Schrödinger, LLC; 2007. [Google Scholar]
- 53.Prime. [1.6] Schrödinger, LLC; 2007. [Google Scholar]
- 54.Strader CD, Fong TM, Tota MR, Underwood D, Dixon RAF. Structure and Function of G-Protein-Coupled Receptors. Annual Review of Biochemistry. 1994;63:101–132. doi: 10.1146/annurev.bi.63.070194.000533. [DOI] [PubMed] [Google Scholar]
- 55.Swaminath G, Xiang Y, Lee TW, Steenhuis J, Parnot C, Kobilka BK. Sequential binding of agonists to the beta(2) adrenoceptor - Kinetic evidence for intermediate conformational states. Journal of Biological Chemistry. 2004;279:686–691. doi: 10.1074/jbc.M310888200. [DOI] [PubMed] [Google Scholar]
- 56.Freddolino PL, Kalani MYS, Vaidehi N, Floriano WB, Hall SE, Trabanino RJ, Kam VWT, Goddard WA. Predicted 3D structure for the human beta 2 adrenergic receptor and its binding site for agonists and antagonists. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2736–2741. doi: 10.1073/pnas.0308751101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xhaard H, Rantanen VV, Nyronen T, Johnson MS. Molecular evolution of adrenoceptors and dopamine receptors: Implications for the binding of catecholamines. Journal of Medicinal Chemistry. 2006;49:1706–1719. doi: 10.1021/jm0511031. [DOI] [PubMed] [Google Scholar]
- 58.Yao XJ, Parnot C, Deupi X, Ratnala VRP, Swaminath G, Farrens D, Kobilka B. Coupling ligand structure to specific conformational switches in the beta(2)-adrenoceptor. Nature Chemical Biology. 2006;2:417–422. doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- 59.Lei S, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. beta(2) adrenergic receptor activation - Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. Journal of Biological Chemistry. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- 60.Javitch JA, Ballesteros JA, Weinstein H, Chen JY. A cluster of aromatic residues in the sixth membrane-spanning segment of the dopamine D2 receptor is accessible in the binding-site crevice. Biochemistry. 1998;37:998–1006. doi: 10.1021/bi972241y. [DOI] [PubMed] [Google Scholar]
- 61.Sato T, Kobayashi H, Nagao T, Kurose H. Ser(203) as well as Ser(204) and Ser(207) in fifth transmembrane domain of the human beta(2)-adrenoceptor contributes to agonist binding and receptor activation. British Journal of Pharmacology. 1999;128:272–274. doi: 10.1038/sj.bjp.0702813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang G, Guida WC, Still WC. An Internal Coordinate Monte-Carlo Method for Searching Conformational Space. Journal of the American Chemical Society. 1989;111:4379–4386. [Google Scholar]
- 63.Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel - An Integrated Software System for Modeling Organic and Bioorganic Molecules Using Molecular Mechanics. Journal of Computational Chemistry. 1990;11:440–467. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.