

Abstract
Previous work suggests the γ-aminobutyric acid (GABA)ergic system may be dynamically regulated during emotional learning. In the current study we examined training-induced changes in the expression of GABAA-related genes and the binding of GABA receptor radioligands in the amygdala after the acquisition and extinction of Pavlovian fear. Using in situ hybridization, we examined the expression pattern changes of mRNAs for GABAergic markers in the lateral, basolateral and central subdivisions of the amygdala in C57Bl/6J mice. These markers included GABA-synthesizing enzymes (GAD67 and GAD65), major GABAA receptor subunits (α1, α2, α3, α5, β2 and γ2) and the expression of mRNAs that are involved in a variety of GABA-related intracellular processes, including GABA transporter-1 (GAT1), GABAA receptor-associated protein and the GABAA clustering protein, gephyrin. With fear conditioning, we found decreased mRNA levels of α1, α5 and GAD67, as well as deceased benzodiazepine binding in the amygdala. Fear extinction induced an increase in mRNA levels of α2, β2, GAD67 and gephyrin, as well as a decrease in GAT1. Together, these findings indicate that the acquisition of fear induced a downregulation of mRNA markers related to a decrease in amygdala GABAergic function, whereas the acquisition of fear extinction produced an upregulation of GABAergic markers related to enhanced GABAergic transmission.
Keywords: benzodiazepines, C57Bl/6J mice, fear-potentiated startle, GABA
Introduction
Fear is a normal emotional response an animal produces when it anticipates or encounters danger, threat or an aversive situation. In the laboratory, fear can be studied using Pavlovian conditioning procedures in which an initially neutral conditioned stimulus (CS), such as a tone, is repeatedly paired with an aversive unconditioned stimulus (US), such as a shock. After a few such CS + US pairings, the CS comes to elicit a constellation of learned behavioral and physiological responses that are characteristic of a central state of fear (McAllister & McAllister, 1971). In addition to the acquisition of fear, Pavlovian conditioning procedures can also be used to study the extinction of fear. The extinction process involves repeatedly presenting a previously established CS in the absence of the US. This non-reinforced presentation of the CS leads to the reduction of conditioned fear responses and is thought to involve the formation of new associations that compete with or inhibit the expression of the still intact fear-producing associations (Davis et al., 2000; Myers & Davis, 2007).
The results of numerous studies demonstrate that the amygdala is a crucial site for synaptic plasticity that occurs during the acquisition, retention and extinction of conditioned fear (Falls et al., 1992; Santini et al., 2001; Walker & Davis, 2002; Walker et al., 2002; Hobin et al., 2003; Szapiro et al., 2003). Long-term potentiation (LTP) and long-term depression (LTD) are two forms of synaptic plasticity that have been proposed to represent the cellular mechanisms underlying the processes of learning and memory. Changes in the synaptic efficacy associated with LTP and LTD are initiated through the influx of Ca2+ into the neuron, which is generated through the activation of N-methyl-D-aspartate (NMDA) receptors and voltage-gated calcium channels (Chapman et al., 2003).
Much of the research aimed at elucidating LTP mechanisms associated with conditioned fear has focused on the enhancement of glutamatergic synaptic transmission associated with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and NMDA receptors in pyramidal cells (Bauer & LeDoux, 2004; Rumpel et al., 2005; Yeh et al., 2006). However, there are converging behavioral and physiological lines of evidence suggesting that changes in γ-aminobutyric acid (GABA)ergic transmission may also be involved in these processes. For example, in mice, tone fear conditioning results in a significant reduction of extracellular GABA levels in the amygdala during subsequent re-exposure to the tone CS (Stork et al., 2002). Likewise, the expression of the GABA-synthesizing enzyme GAD65 is significantly reduced in the amygdala 24 h after fear-conditioning training (Pape & Stork, 2003). In contrast, extinction training increases the expression of gephryin, a GABAA receptor clustering protein, in the amygdala (Chhatwal et al., 2005). Studies that investigated in vitro LTP in amygdala GABAergic interneurons suggest that both excitatory glutamatergic synapses onto interneurons and inhibitory synapses onto pyramidal cells can undergo LTP that are dependent on postsynaptic calcium influx (Mahanty & Sah, 1998; Bauer & LeDoux, 2004). Both forms of GABAergic LTP induce a lasting potentiation of inhibitory postsynaptic potentials recorded from lateral amygdala (LA) pyramidal neurons, and are likely to contribute to the strengthening of the inhibitory network in the LA.
The involvement of GABAA receptors in the regulation of fear and anxiety is highlighted by the fact that patients suffering from anxiety disorders are commonly treated by the administration of benzodiazepines, which mediate their actions via GABAA receptors (Hevers & Luddens, 1998; Teuber et al., 1999; Smith, 2001). The amygdala is particularly rich in GABAA receptors that have high affinity to benzodiazepines and other GABAA receptor agonists that are important targets for the treatment of fear and anxiety. In a number of behavioral tests designed to examine fear and anxiety in rats, intra-amygdala infusions of benzodiazepine agonists produce anxiolytic effects (Green & Vale, 1992; Helmstetter, 1993; Harris & Westbrook, 1995; Pesold & Treit, 1995; Zangrossi et al., 1999). On the other hand, microinjections of benzodiazepine antagonists into the amygdala induce anxiogenic-like behaviors (Shibata et al., 1989; Da Cunha et al., 1992; Sanders & Shekhar, 1995). Intra-amygdala infusions of benzodiazepine antagonists can also prevent the anxiolytic effects of peripheral injections of benzodiazepine agonists (Petersen et al., 1985). Furthermore, in a recent study that examined the physiological responses of the basolateral (BL) and central (CE) amygdala nuclei, Kang-Park et al. (2004) found that acute application of the benzodiazepine diazepam enhanced GABAA receptor-mediated inhibition in both amygdala nuclei.
Together, these observations suggest that GABAergic mechanisms in the amygdala physiologically mediate fear and anxiety. This study was undertaken to examine whether alterations in GABAergic function within the amygdala are associated with emotional learning. Specifically, we evaluated whether the acquisition of fear was associated with a downregulation of GABAergic markers related to decreased GABAergic transmission in the amygdala, whereas the acquisition of fear extinction was associated with an upregulation of amygdala GABAergic markers related to enhanced GABAergic function. To accomplish this, we examined training-induced changes in the GABAergic system by measuring the mRNA levels of GABA-related genes and the binding of GABA receptor radioligands in the amygdala after the acquisition and extinction of Pavlovian fear. In particular, we examined changes in mRNAs levels of six GABAA receptor subunits (α1, α2, α3, α5, β2 and γ2) and a variety of GABA-related proteins, including GABA transporter-1 (GAT1), GABAA receptor-associated protein (GABARAP) and the GABAA clustering protein, gephyrin.
Materials and methods
Animals
Adult male C57Bl/6J mice, 42–50 days old at the beginning of experiments and weighing 20–30 g (Jackson Laboratories, Bar Harbor, ME, USA), were group housed four per cage in standard plastic cages (30 × 20 × 16 cm) on corn dust bedding. Mice were given ad libitum access to food and water on a 12: 12 h light: dark schedule (lights on at 07.00 h). All experiments were performed during the light cycle, and were approved by Emory University Institutional Review Board following Institutional Animal Care and Use Committee (IACUC) standards with accordance to the Yerkes Primate Research Center regulations.
Behavioral and histological group assignments
Separate animals were used for behavioral validation of training protocols, and histological examination of mRNA and receptor binding profiles. To behaviorally confirm that our fear acquisition training protocol produced conditioned fear, mice were first given training that consisted of five tone + shock paired trials (Paired, n = 20) or five tone + shock unpaired trials (Unpaired, n = 20). One day after training, mice were tested for conditioned fear. For the extinction of conditioned fear, mice were first given tone + shock training (n = 40) followed 24 h later by CS extinction training (see below; Extinction, n = 20) or exposed to the extinction context without CS presentations (Context, n = 20). One day after extinction training, mice were tested for conditioned fear.
Fear was measured using two widely used measures of conditioned fear: fear-potentiated startle and freezing (Blanchard & Blanchard, 1969; Fendt & Fanselow, 1999; Davis, 2000). Fear-potentiated startle is defined as an augmented startle response in the presence of an aversive conditioned cue, while conditioned freezing is defined as the absence of activity in the presence of an aversive cue. Numerous studies have demonstrated that both these fear responses are dependent upon the integrity of the amygdala complex (Fanselow & LeDoux, 1999; Davis, 2000; Fendt, 2001).
To examine training-induced changes in mRNA levels and receptor binding, 60 naive mice were first assigned to one of six training groups (n = 10 per group). Mice in the first three acquisition groups received training that consisted of five tone + shock paired trials (Paired), five tone + shock unpaired trials (Unpaired), or they remained in home cages (Home). These mice were killed 2.5 h after training (3 h after start of training). Mice in the remaining three extinction groups all received five tone + shock trials on Day 1, and 24 h later were left in home cages (No Extinction), given an extinction training session (Extinction), or exposed to the extinction context without CS presentations (Context). The three extinction groups were killed 1.5 h after training (3 h after start of training). The mice were killed with an overdose of isoflurane anesthesia.
Apparatus
Eight identical startle reflex chambers were used for acclimation, conditioning and testing sessions (SR-LAB, SDI, San Diego, CA, USA). These chambers were also used for the mRNA expression portion of the study. Each chamber consisted of a non-restrictive Plexiglas cylinder 5.5 cm in diameter and 13 cm long mounted on a Plexiglas platform and housed in a ventilated, sound-attenuated cabinet. The floor of each cylinder contained a cradle-shaped grid that consisted of seven stainless steel bars (3.0 mm diameter) spaced 1 cm apart. A piezoelectric accelerometer mounted under each platform detected cylinder movements that were digitized and stored by an interfacing computer assembly. Startle amplitude was defined as the peak accelerometer voltage that occurred during the 100 ms after the onset of the startle stimulus. Response sensitivities were calibrated (SR-LAB Startle Calibration System) to be nearly identical in all startle cylinders.
The tone CS was a 30-s, 70-dB SPL, 12-kHz tone generated by a Tektronix function generator audio oscillator (Model CFG253, Beaverton, OR, USA) that was delivered through a high-frequency speaker (Motorola, Model 948) located 13 cm from the rear of each cylinder. A high-frequency speaker (Radio Shack, 40-1278B) located 15 cm above the cylinder provided a 50-ms white-noise startle stimulus at intensities of 95, 105 and 115 dB. The US was a 0.4-mA scrambled footshock delivered through the stainless steel bars of the cylinder floor by a constant current shock generator (SDI, San Diego, CA, USA) located outside the isolation chambers. Shock levels were verified by using a 1-kOhm resistor across the bars of the shock grids and measuring the voltage drop between the bars to calculate the constant current. The chamber ventilation fans produced a 55-dB white-noise background that was present during acclimation, conditioning and testing sessions. All sound intensities were measured by an audiometer (Radio Shack, 33-2055). Stimuli presentation and data acquisition were controlled by an IBM PC-compatible computer using SR-Laboratory software.
Procedures
Acclimation
In each experiment, mice were given 5 days of pre-exposure to the conditioning chambers to minimize contextual conditioning and to acclimate animals to handling.
Fear conditioning
A mouse was placed in the cage, and 5 min later presented with the five training trials at an average intertrial interval (ITI) of 3.5 min (2.5–4.5 min). Mice in the paired training group were given five tone + shock training trials consisting of a 30-s tone co-terminating with a 0.25-s, 0.4-mA footshock. The mice in the unpaired training group were given the same number of tone and shock presentations, but the stimuli were explicitly unpaired, with a variable interstimulus interval (CS and US separated by 1–2 min).
Fear extinction conditioning
Mice assessed for extinction of fear were given an extinction training session or context training 24 h after conditioning. For extinction training, each mouse was placed in the cage and 5 min later presented with the first of nine blocks of tone-alone trials. Each block contained 10 tone-alone trials, each consisting of a 30-s tone in the absence of the US. A 1-min ITI separated each trial and each block was separated by 3 min. Mice given context training were exposed to the extinction context without CS presentations for the same duration of time as extinction training (approximately 1.5 h).
Fear-conditioning test
Fear-potentiated startle was assessed 24 h after fear or extinction conditioning. During testing, each mouse was placed in the cylinder and 5 min later presented with four startle stimuli at each of three different startle stimulus intensities (95, 105, 115 dB). These initial 12 startle-alone trials served to minimize the contribution of very high startle responses that often occur with the first few startle stimuli. After these initial trials, mice were presented with 10 additional startle-alone trials and 10 tone + startle trials at each of the three startle stimulus intensities. On tone + startle test trials, the startle stimulus was presented 29.750 s after the onset of the tone CS. All trials were presented in a pseudo-random order with the constraint that each trial type occurred only once in each consecutive six trial block. The ITI was 1 min.
Freezing
Freezing in startle reflex chambers was assessed as described previously (Jones et al., 2005; Heldt & Ressler, 2006). Briefly, activity measurements during the presentation of the CS were first converted to the average voltage output for each second of the 5-s activity window. Based on the voltage output, each mouse was give an immobility score of 1 or 0 (0, moving; 1, immobile) for each 1 s of the 5-s activity window. A mean percent immobility score was computed by averaging the five immobility scores and multiplying by 100. The percent immobility score was used as an index of freezing, and in pilot work has demonstrated a high correlation with observational ratings of freezing (rs > 0.89).
Tissue preparation for histological examination
After isoflurane anesthesia, mice were decapitated and the brains were rapidly dissected and frozen on crushed dry ice. Coronal sections (15 μm) of brains were cut on a cryostat (Leica; Nussloch, Germany) under RNase-free conditions at 20 °C, mounted on gelatin-coated slides and stored at −80 °C. For each brain, sections were placed consecutively on 15 slides such that each slide contained roughly corresponding rostral to caudal sections of the brain. One slide from each brain was used for in situ hybridization analyses of each mRNA or [3H]-binding probe. One slide from each mouse was stained with Cresyl violet Nissl stain.
[3H]-Muscimol and [3H]-flunitrazepam binding
For regional localization of GABAA receptor levels, [3H]-muscimol (88 Ci/mmol; PerkinElmer, Waltham, MA, USA) binding was performed according to the method of Palacios et al. (1981). In brief, tissue sections were thawed, dried at room temperature, then preincubated in 50 mM Tris–citrate buffer (pH 7.1) for 20 min at 4 °C. Tissue sections were incubated in 50 mM Tris–citrate buffer containing 10 nM [3H]-muscimol for 40 min at 4 °C. At the end of the incubation, the tissues were rinsed with ice-cold 50 mM Tris–citrate buffer three times for 5 s and subsequently washed with one dip in cold distilled water. Slides were immediately dried in a stream of cool air. Regional localization of benzodiazepine receptor was assessed by means of [3H]-flunitrazepam (85 Ci/mmol; Amersham, Piscataway, NJ, USA) binding using a method described previously (Chhatwal et al., 2005), with modifications. Briefly, tissue sections were thawed and dried at room temperature. Next, sections were preincubated in 50 mM Tris–HCl buffer containing 120 mM NaCl, and 5 mM KCl at pH 7.4. Tissue sections were incubated in above Tris–citrate buffer containing 15 nM [3H]-flunitrazepam for 60 min at 4 °C, rinsed with cold buffer twice for 5 min each, dipped once in ice-cold distilled water, and immediately dried in a stream of cool air. Dried tissue sections were apposed to Biomax MR autoradiography film (Eastman Kodak, Rochester, NY, USA) with a set of tritium standards ([3H]-Micro-scale RPA 510, Amersham) and exposed for 3 weeks ([3H]-flunitrazepam) or 10 weeks ([3H]-muscimol).
Hybridization
In preparation for the hybridization experiments, slides were thawed, air-dried and fixed in a 4% paraformaldehyde solution for 30 min. After washing twice in phosphate-buffered saline (PBS) for 5 min, slides were treated for 7.5 min with Proteinase K (20 μg/mL) dissolved in PK buffer (10 mM Tris, 5 mM EDTA), rinsed for 5 min in PBS, then fixed with 4% paraformaldehyde for 30 min. Next, slides were dipped in H2O, then immersed for 10 min in 0.1 M triethanolamine containing 0.25% acetic anhydride. After 5 min washes in PBS and 0.85% saline, sections were dehydrated for 2 min in 30%, 50%, 70%, 85%, 95% and 100% ethanol, air-dried, and stored in a desiccator at 4 °C.
Hybridization riboprobes were prepared from linearized clones (Table 1) using T7, T3 or SP6 polymerase at high specific activity by only using radioactive 35S-UTP (1250 Ci/mmol, Amersham) in the polymerase reaction. The alpha2 subunit plasmid was constructed by TOPO subcloning (Invitrogen Life Technologies Carlsbad, CA, USA) of a polymerase chain reaction product using mouse cDNA and the following customized primers: Alpha2 sense, AGCCACTGGAGGAAAACATCT; Alpha2 antisense, TCATGGACTGACCCCTAATACAG (Sigma Chemical, St Louis, MO, USA). Following preparation of full-length antisense or sense RNA strands, the RNA was base hydrolysed to average lengths of 100–300 bp. Probes were isolated using G50 Sephadex Quick Spin Columns (Roche Diagnostics, Indianapolis, IN, USA) and diluted to a concentration of 100 000 cpm/μL in hybridization buffer consisting of 50% deionized formamide, 10 mM dithiothreitol (DTT), 20 mM Tris, 300 mM sodium chloride, 5 mM EDTA, 10% dextran sulfate, 1 × Denhardt’s solution, 0.5 mg/mL yeast RNA and 10 mM NaH2PO4. Sections were then incubated overnight in humid chambers at 52 °C with 75 mL/slide of hybridization buffer covered with a Parafilm coverslip.
Table 1.
Source and identification of plasmid clones used to prepare riboprobes
| Clone source | GI number | IMAGE number | |
|---|---|---|---|
| GABAA α1 | ATCC | 15710358 | 5360723 |
| GABAA α2 | cDNA subclone | N/A | N/A |
| GABAA α3 | Open Biosystems | 31560694 | N/A |
| GABAA α5 | Open Biosystems | 38328407 | 6815819 |
| GABAA β2 | Open Biosystems | 29359843 | 6847737 |
| GABAA γ2 | ATCC | 18490287 | 4504205 |
| GAD65 | Invitrogen | 182933 | N/A |
| GAD67 | Open Biosystems | 20073331 | 5358787 |
| GAT-1 | ATCC | 37590748 | 6827121 |
| Gephyrin | ATCC | 20072568 | 5358943 |
| GABARAP | ATCC | 15562144 | 5316477 |
ATCC, American Type Culture Collection; GI, GenInfo Identifier; IMAGE, Integrated Molecular Analysis of Genomes and their Expression.
Following hybridization, slides were incubated in prewarmed 5 × standard sodium citrate (SSC) until coverslips floated off. Sections were washed in 5 × SSC/10 mM DTT at 50 °C for 30 min and treated with 50% formamide/2 × SSC/10 mM DTT at 55 °C for 30 min. Next, slides were washed twice in wash buffer (in mM: Tris, 10; EDTA, 5; NaCl, 100) for 10 min at 37 °C, then treated with RNase A (20 mg/mL) in wash buffer for 30min at 37 °C. Sections were dehydrated for 2 min in 30%, 50%, 70%, 85% and 95% ethanol containing 300 mM ammonium acetate, followed by 100% ethanol. Slides were air dried and apposed to Biomax MR autoradiographic film (Eastman Kodak, Rochester, NY, USA) for 1–5 days. For each riboprobe, a slide from each brain was processed in parallel and handled identically across all time points.
In situ hybridization signal specificities and intensities
Signal specificities and intensities for each probe were assessed simultaneously by comparing brain-paste calibration standards with brain sections treated with antisense or sense radiolabeled probes developed on the same film (Fig. 1). Brain sections hybridized with sense RNA probes resulted in virtually blank autoradiograms (Fig. 1B). Calibration spots were prepared by a modification of the method described for [35S] and [125I] standards (Davenport & Hall, 1988; Palfi et al., 1998; Vizi et al., 2001). Briefly, three whole brains were rinsed to remove blood, blotted dry, and homogenized with 300 μL RNA-free water at 4 °C. Known amounts of [35S]cRNA probe in 10 μL RNA-free water were added to homogenized tissue paste in microcentrifuge tubes and vortexed to ensure the homogenous distribution of the radiolabeled probe. Two rows of six circles (2-mm diameter) were drawn with a felt pen on the back of microscope slide. A 1-μL drop of the 35S-containing paste calibration solution containing a distinct amount of radioactivity was evenly deposited over each circle. The radioactivity (cpm) of a 1-μL sample of brain paste was determined by liquid scintillation to estimate the amount of radioactivity applied on the slides for each probe. The resulting scales covered the range 10–20 000 cpm/cm2, at intervals of approximately halving activity levels. After application of paste on the slides, they were air dried for 20 min and affixed to the microscope slides overnight at 37 °C. Slides were air dried and apposed to autoradiography film along with brain sections treated with radiolabeled probes for 1–5 days.
Fig. 1.
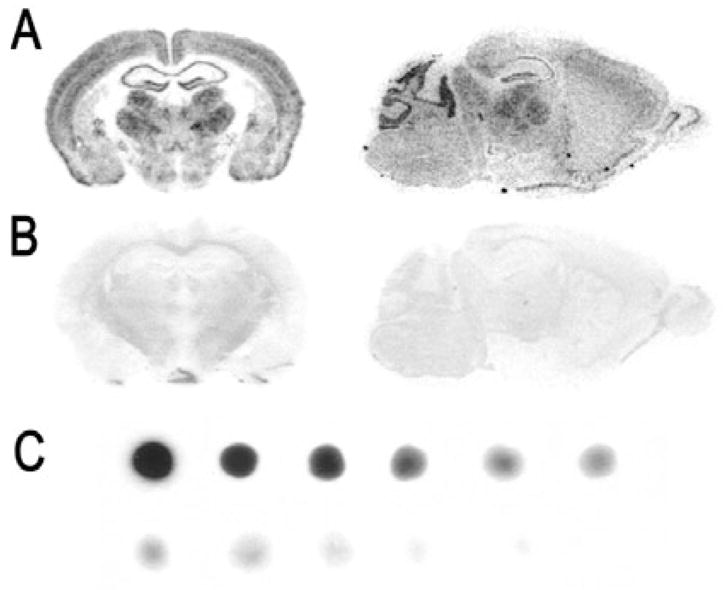
In situ hybridization signal specificities and density distribution. Representative coronal and sagittal autoradiographic images showing the specificity of mRNAs probes. Sections shown were hybridized with (A) antisense or (B) sense [35S]-RNA probes for β2 subunit mRNAs. (C) Representative autoradiographic images of [35S]-tissue paste standard scales.
Image analysis
Each film was scanned using a high-resolution Epson 3700 flat-bed scanner (3000 dpi) and was saved in JPEG format at a size of 32 000 × 18 000 pixels. The hybridization signal intensities of brain regions were calculated on the basis of gray values (GVs) between 0 (brightest) and 255 (darkest) obtained from the luminosity histogram feature of Adobe Photo. Log transformations of GVs [log10 (256/256–GV)] from scanned autoradiographic images of brain paste standards were plotted against known radioactivity values of paste standards (cpm/cm2) to insure signal intensities fell within the linear range of radioactivity, which is reliable for the quantification of autoradiographic films (Davenport & Hall, 1988; Jonker et al., 1997; Palfi et al., 1998). For each section, hybridization signal was determined for the regions of interest (ROI), as well as an adjacent background area with little or no hybridization signal (e.g. internal capsule). To reduce the possible contribution of a signal from neighboring areas, ROI delineation was limited to the innermost 90% of the total area. Measures from each ROI typically fell within the 10–240 gray-scale range. Once established, the settings remained constant for each probe. The anatomical level of analysis was verified using the mouse brain atlas of Paxinos & Franklin (2001) and sections Nissl stained with Cresyl violet from the same mouse.
Data and statistical analysis
Mean startle amplitudes were computed for the 30 trials in the presence of the tone (tone + startle) and the 30 trials in the absence of the tone (startle-alone) by averaging the startle amplitude of each trial type across the three different startle intensities. The percentage change in startle was computed for each mouse by dividing the difference between the mean startle amplitudes of these two trial types (response to tone + startle stimulus minus response to startle stimulus alone) by the mean startle amplitude on startle stimulus-alone trials:
For the quantification, signal intensities were converted to signal to noise ratios by dividing signal intensity by background area. Statistical comparisons were carried out for each analysed amygdala region (LA, BL, CE) and training protocol (acquisition, extinction) independently. Statistical analyses were performed by ANOVA with Dunnett’s t-test for pairwise comparisons against a single control (home or no extinction) to maintain a 0.05 family wise error rate for each analysis. Unplanned comparisons were adjusted with Bonferonni corrections. For ease of presentation, figures are presented as percentages of control group (home or no extinction).
Results
Behavioral results
Retention of conditioned fear
To validate the fear-conditioning training protocol resulted in conditioned fear, mice were tested for either fear-potentiated startle or conditioned freezing 24 h after training. As seen in Fig. 2A, with mice tested for conditioned freezing, paired mice (n = 10) showed significantly more freezing behavior than unpaired animals (n = 10). Likewise, as seen in Fig. 2B, 1 day after training, paired animals (n = 10) displayed significantly more fear-potentiated startle than unpaired animals (n = 10). Independent sample t-tests confirmed that paired animals displayed more conditioned freezing, t18 = 6.82, P < 0.01, or fear-potentiated startle, t18 = 2.37, P < 0.03, when compared with respective unpaired controls
Fig. 2.
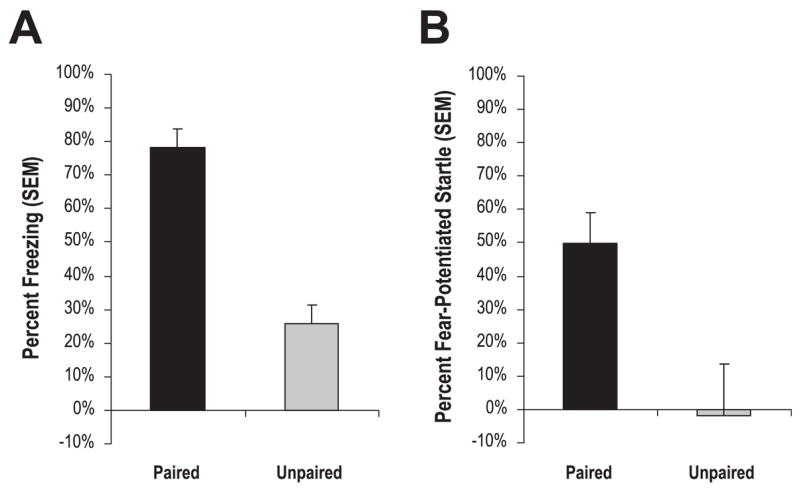
Retention of conditioned fear. One day after training, mice given paired tone + shock training (n = 10) displayed significantly more conditioned freezing (A) or fear-potentiated startle (B) than animals given unpaired tone + shock training (n = 10).
Retention of fear extinction
To validate the fear extinction training protocol, mice were given either CS extinction training (n = 20) or exposure to the extinction context in the absence of the CS (n = 20) 1 day after tone + shock training. Twenty-four hours later, mice were given a post-extinction test. Mice given extinction training showed significantly less conditioned freezing, t18 = 3.67, P < 0.01, and fear-potentiated startle, t18 = 2.69, P < 0.02, than respective extinction context controls (Fig. 3A and B). Together with the retention of fear results, these behavioral data indicate our training protocols are effective in producing ‘long-term’ conditioned fear and extinction, and may be employed to induce and measure genetic markers associated with learned fear and extinction of fear.
Fig. 3.
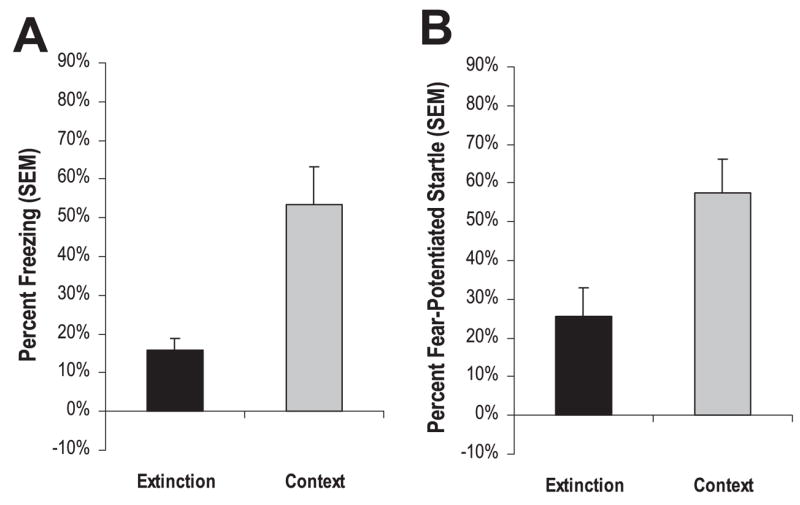
Retention of fear extinction. One day after tone + shock training, mice were given either an extinction training session (Extinction, n = 10) or exposed to the extinction context without CS presentations (Context, n = 10). When tested 24 h later, extinction animals displayed significantly less conditioned freezing (A) or fear-potentiated startle (B) than context animals, indicative of fear extinction.
Histological results
Amygdala mRNA and receptor binding distribution profiles
The expression patterns of GABAA subunits in the amygdala complex are in good general agreement with previous in situ hybridization studies (Araki & Tohyama, 1992; Wisden et al., 1992; Marowsky et al., 2004). As seen in Fig. 4, particularly strong mRNA labeling was observed for the α2 and α3 subunits in the LA and BL nuclei, with more moderate signals from α1, β2 and γ2, and weak α5 expression. The central nucleus was characterized by strong α2 and β3 labeling, and weak to moderate α3, α5, β2 and γ2 hybridization. The presence of α1 was undetected in the CE nucleus, and the expression of α3 was limited to the medial division of the CE nucleus. Representative bright-field photomicrographs of GABAergic associated genes are presented in Fig. 5. GAD67 mRNA was conspicuously labeled in the LA and BL, but with slightly weaker expression in CE. In contrast, the GAD65 signal was abundant in CE but relatively weak in the LA and BL. Similarly, the expression of GAT1 and GABARAP transcripts was more prominent in the CE than the other amygdala nuclei. For gephyrin, hybridization of mRNA was clearly evident in the LA and BL nuclei, and less prominent in the CE, as we have seen in rats previously(Chhatwal et al., 2005). Within the amygdala, the distribution profiles of [3H]-flunitrazepam and [3H]-muscimol binding were consistent with past reports (Canonaco et al., 1997; Luthman & Humpel, 1997; Chacur et al., 1999). As shown in Fig. 6, both GABAA and benzodiazepine-GABAA receptor levels were high in the LA and BL relative to the CE, as noted by [3H]-muscimol and [3H]-flunitrazepam binding, respectively.
Fig. 4.
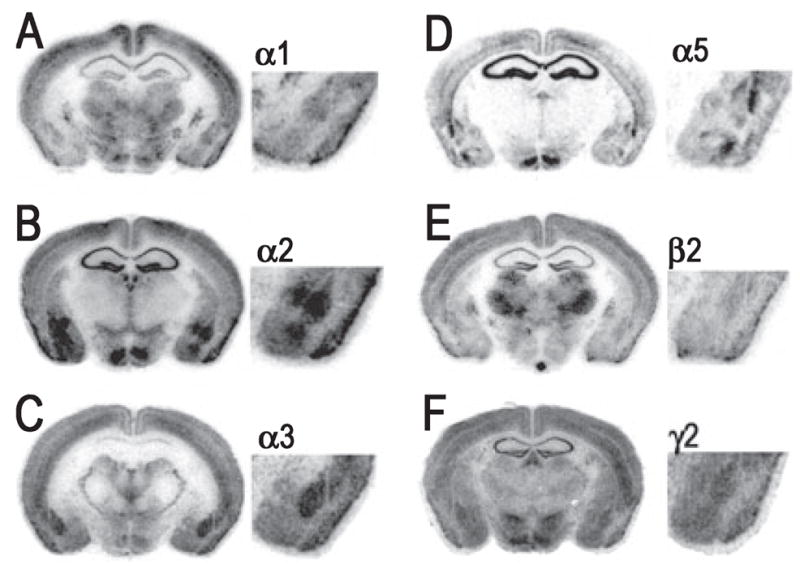
Representative bright-field photomicrograph showing hybridization signals of GABAA receptor subunit mRNAs in the mouse brain. Photomicrographs correspond roughly to coronal sections 1.4 mm posterior to Bregma as referenced by Paxinos & Franklin (2001). (A–F) Sections correspond to GABAA receptor subunits α1, α2, α3, α5, β2 and γ2, respectively.
Fig. 5.
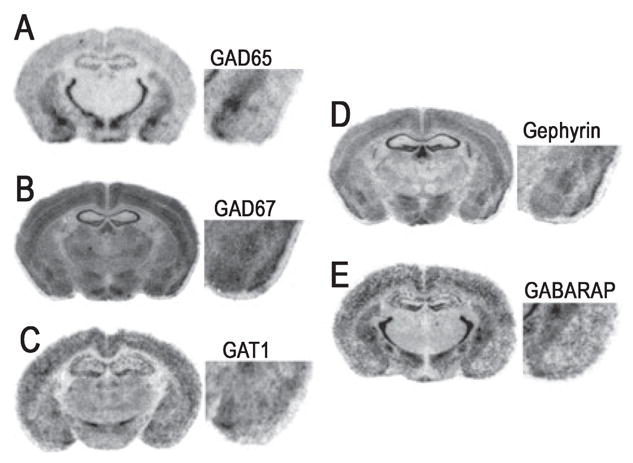
Representative bright-field photomicrographs showing mRNA hybridization signals for GABAergic-associated mRNA in the mouse brain. Photomicrographs correspond roughly to coronal sections 1.4 mm posterior to Bregma as referenced by Paxinos & Franklin (2001). (A–E) Sections correspond to GAD65, GAD67, GABA transporter-1 (GAT1), gephyrin, and GABAA receptor-associated protein (GABARAP), respectively.
Fig. 6.
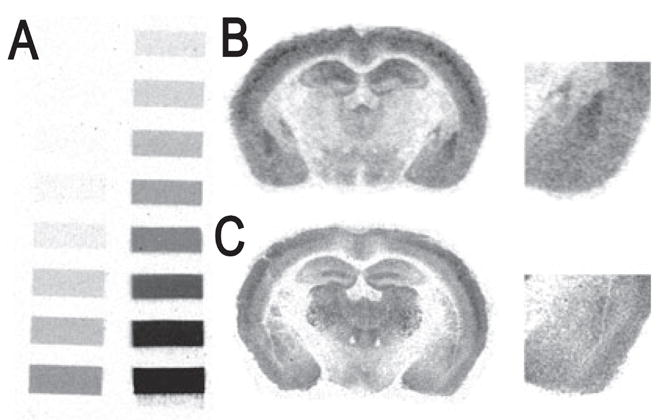
Representative bright-field photomicrographs showing GABAA receptor binding signals at the level of the amygdala in the mouse brain. Photomicrographs correspond roughly to coronal sections 1.4 mm posterior to Bregma as referenced by Paxinos & Franklin (2001). (A) Tritium standards. (B) [3H]-Flunitrazepam binding signal. (C) [3H]-Muscimol binding signal.
Hybridization acquisition results
For the acquisition of fear, one-way ANOVAs revealed a significant group difference in mRNA levels for subunits α1, α5 and β2, as well as levels of GAD67 and GAT1 (Fig. 7). No significant group differences were identified for subunits α2, α3 and γ2, or mRNA levels of GAD65, gephyrin or GABARAP (Table 2).
Fig. 7.

Levels of mRNA in the amygdala following the acquisition of conditioned fear. Mice received training that consisted of five tone + shock paired trials (Paired, n = 10), five tone + shock unpaired trials (Unpaired, n = 10) or remained in home cages (Home, n = 10), and were killed 2.5 h after training (3 h after start of training). Shown are group mRNA transcripts levels deemed significant in the (A) LA, (B) BL and (C) CE amygdala (*P < 0.05).
Table 2.
mRNA signal intensities of amygdala nuclei in animals that remained in home cages, or received training that consisted of tone + shock paired trials or tone + shock unpaired trials
| Subregions and conditions | a1 | a2 | a3 | a5 | b2 | g2 | GAD65 | GAD67 | GAT1 | Gephyrin | RAP |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LA | |||||||||||
| Home | 2.54 ± 0.14 | 2.16 ± 0.16 | 2.67 ± 0.19 | 2.08 ± 0.08 | 2.00 ± 0.11 | 1.93 ± 0.16 | 1.08 ± 0.05 | 1.62 ± 0.08 | 1.26 ± 0.03 | 1.69 ± 0.03 | 1.08 ± 0.10) |
| Unpaired | 2.41 ± 0.15 | 2.29 ± 0.13 | 2.95 ± 0.38 | 2.11 ± 0.09 | 2.75 ± 0.16 | 1.68 ± 0.05 | 1.11 ± 0.05 | 1.45 ± 0.05 | 1.14 ± 0.04 | 1.66 ± 0.07 | 1.08 ± 0.12) |
| Paired | 2.04 ± 0.08 | 2.13 ± 0.14 | 2.76 ± 0.29 | 1.98 ± 0.13 | 2.37 ± 0.21 | 1.81 ± 0.08 | 1.12 ± 0.04 | 1.37 ± 0.04 | 1.25 ± 0.04 | 1.55 ± 0.02 | 1.15 ± 0.05) |
| P-value | 0.028 | 0.589 | 0.683 | 0.654 | 0.005 | 0.296 | 0.775 | 0.020 | 0.036 | 0.110 | 0.750 |
| BL | |||||||||||
| Home | 2.37 ± 0.10 | 2.89 ± 0.22 | 3.36 ± 0.36 | 1.15 ± 0.04 | 2.28 ± 0.12 | 2.08 ± 0.13 | 1.16 ± 0.04 | 1.90 ± 0.11 | 1.44 ± 0.06 | 1.98 ± 0.08 | 1.30 ± 0.06) |
| Unpaired | 2.22 ± 0.10 | 2.79 ± 0.17 | 3.72 ± 0.59 | 1.24 ± 0.06 | 3.39 ± 0.38 | 1.88 ± 0.09 | 1.25 ± 0.04 | 1.89 ± 0.07 | 1.31 ± 0.04 | 1.87 ± 0.05 | 1.38 ± 0.07) |
| Paired | 1.92 ± 0.07 | 2.73 ± 0.15 | 3.67 ± 0.35 | 1.23 ± 0.13 | 2.51 ± 0.23 | 2.03 ± 0.12 | 1.18 ± 0.03 | 1.72 ± 0.03 | 1.32 ± 0.02 | 1.77 ± 0.04 | 1.45 ± 0.12) |
| P-value | 0.005 | 0.716 | 0.773 | 0.738 | 0.007 | 0.446 | 0.176 | 0.173 | 0.079 | 0.057 | 0.312 |
| CE | |||||||||||
| Home | 1.12 ± 0.08 | 2.62 ± 0.17 | 1.78 ± 0.16 | 1.93 ± 0.07 | 1.60 ± 0.08 | 1.74 ± 0.08 | 2.18 ± 0.17 | 1.45 ± 0.08 | 1.20 ± 0.04 | 1.58 ± 0.08 | 2.07 ± 0.14) |
| Unpaired | 1.02 ± 0.06 | 2.64 ± 0.21 | 1.95 ± 0.22 | 1.79 ± 0.07 | 2.19 ± 0.22 | 1.66 ± 0.09 | 2.41 ± 0.20 | 1.45 ± 0.09 | 1.12 ± 0.04 | 1.51 ± 0.09 | 2.06 ± 0.11) |
| Paired | 0.92 ± 0.07 | 2.56 ± 0.16 | 2.02 ± 0.16 | 1.64 ± 0.05 | 1.88 ± 0.16 | 1.74 ± 0.08 | 2.25 ± 0.19 | 1.33 ± 0.06 | 1.17 ± 0.04 | 1.55 ± 0.06 | 2.09 ± 0.17) |
| P-value | 0.135 | 0.914 | 0.543 | 0.009 | 0.027 | 0.685 | 0.668 | 0.432 | 0.395 | 0.821 | 0.979 |
Data are mean ± SEM of intensities defined as signal to noise ratios. Numbers of animals exposed to each condition: Home, n = 10; training consisting of tone + shock paired trials (Paired) n = 10; training consisting of tone + shock unpaired trials (Unpaired) n = 10.
In particular, reliable mRNA changes were noted for α1 within both the LA (F2,27 = 4.09, P < 0.03) and BL (F2,27 = 6.42, P < 0.01). Contrasts on the mean signal ratios indicated that α1 levels in the paired group were significantly less than the home group, P < 0.02, and indicated that paired tone + shock induced a reduction in α1. Significant changes in α5 and GAD67 mRNA levels were also limited to animals receiving paired tone + shock. For α5, mRNA levels were denoted as significant in the CE, F2,27 = 5.57, P < 0.01, which was governed by a reliable mRNA reduction in paired animals when contrasted to the home cage group, P < 0.01. GAD67 mRNA levels were significantly different in the LA (F2,27 = 4.55, P < 0.03), which was also driven by a significant reduction of GAD67 mRNA in the LA of paired animals, P < 0.02. An ANOVA also denoted reliable mRNA changes in levels of β2 mRNA in all nuclei of the amygdala (F2,27 > 4.15, P < 0.02), and contrasts disclosed an increased β2 level in all nuclei of unpaired animals, P < 0.05. The reliable difference in GAT1 mRNA levels (F2,27 = 4.16, P < 0.03) was driven by a significant decrease of LA mRNA levels in unpaired animals when compared with the home group, P < 0.02.
Hybridization extinction results
With extinction of fear, one-way ANOVAs revealed a significant group difference in mRNA levels for subunits α1, α2 and β2, as well as levels of GAD67, GAT1 and gephyrin (Fig. 8). No significant group differences were identified for subunits α3 and γ2, or GAD65 and GABARAP mRNA levels (Table 3).
Fig. 8.
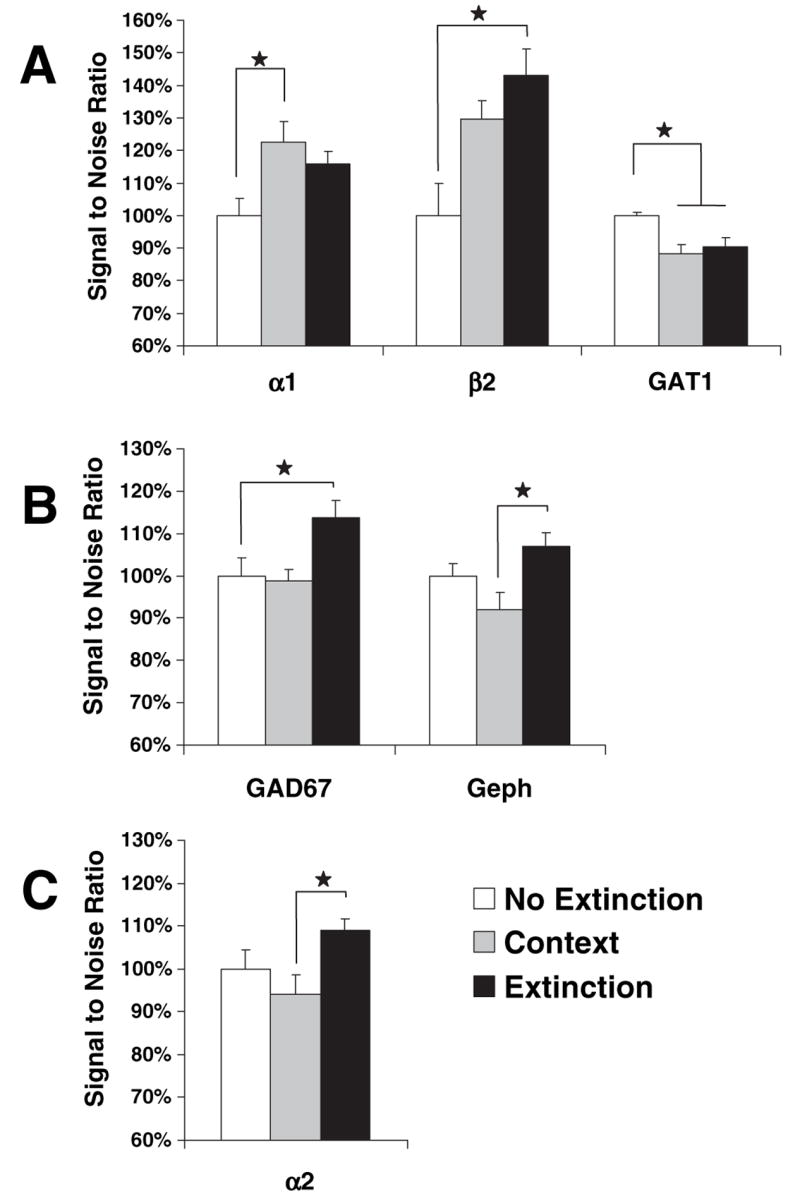
Levels of mRNA in the amygdala following the extinction of conditioned fear. All mice received five tone + shock trials and 24 h later were left in the home cage (No Extinction, n = 10), given an extinction training session (Extinction, n = 10), or exposed to the extinction context alone without CS presentations (Context, n = 10). Groups were killed 1.5 h after training (3 h after start of training). Shown are group mRNA transcripts levels deemed significant in the (A) LA, (B) BL and (C) CE amygdala (*P < 0.05).
Table 3.
mRNA signal intensities of amygdala nuclei in fear-conditioned animals that subsequently received no extinction, exposure to the extinction context without the CS or exposure to extinction training
| Amygdala nucleus and conditions* | a1 | a2 | a3 | a5 | b2 | g2 | GAD65 | GAD67 | GAT1 | Gephyrin | RAP |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LA | |||||||||||
| No extinction | 1.98 ± 0.11 | 2.18 ± 0.13 | 2.86 ± 0.30 | 1.93 ± 0.12 | 3.28 ± 0.36 | 1.73 ± 0.08 | 1.01 ± 0.03 | 1.54 ± 0.09 | 1.40 ± 0.02 | 1.68 ± 0.05 | 1.20 ± 0.08) |
| Context | 2.43 ± 0.16 | 2.23 ± 0.14 | 2.92 ± 0.24 | 1.94 ± 0.12 | 4.25 ± 0.27 | 1.87 ± 0.10 | 1.01 ± 0.04 | 1.44 ± 0.05 | 1.23 ± 0.04 | 1.59 ± 0.07 | 1.05 ± 0.06) |
| Extinction | 2.30 ± 0.09 | 2.49 ± 0.10 | 2.79 ± 0.26 | 1.79 ± 0.08 | 4.69 ± 0.43 | 1.90 ± 0.09 | 1.05 ± 0.05 | 1.60 ± 0.09 | 1.27 ± 0.03 | 1.65 ± 0.01 | 1.23 ± 0.14) |
| P-value | 0.045 | 0.078 | 0.947 | 0.537 | 0.014 | 0.404 | 0.694 | 0.349 | 0.001 | 0.429 | 0.387 |
| BL | |||||||||||
| No extinction | 1.978 ± 0.09 | 2.75 ± 0.15 | 3.65 ± 0.42 | 1.12 ± 0.07 | 4.38 ± 0.59 | 1.93 ± 0.10 | 1.11 ± 0.03 | 1.84 ± 0.08 | 1.57 ± 0.03 | 1.83 ± 0.05 | 1.45 ± 0.07) |
| Context | 2.19 ± 0.11 | 2.74 ± 0.17 | 3.81 ± 0.39 | 1.23 ± 0.07 | 5.12 ± 0.42 | 2.00 ± 0.12 | 1.16 ± 0.02 | 1.81 ± 0.05 | 1.45 ± 0.06 | 1.69 ± 0.07 | 1.56 ± 0.08) |
| Extinction | 2.05 ± 0.11 | 3.07 ± 0.10 | 3.55 ± 0.29 | 1.17 ± 0.06 | 5.95 ± 0.72 | 2.08 ± 0.13 | 1.19 ± 0.05 | 2.09 ± 0.08 | 1.44 ± 0.06 | 1.96 ± 0.06 | 1.53 ± 0.17) |
| P-value | 0.367 | 0.083 | 0.883 | 0.512 | 0.131 | 0.655 | 0.377 | 0.017 | 0.143 | 0.016 | 0.793 |
| CE | |||||||||||
| No extinction | 0.93 ± 0.04 | 2.67 ± 0.15 | 1.88 ± 0.12 | 1.90 ± 0.04 | 2.75 ± 0.33 | 1.75 ± 0.08 | 2.12 ± 0.17 | 1.46 ± 0.06 | 1.30 ± 0.03 | 1.57 ± 0.06 | 2.34 ± 0.12) |
| Context | 1.00 ± 0.05 | 2.50 ± 0.14 | 1.96 ± 0.14 | 1.87 ± 0.11 | 3.08 ± 0.29 | 1.66 ± 0.09 | 2.36 ± 0.22 | 1.47 ± 0.08 | 1.22 ± 0.03 | 1.53 ± 0.08 | 2.22 ± 0.11) |
| Extinction | 0.94 ± 0.09 | 2.91 ± 0.10 | 1.96 ± 0.14 | 1.71 ± 0.07 | 3.37 ± 0.40 | 1.74 ± 0.08 | 2.26 ± 0.19 | 1.57 ± 0.09 | 1.21 ± 0.06 | 1.51 ± 0.05 | 2.30 ± 0.13) |
| P-value | 0.635 | 0.036 | 0.836 | 0.195 | 0.372 | 0.539 | 0.694 | 0.532 | 0.292 | 0.843 | 0.783 |
Data are mean ± SEM of intensities defined as signal to noise ratios.
Numbers of animals exposed to each condition: No extinction, n = 10; exposure to the extinction context without the CS (Context), n = 10; extinction training (Extinction), n = 10.
Group differences in mRNA expression were noted for GAD67 (F2,27 = 4.72, P < 0.02). For this transcript, BL increases were reliably significant in mice receiving extinction training compared with non-extinguished controls, P < 0.03. A group difference in mRNA levels for the β2 subunit was limited to the LA (F2,27 = 3.47, P < 0.05). For mice receiving tone extinction training, the level of β2 was significantly increased, P < 0.05. An ANOVA disclosed training-induced changes in the levels of GAT1 (F2,27 = 4.21, P < 0.05). For this transcript, both extinction and context groups displayed decreased expression levels of GAT1, P < 0.05 in the LA. For α1, mRNA levels were denoted as significant in the LA (F2,27 = 3.52, P < 0.05), which was governed by a reliable mRNA increase in context animals when contrasted to the home group, P < 0.01. Also of note was a significant one-way ANOVA indicating group mRNA levels for the subunit α2 were different in the CE (F2,27 = 4.21, P < 0.05). Dunnett’s t-tests revealed no significant differences when no extinction controls were compared with extinction or context groups, P > 0.05. However, Bonferonni t-tests uncovered an elevated level of α2 mRNA in the CE of tone extinction animals relative to animals exposed to the context alone, P < 0.04. Likewise, significant group differences identified in gephyrin (F2,27 = 4.83, P < 0.02) were driven by an elevated level of gephyrin mRNA in the BL of extinction animals relative to context animals, P < 0.01.
Binding results
An analysis of the [3H]-flunitrazepam binding uncovered a significant group difference in the BL and CE (F2,27 > 5.11, P < 0.02). Within these nuclei, the level of [3H]-flunitrazepam binding was significantly lower in animals receiving tone + shock training compared with the home group, P < 0.02. No other analyses were deemed significant. Likewise, no reliable group differences were revealed in analysing the [3H]-muscimol binding (F2,27 < 1.00, P > 0.05).
Discussion
In the current study, we report changes in levels of different GABAA receptor subunit mRNA and GABAA receptor expression in response to conditioned fear and extinction of fear training protocols. Behavioral results revealed that protocols used to examine training-induced mRNA changes produced reliable acquisition, retention and extinction of tone-CS fear conditioning as measured by two widely used indices of conditioned fear: fear-potentiated startle and freezing (Blanchard & Blanchard, 1969; Fendt & Fanselow, 1999; Davis, 2000).
Irrespective of individual amygdala nuclei, our results showed that significant decreases in α1, α5 and GAD67 mRNA levels were limited to animals receiving paired tone + shock training. In contrast, unpaired tone + shock training induced a significant increase in β2 and a decrease in GAT1 mRNA levels. In the extinction phase of our experiment, we found that tone extinction training produced specific increases in GAD67 and β2 mRNA levels compared with non-extinguished controls. In addition, extinction training induced elevated levels of α2 and gephyrin mRNA relative to animals exposed to the context alone. Both extinction and context groups displayed decreased expression levels of GAT1, and an increase in α1 expression was limited to animals exposed to the context alone. A graphic summary of significant changes found in the current study is illustrated in Fig. 9.
Fig. 9.

Summary of significant mRNA and binding changes associated with fear acquisition and extinction. (A) Significant changes associated with paired tone + shock, P; unpaired tone + shock, U; extinction, E; and context training, C, in LA, BL and CE nucleus of the amygdala. (B) Significant changes in paired and unpaired groups as percentages of the home control group. (C) Significant changes in extinction and context groups as percentages of the no extinction control group.
If confirmed at the protein level, the changes detected in this study suggest that Pavlovian conditioning is capable of altering GABAA receptor subunit composition to potentially modify the number and/or ratio of discrete functional benzodiazepine-GABAA subtype receptors in the amygdala. The distinctive changes likely result from the types of neural associative processes engaged during learning and underlie the long-term associative plasticity that constitutes memory of the conditioning experience. For fear conditioning, associated changes in protein levels would likely decrease the level of phasic and/or tonic inhibition resulting in a state of heightened excitatory drive. Such a shift in the excitation–inhibition balance would support LTP of excitatory CS–US association formed during fear acquisition or represent LTP-induced alterations associated with the consolidation or retention of CS–US associations. The associative processes of extinction are thought to involve the formation of inhibitory connections that actively oppose CS-eliciting responses (Bouton, 2002). Neural network models propose that the inhibition of CS-elicited conditioned responses associated with extinction is, at least in part, mediated by circuits activating and/or strengthening GABAergic transmission in the amygdala (Maren, 2005; Akirav & Maroun, 2007; Myers & Davis, 2007). The extinction-induced alterations in amygdala mRNAs are associated with increased GABAergic transmission and are compatible with the notion that extinction increases inhibitory tone.
The associative processes underlying unpaired tone + shock and context-alone learning rely predominantly upon foreground contextual cues. Explicit unpairing of the CS and US favors contextual cues entering into excitatory associations with the US (Phillips & LeDoux, 1992; Desmedt et al., 1998). Likewise, exposure to the extinction context alone likely results in conditioned inhibitory associations involving foreground contextual cues to the US. The amygdala is critically involved in the acquisition of both CS–US and context–US fear associations (Davis, 2000; Goosens & Maren, 2001; Gale et al., 2004), while extensive evidence indicates that the hippocampus plays a prominent role in encoding the contextual representations that associate with the US in the amygdala (Phillips & LeDoux, 1992; Maren et al., 1997; Desmedt et al., 1998). The hippocampus is also essential for encoding CS-context representations that yield the context dependence of extinction (Bouton et al., 2006). Thus, the different patterns of amygdala mRNA alterations may in part be a function of differential requirement of the hippocampus or other areas (e.g. prefrontal cortex) during associative learning.
In most cases, significant changes of specific GABAergic markers were limited to discrete amygdala nuclei. Distinct subregional changes of mRNA markers relative to training suggest that the contribution of each nucleus to the different types of training protocols was different. A detailed understanding of the contribution of each nucleus to various training protocols remains to be determined; however, past data have shown dissociated roles for the LA, BL and CE nuclei in fear conditioning, depending on the nature of the CS, US and CS–US association (e.g. Nader et al., 2001; Calandreau et al., 2005). Clearly, further investigation will be required to determine how potential changes in the GABAergic circuitry within and among amygdala nuclei contribute to the neural substrates underlying different training protocols.
It should be noted that the current results differ to some degree from a previous report that examined the expression of gephyrin and [3H]-flunitrazepam binding levels at various time points following fear acquisition and extinction in rats (Chhatwal et al., 2005). In that study, fear conditioning significantly decreased in gephyrin mRNA and protein in the BL 2 h after training, and [3H]-flunitrazepam binding in the BL was decreased 6 h after training. In contrast, gephyrin mRNA and protein levels as well as [3H]-flunitrazepam binding significantly increased at similar time points following extinction training. Differences in species (i.e. rat vs mouse) and training protocols between the two studies may account for discrepancies between the two studies. Furthermore, in the current study, the effects of conditioning on mRNA levels were studied 3 h after the start of the training protocol, because marked alterations of amygdala mRNA levels have been reliably reported at this time (Ressler et al., 2002; Rattiner et al., 2004). However, it is also possible that induction of some genes may occur more slowly and thus were not detected in the present study. While the overall analysis of gephyrin in the BL was marginally non-significant (P = 0.056) in the current study, a direct comparison revealed paired animals expressed less mRNA levels of gephyrin than unpaired animals (P < 0.05), thus providing some support for a paired tone + shock-induced reduction of gephyrin in the amygdala.
To our knowledge, no previous studies report the effects of conditioned fear or extinction on GABAA receptor isoform mRNA levels. However, several groups have demonstrated changes in GABAA receptor function and specific GABAA receptor subunit transcript levels that follow stress exposure in rodents (Medina et al., 1983; Rago et al., 1989). For example, Zhang et al. (1998) have demonstrated selective changes in GABAA α1 and α2 mRNA levels in several brain areas following acute stressors, including forced swimming (Schwartz et al., 1987; Park et al., 1993; Avital et al., 2001; Amitani et al., 2005), electroconvulsive shock (Kang et al., 1991a; Pratt et al., 1993) and exposure to social stress (Miller et al., 1987; Kang et al., 1991b). Likewise, more prolonged treatments also induce complex changes in subunit mRNA levels, pharmacological properties of GABAA receptors and total receptor number (Montpied et al., 1993). Changes in the levels of various subunit mRNAs have been associated with alcohol tolerance and withdrawal (Cagetti et al., 2003; Follesa et al., 2003), animal models of epilepsy (Corda et al., 1992; Nishimura et al., 2005), social isolation (Serra et al., 2000, 2007) and benzodiazepine administration (Liu & Glowa, 1999; Tietz et al., 1999).
Changes in α1 and α2 subunit expression
Because α1 subtype receptors are important for the binding of benzodiazepine agonists, this observed decrease may also contribute to the corresponding decreases in benzodiazepine-GABAA receptor as revealed by [3H]-flunitrazepam binding. Indeed, the regulation of the α1 mRNA subunit expression has been implicated in mediating both receptor downregulation and decreased allosteric coupling between GABA and benzodiazepine binding sites (Klein et al., 1994; Ali & Olsen, 2001; Pericic et al., 2007).
The higher levels of CE α2 mRNA in extinction animals, relative to context animals, may well represent a mechanism that enables enhanced inhibitory control of fear responses. The GABAA α2 subtype is the prevailing subtype in the CE (Kaufmann et al., 2003), and plays a major role in modulating anxiety. Direct application of diazepam into the CE enhances GABAA receptor-mediated inhibitory postsynaptic currents (Kang-Park et al., 2004), and may generate anxiolytic effects by influencing activity in the direct projections from the CE to areas known to be involved in anxiety. Studies in mice with point-mutated α2 GABAA receptor subunits are resistant to the anxiolytic-like effects of benzodiazepines in unconditioned and conditioned models of anxiety (Low et al., 2000; Morris et al., 2006). In addition, the enhanced inhibitory transmission induced by diazepam is dramatically reduced in the CE of mice with point-mutated α2 receptors (Marowsky et al., 2004).
Changes in β2 subunit expression
Increases in the expression of β2 receptor subunit mRNA transcripts were induced in groups of mice that received unpaired tone-shock training and CS extinction training. Changes in the level of β2 subunits are of particular interest because the β subunit class contains the GABA binding site and changes in the GABA receptor β isoform can alter GABA affinity and the maximal current amplitude of receptors (Ducic et al., 1995). Furthermore, the β subunit plays a critical role in subcellular delivery and targeting of assembled receptors to distinct cellular locations (Perez-Velazquez & Angelides, 1993; Connolly et al., 1996). Thus, spatial and temporal changes in the expression of β subunit isoforms may provide a mechanism for reorganization of GABAA receptors and alterations capable of shifting the response properties of amygdala neurons to GABA.
The increased level of β2 mRNA expression seen in unpaired tone-shock animals was surprisingly robust in all amygdala nuclei. Interestingly, the expression levels of β-subunits tend to selectively increase in animal epilepsy models (Kamphuis et al., 1995; Tsunashima et al., 1997; Lauren et al., 2003; Nishimura et al., 2005). Thus, the selective β2 increase seen in unpaired animals may be akin to the hyperexcitability found in seizure-evoked animal models of epilepsy. Alternatively, increased β2 subunit expression suggests that other αβ2γ2 receptors might subsequently be upregulated. Whether the increased β2 mRNA expression seen in the current study was associated with the altered expression of other β transcripts remains to be determined. While we did not examine β1 or β3 mRNA levels in the in the present study, the presence of compensatory regulation of these subunits may support such a notion.
Changes in α5 subunit expression
The acquisition of fear was associated with a decrease in the expression levels of α5 subunit mRNA in the CE. In the CA1 area of the hippocampus, α5-GABAA receptors are known to mediate slow tonic inhibition and are important for controlling basal neuronal excitability (Cope et al., 2005; Mtchedlishvili & Kapur, 2006; Prenosil et al., 2006). Specific reduction of α5-GABAA extrasynaptic receptors in the CE may likely decrease tonic inhibition and tend to make the CE more excitable during or after the acquisition of Pavlovian-conditioned fear. The possibility that a decrease in α5-containing GABAA receptors could be associated with the acquisition of conditioned fear is supported by the observation that α5 subunit knockout mice show better performance in various learning and memory tests (Collinson et al., 2002; Crestani et al., 2002).
Changes in GAT1 and GAD67
In the current study, both context and extinction training were associated with decreased mRNA levels of GAT1, which mediates GABA reuptake into presynaptic terminals. A reduction of GAT1-mediated reuptake is known to increase postsynaptic GABA levels and prolong postsynaptic inhibitory currents. Thus, a decrease in GAT1 levels in the amygdala would tend to decrease amygdala excitation and could underlie the means by which fear is inhibited in context and extinction animals. Interestingly, reduced anxiety-like behaviors have been identified in mice lacking GAT1 (Liu et al., 2007).
Changes in mRNA levels of the GABA-synthesizing enzyme GAD67 were detected in amygdala following both acquisition and extinction. GAD67 levels are markers of the GABAergic activity and are believed to reflect biosynthesis keyed to neuronal activity (Bowers et al., 1998). Ultimately, changes in GAD67 would impact GABA synthesis and the resulting amygdala output. The reduced GAD67 expression during acquisition may be associated with loss of inhibitory control of the amygdala, contributing to hyperactivity or prolonged activation of the axis. Conversely, the elevated GAD67 seen during extinction is consistent with enhanced GABA synthesis, as tonically firing GABA neurons express high amounts of GAD67 mRNA (Soghomonian & Martin, 1998).
Changes in benzodiazepine-GABAA binding
After fear acquisition, paired mice demonstrated significantly decreased binding of the benzodiazepine site ligand, [3H]-flunitrazepam. This finding parallels previous data from our lab that showed the acquisition of fear in rats is associated with a decrease in [3H]-flunitrazepam binding (Chhatwal et al., 2005). The reduction in [3H]-flunitrazepam binding can be interpreted as a decrease in the total number or density of the benzodiazepine-GABAA receptors. In contrast, [3H]-muscimol binding, which provides a marker of the total number of functional GABA receptors, was not affected. We therefore conclude that the reduction of [3H]-flunitrazepam binding is not due to a loss of GABAA receptors per se, but rather to the effects of training-induced decrease in benzodiazepine-binding sites associated with GABA receptors. Besides physiological effects on the local excitability of amygdala neurons, there may be also consequences for the pharmacological treatment of conditioned animals. The sensitivity for anxiolytic drugs such as barbiturates and benzodiazepines may be significantly altered.
In addition to changes in gene transcription, the downregulation in benzodiazepine binding seen in mice after fear conditioning may have been triggered by activity-dependent modification of signaling pathways associated with post-translational modification of existing proteins (Luscher & Keller, 2004; Routtenberg & Rekart, 2005). Dynamic changes in the phosphorylation state of GABA receptor subunits correlate temporally with changes in postsynaptic GABAA receptor function and reflect mechanisms underlying short- and long-lasting activity-dependent plasticity (Brunig et al., 2001; Herring et al., 2003; Luscher & Keller, 2004; Goto et al., 2005). Using phosphorylation state-specific antibodies, a future goal will be to determine if changes in the phosphorylation levels of GABA subunits and associated proteins underlie some of the activity-dependent GABAergic plasticity in benzodiazepine binding seen in the current study.
Summary
This study examined training-induced changes in the GABAergic system by measuring the mRNA levels of GABA-related genes and the binding of GABA receptor radioligands in the amygdala after the acquisition and extinction of Pavlovian fear. Following fear conditioning, we found decreased mRNA levels of α1, α5 and GAD67, as well as deceased benzodiazepine binding in the amygdala. Fear extinction induced an increase in mRNA levels of α2, β2, GAD67 and gephyrin, as well as a decrease in GAT1. If associated with changes in protein levels, these patterns of training-induced amygdala changes are in line with a model suggesting that decreased amygdala GABAergic function is induced after fear conditioning, whereas the opposite pattern of activity is induced after extinction. These findings are also particularly relevant in the light of past studies showing pharmacological treatments that increase the excitability of amygdala by decreasing GABA transmission facilitate aversive conditioning, whereas treatments that decrease excitability by increasing GABA transmission inhibit conditioning and produce anxiolytic-like effects (Davis et al., 1994). Although further experiments are needed to determine how changes in the GABAergic circuitry within amygdala nuclei contribute to the neural substrates underlying fear acquisition and extinction, the present findings highlight GABAergic contributions as a function of the particular learning situation.
Acknowledgments
This research was supported by NARSAD, Burroughs Wellcome Foundation, NIH (DA-019624 and F32 MH073389-01), and Center for Behavioral Neuroscience (NSF agreement IBN-98767).
Abbreviations
- BL
basolateral amygdala
- CE
central amygdala
- CS
conditioned stimulus
- DTT
dithiothreitol
- GABA
γ-aminobutyric acid
- GABARAP
GABAA receptor-associated protein
- GVs
gray values
- ITI
intertrial interval
- LA
lateral amygdala
- LTD
long-term depression
- LTP
long-term potentiation
- NMDA
N-methyl-D-aspartate
- PBS
phosphate-buffered saline
- ROI
region of interest
- SSC
standard sodium citrate
- US
unconditioned stimulus
References
- Akirav I, Maroun M. The role of the Medial Prefrontal Cortex-Amygdala Circuit in Stress Effects on the Extinction of Fear. [Retrieved July 18, 2007];Neural Plasticity. 2007 doi: 10.1155/2007/30873. from http://www.hindawi.com/GetArticle.aspx?doi=10.1155/2007/30873&e=cta. [DOI] [PMC free article] [PubMed]
- Ali NJ, Olsen RW. Chronic benzodiazepine treatment of cells expressing recombinant GABAA receptors uncouples allosteric binding: studies on possible mechanisms. J Neurochem. 2001;79:1100–1108. doi: 10.1046/j.1471-4159.2001.00664.x. [DOI] [PubMed] [Google Scholar]
- Amitani M, Umetani Y, Hosoi R, Kobayashi K, Abe K, Inoue O. Changes in in vivo [3H]-Ro15–4513 binding induced by forced swimming in mice. Synapse. 2005;58:23–29. doi: 10.1002/syn.20180. [DOI] [PubMed] [Google Scholar]
- Araki T, Tohyama M. Region-specific expression of GABAA receptor [alpha]3 and [alpha]4 subunits mRNAs in the rat brain. Mol Brain Res. 1992;12:293–307. doi: 10.1016/0169-328x(92)90132-u. [DOI] [PubMed] [Google Scholar]
- Avital A, Richter-Levin G, Leschiner S, Spanier I, Veenman L, Weizman A, Gavish M. Acute and repeated swim stress effects on peripheral benzodiazepine receptors in the rat hippocampus, adrenal, and kidney. Neuropsychopharmacology. 2001;25:669–678. doi: 10.1016/S0893-133X(01)00286-X. [DOI] [PubMed] [Google Scholar]
- Bauer EP, LeDoux JE. Heterosynaptic long-term potentiation of inhibitory interneurons in the lateral amygdala. J Neurosci. 2004;24:9507–9512. doi: 10.1523/JNEUROSCI.3567-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard DC, Blanchard RJ. Crouching as an index of fear. J Comp Physiol Psychol. 1969;67:370–375. doi: 10.1037/h0026779. [DOI] [PubMed] [Google Scholar]
- Bouton ME. Context, ambiguity, and unlearning: sources of relapse after behavioural extinction. Biological Psychiatry. 2002;52:976–986. doi: 10.1016/s0006-3223(02)01546-9. [DOI] [PubMed] [Google Scholar]
- Bouton ME, Westbrook RF, Corcoran KA, Maren S. Contextual and temporal modulation of extinction: behavioral and biological mechanisms. Biol Psychiatry. 2006;60:352–360. doi: 10.1016/j.biopsych.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Bowers G, Cullinan WE, Herman JP. Region-specific regulation of glutamic acid decarboxylase (GAD) mRNA expression in central stress circuits. J Neurosci. 1998;18:5938–5947. doi: 10.1523/JNEUROSCI.18-15-05938.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. Eur J Neurosci. 2001;13:1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Cagetti E, Liang J, Spigelman I, Olsen RW. Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol Pharmacol. 2003;63:53–64. doi: 10.1124/mol.63.1.53. [DOI] [PubMed] [Google Scholar]
- Calandreau L, Desmedt A, Decorte L, Jaffard R. A different recruitment of the lateral and basolateral amygdala promotes contextual or elemental conditioned association in Pavlovian fear conditioning. Learn Mem. 2005;12:383–388. doi: 10.1101/lm.92305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canonaco M, Tavolaro R, Facciolo RM. Dimorphic distribution of the two main GABA(A) binding sites in cortical and limbic areas of a rodent living in natural environmental conditions. J Comp Neurol. 1997;380:423–434. [PubMed] [Google Scholar]
- Chacur C, Raymond R, Hipolide DC, Giugliano EB, Leite JR, Nobrega JN. Immediate increase in benzodiazepine binding in rat brain after a single brief experience in the plus maze: a paradoxical effect. Neurosci Lett. 1999;269:29–32. doi: 10.1016/s0304-3940(99)00425-5. [DOI] [PubMed] [Google Scholar]
- Chapman PF, Ramsay MF, Krezel W, Knevett SG. Synaptic plasticity in the amygdala: comparisons with hippocampus. Ann NY Acad Sci. 2003;985:114–124. doi: 10.1111/j.1749-6632.2003.tb07076.x. [DOI] [PubMed] [Google Scholar]
- Chhatwal JP, Myers KM, Ressler KJ, Davis M. Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci. 2005;25:502–506. doi: 10.1523/JNEUROSCI.3301-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, Sur C, Smith A, Otu FM, Howell O, Atack JR, McKernan RM, Seabrook GR, Dawson GR, Whiting PJ, Rosahl TW. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci. 2002;22:5572–5580. doi: 10.1523/JNEUROSCI.22-13-05572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly CN, Wooltorton JRA, Smart TG, Moss SJ. Subcellular localization of gamma-aminobutyric acid type A receptors is determined by receptor beta subunits. PNAS. 1996;93:9899–9904. doi: 10.1073/pnas.93.18.9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Hughes SW, Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosci. 2005;25:11553–11563. doi: 10.1523/JNEUROSCI.3362-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corda M, Orlandi M, Lecca D, Giorgi O. Decrease in GABAergic function induced by pentylenetetrazol kindling in rats: antagonism by MK-801. J Pharmacol Exp Ther. 1992;262:792–800. [PubMed] [Google Scholar]
- Crestani F, Keist R, Fritschy JM, Benke D, Vogt K, Prut L, Bluthmann H, Mohler H, Rudolph U. Trace fear conditioning involves hippocampal alpha 5 GABAA receptors. PNAS. 2002;99:8980–8985. doi: 10.1073/pnas.142288699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Cunha C, Wolfman C, Levi de Stein M, Ruschel AC, Izquierdo I, Medina JH. Anxiogenic effects of the intraamygdala injection of flumazenil, a benzodiazepine receptor antagonist. Funct Neurol. 1992;7:401–405. [PubMed] [Google Scholar]
- Davenport AP, Hall MD. Comparison between brain paste and polymer [125I]standards for quantitative receptor autoradiography. J Neurosci Meth. 1988;25:75–82. doi: 10.1016/0165-0270(88)90122-7. [DOI] [PubMed] [Google Scholar]
- Davis M. The role of the amygdala in conditioned and unconditioned fear and anxiety. In: Aggleton JP, editor. The Amygdala: a Functional Analysis. Oxford University Press; New York: 2000. pp. 213–287. [Google Scholar]
- Davis M, Falls WA, Gerwirtz J. Neural systems involved in fear inhibition: extinction and conditioned inhibition. In: Myslobodsky M, Weiner I, editors. Contemporary Issues in Modeling Psychopathology. Kluwer Academic; Boston: 2000. pp. 113–141. [Google Scholar]
- Davis M, Rainnie D, Cassell M. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 1994;17:208–214. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Desmedt A, Garcia R, Jaffard R. Differential modulation of changes in hippocampal-septal synaptic excitability by the amygdala as a function of either elemental or contextual fear conditioning in mice. J Neurosci. 1998;18:480–487. doi: 10.1523/JNEUROSCI.18-01-00480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducic I, Caruncho HJ, Zhu Wei J, Vicini S, Costa E. gamma-Aminobutyric acid gating of Cl-channels in recombinant GABAA receptors. J Pharmacol Exp Ther. 1995;272:438–445. [PubMed] [Google Scholar]
- Falls WA, Miserendino MJD, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an excitatory amino acid antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, LeDoux JE. Why we think plasticity underlying Pavlovian fear conditioning occurs in the basolateral amygdala. Neuron. 1999;23:229–232. doi: 10.1016/s0896-6273(00)80775-8. [DOI] [PubMed] [Google Scholar]
- Fendt M. Injections of the NMDA receptor antagonist aminophosphonopentanoic acid into the lateral nucleus of the amygdala block the expression of fear-potentiated startle and freezing. J Neurosci. 2001;21:4111–4115. doi: 10.1523/JNEUROSCI.21-11-04111.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt M, Fanselow MS. The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev. 1999;23:743–760. doi: 10.1016/s0149-7634(99)00016-0. [DOI] [PubMed] [Google Scholar]
- Follesa P, Mancuso L, Biggio F, Mostallino MC, Manca A, Mascia MP, Busonero F, Talani G, Sanna E, Biggio G. gamma-Hydroxybutyric acid and diazepam antagonize a rapid increase in GABAA receptors alpha 4 subunit mRNA abundance induced by ethanol withdrawal in cerebellar granule cells. Mol Pharmacol. 2003;63:896–907. doi: 10.1124/mol.63.4.896. [DOI] [PubMed] [Google Scholar]
- Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR, Wiltgen B, Fanselow MS. Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J Neurosci. 2004;24:3810–3815. doi: 10.1523/JNEUROSCI.4100-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goosens KA, Maren S. Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learn Mem. 2001;8:148–155. doi: 10.1101/lm.37601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Terunuma M, Kanematsu T, Misumi Y, Moss SJ, Hirata M. Direct interaction of N-ethylmaleimide-sensitive factor with GABAA receptor [beta] subunits. Mol Cell Neurosci. 2005;30:197–206. doi: 10.1016/j.mcn.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Green S, Vale AL. Role of amygdaloid nuclei in the anxiolytic effects of benzodiazepines in rats. Behav Pharmacol. 1992;3:261–264. [PubMed] [Google Scholar]
- Harris JA, Westbrook RF. Effects of benzodiazepine microinjection into the amygdala or periaqueductal gray on the expression of conditioned fear and hypoalgesia in rats. Behav Neurosci. 1995;109:295–304. doi: 10.1037//0735-7044.109.2.295. [DOI] [PubMed] [Google Scholar]
- Heldt SA, Ressler KJ. Lesions of the habenula produce stress- and dopamine-dependent alterations in prepulse inhibition and locomotion. Brain Res. 2006:1073–1074. 229–239. doi: 10.1016/j.brainres.2005.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstetter FJ. Stress-induced hypoalgesia and defensive freezing are attenuated by application of diazepam to the amygdala. Pharmacol Biochem Behav. 1993;44:433–438. doi: 10.1016/0091-3057(93)90487-e. [DOI] [PubMed] [Google Scholar]
- Herring D, Huang R, Singh M, Robinson LC, Dillon GH, Leidenheimer NJ. Constitutive GABAA receptor endocytosis is dynamin-mediated and dependent on a dileucine AP2 adaptin-binding motif within the {beta}2 subunit of the receptor. J Biol Chem. 2003;278:24046–24052. doi: 10.1074/jbc.M301420200. [DOI] [PubMed] [Google Scholar]
- Hevers W, Luddens H. The diversity of GABAA receptors. Pharmacological and electrophysiological properties of GABAA channel subtypes. Mol Neurobiol. 1998;18:35–86. doi: 10.1007/BF02741459. [DOI] [PubMed] [Google Scholar]
- Hobin JA, Goosens KA, Maren S. Context-dependent neuronal activity in the lateral amygdala represents fear memories after extinction. J Neurosci. 2003;23:8410–8416. doi: 10.1523/JNEUROSCI.23-23-08410.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SV, Heldt SA, Davis M, Ressler KJ. Olfactory-mediated fear conditioning in mice: simultaneous measurements of fear-potentiated startle and freezing. Behav Neurosci. 2005;119:329–335. doi: 10.1037/0735-7044.119.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker A, de Boer PAJ, van den Hoff MJB, Lamers WH, Moorman AFM. Towards quantitative in situ hybridization. J Histochem Cytochem. 1997;45:413–424. doi: 10.1177/002215549704500309. [DOI] [PubMed] [Google Scholar]
- Kamphuis W, De Rijk TC, Lopes da Silva FH. Expression of GABAA receptor subunit mRNAs in hippocampal pyramidal and granular neurons in the kindling model of epileptogenesis: an in situ hybridization study. Brain Res Mol Brain Res. 1995;31:33–47. doi: 10.1016/0169-328x(95)00022-k. [DOI] [PubMed] [Google Scholar]
- Kang I, Miller LG, Moises J, Bazan N. GABAA receptor mRNAs are increased after electroconvulsive shock. Psychopharmacol Bull. 1991a;27:359–363. [PubMed] [Google Scholar]
- Kang I, Thompson ML, Heller J, Miller LG. Persistent elevation in GABAA receptor subunit mRNAs following social stress. Brain Res Bull. 1991b;26:809–812. doi: 10.1016/0361-9230(91)90179-n. [DOI] [PubMed] [Google Scholar]
- Kang-Park MH, Wilson WA, Moore SD. Differential actions of diazepam and zolpidem in basolateral and central amygdala nuclei. Neuropharmacology. 2004;46:1–9. doi: 10.1016/s0028-3908(03)00340-x. [DOI] [PubMed] [Google Scholar]
- Kaufmann WA, Humpel C, Alheid GF, Marksteiner J. Compartmentation of alpha 1 and alpha 2 GABA(A) receptor subunits within rat extended amygdala: implications for benzodiazepine action. Brain Res. 2003;964:91–99. doi: 10.1016/s0006-8993(02)04082-9. [DOI] [PubMed] [Google Scholar]
- Klein RL, Whiting PJ, Harris RA. Benzodiazepine treatment causes uncoupling of recombinant GABAA receptors expressed in stably transfected cells. J Neurochem. 1994;63:2349–2352. doi: 10.1046/j.1471-4159.1994.63062349.x. [DOI] [PubMed] [Google Scholar]
- Lauren HB, Pitkanen A, Nissinen J, Soini SL, Korpi ER, Holopainen IE. Selective changes in gamma-aminobutyric acid type A receptor subunits in the hippocampus in spontaneously seizing rats with chronic temporal lobe epilepsy. Neurosci Lett. 2003;349:58–62. doi: 10.1016/s0304-3940(03)00735-3. [DOI] [PubMed] [Google Scholar]
- Liu GX, Cai GQ, Cai YQ, Sheng ZJ, Jiang J, Mei Z, Wang ZG, Guo L, Fei J. Reduced anxiety and depression-like behaviors in mice lacking GABA transporter subtype 1. Neuropsychopharmacology. 2007;32:1531–1539. doi: 10.1038/sj.npp.1301281. [DOI] [PubMed] [Google Scholar]
- Liu M, Glowa JR. Alterations of GABAA receptor subunit mRNA levels associated with increases in punished responding induced by acute alprazolam administration: an in situ hybridization study. Brain Res. 1999;822:8–16. doi: 10.1016/s0006-8993(98)01205-0. [DOI] [PubMed] [Google Scholar]
- Low K, Crestani F, Keist R, Benke D, Brunig I, Benson JA, Fritschy JM, Rulicke T, Bluethmann H, Mohler H, Rudolph U. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290:131–134. doi: 10.1126/science.290.5489.131. [DOI] [PubMed] [Google Scholar]
- Luscher B, Keller CA. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther. 2004;102:195–221. doi: 10.1016/j.pharmthera.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Luthman J, Humpel C. Pentylenetetrazol kindling decreases N-methyl-aspartate and kainate but increases gamma-aminobutyric acid-A receptor binding in discrete rat brain areas. Neurosci Lett. 1997;239:9–12. doi: 10.1016/s0304-3940(97)00880-x. [DOI] [PubMed] [Google Scholar]
- Mahanty NK, Sah P. Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature. 1998;394:683–687. doi: 10.1038/29312. [DOI] [PubMed] [Google Scholar]
- Maren S. Building and burying fear memories in the brain. Neuroscientist. 2005;11:89–99. doi: 10.1177/1073858404269232. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Fanselow MS. Neurotoxic lesions of the dorsal hippocampus and Pavlovian fear conditioning in rats. Behav Brain Res. 1997;88:261–274. doi: 10.1016/s0166-4328(97)00088-0. [DOI] [PubMed] [Google Scholar]
- Marowsky A, Fritschy JM, Vogt KE. Functional mapping of GABA A receptor subtypes in the amygdala. Eur J Neurosci. 2004;20:1281–1289. doi: 10.1111/j.1460-9568.2004.03574.x. [DOI] [PubMed] [Google Scholar]
- McAllister WR, McAllister DE. Behavioral measurements of conditioned fear. In: Brush FR, editor. Aversive Conditioning and Learning. Academic Press; New York: 1971. pp. 105–179. [Google Scholar]
- Medina JH, Novas ML, Wolfman CN, Levi de Stein M, De Robertis E. Benzodiazepine receptors in rat cerebral cortex and hippocampus undergo rapid and reversible changes after acute stress. Neuroscience. 1983;9:331–335. doi: 10.1016/0306-4522(83)90298-1. [DOI] [PubMed] [Google Scholar]
- Miller LG, Thompson ML, Greenblatt DJ, Deutsch SI, Shader RI, Paul SM. Rapid increase in brain benzodiazepine receptor binding following defeat stress in mice. Brain Res. 1987;414:395–400. doi: 10.1016/0006-8993(87)90023-0. [DOI] [PubMed] [Google Scholar]
- Montpied P, Weizman A, Weizman R, Kook KA, Morrow AL, Paul SM. Repeated swim-stress reduces GABAA receptor [alpha] subunit mRNAs in the mouse hippocampus. Mol Brain Res. 1993;18:267–272. doi: 10.1016/0169-328x(93)90199-y. [DOI] [PubMed] [Google Scholar]
- Morris HV, Dawson GR, Reynolds DS, Atack JR, Stephens DN. Both α2 and α3 GABAA receptor subtypes mediate the anxiolytic properties of benzodiazepine site ligands in the conditioned emotional response paradigm. Eur J Neurosci. 2006;23:2495–2504. doi: 10.1111/j.1460-9568.2006.04775.x. [DOI] [PubMed] [Google Scholar]
- Mtchedlishvili Z, Kapur J. High-affinity, slowly desensitizing GABAA receptors mediate tonic inhibition in hippocampal dentate granule cells. Mol Pharmacol. 2006;69:564–575. doi: 10.1124/mol.105.016683. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2007;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- Nader K, Majidishad P, Amorapanth P, LeDoux JE. Damage to the lateral and central, but not other, amygdaloid nuclei prevents the acquisition of auditory fear conditioning. Learn Mem. 2001;8:156–163. doi: 10.1101/lm.38101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Schwarzer C, Gasser E, Kato N, Vezzani A, Sperk G. Altered expression of GABAa and GABAb receptor subunit mRNAs in the hippocampus after kindling and electrically induced status epilepticus. Neuroscience. 2005;134:691–704. doi: 10.1016/j.neuroscience.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Palacios JM, Wamsley JK, Kuhar MJ. High affinity GABA receptors – Autoradiographic localization. Brain Res. 1981;222:285–307. doi: 10.1016/0006-8993(81)91034-9. [DOI] [PubMed] [Google Scholar]
- Palfi A, Hatvani L, Gulya K. A new quantitative film autoradiographic method of quantifying mRNA transcripts for in situ hybridization. J Histochem Cytochem. 1998;46:1141–1150. doi: 10.1177/002215549804601006. [DOI] [PubMed] [Google Scholar]
- Pape HC, Stork O. Genes and mechanisms in the amygdala involved in the formation of fear memory. Ann N Y Acad Sci. 2003;985:92–105. doi: 10.1111/j.1749-6632.2003.tb07074.x. [DOI] [PubMed] [Google Scholar]
- Park CH, Hitri A, Lukacs LG, Deutsch SI. Swim stress selectively alters the specific binding of a benzodiazepine antagonist in mice. Pharmacol Biochem Behav. 1993;45:299–304. doi: 10.1016/0091-3057(93)90242-l. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. Academic Press; New York: 2007. [Google Scholar]
- Perez-Velazquez JL, Angelides KJ. Assembly of GABAA receptor subunits determines sorting and localization in polarized cells. Brain Res. 1993;361:457–460. doi: 10.1038/361457a0. [DOI] [PubMed] [Google Scholar]
- Pericic D, Strac DS, Jembrek MJ, Vlainic J. Allosteric uncoupling and up-regulation of benzodiazepine and GABA recognition sites following chronic diazepam treatment of HEK 293 cells stably transfected with alpha(1)beta(2)gamma(2S) subunits of GABA(A) receptors. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:177–187. doi: 10.1007/s00210-007-0152-z. [DOI] [PubMed] [Google Scholar]
- Pesold C, Treit D. The central and basolateral amygdala differentially mediate the anxiolytic effects of benzodiazepines. Brain Res. 1995;671:213–221. doi: 10.1016/0006-8993(94)01318-c. [DOI] [PubMed] [Google Scholar]
- Petersen EN, Braestrup C, Scheel-Kruger J. Evidence that the anticonflict effect of midazolam in amygdala is mediated by the specific benzodiazepine receptors. Neurosci Lett. 1985;53:285–288. doi: 10.1016/0304-3940(85)90552-x. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Pratt JS, Kang I, Bazan NG, Miller LG. Electroconvulsive shock alters GABAA receptor subunit mRNAs: use of quantitative PCR methodology. Brain Res Bull. 1993;30:691–693. doi: 10.1016/0361-9230(93)90101-g. [DOI] [PubMed] [Google Scholar]
- Prenosil GA, Schneider Gasser EM, Rudolph U, Keist R, Fritschy JM, Vogt KE. Specific subtypes of GABAA receptors mediate phasic and tonic forms of inhibition in hippocampal pyramidal neurons. J Neurophysiol. 2006;96:846–857. doi: 10.1152/jn.01199.2005. [DOI] [PubMed] [Google Scholar]
- Rago L, Kiivet RA, Harro J, Pold M. Central-and peripheral-type benezodiazepine receptors: similar regulation by stress and GABA recptor agonists. Phamacol Biochem Behav. 1989;32:879–883. doi: 10.1016/0091-3057(89)90052-x. [DOI] [PubMed] [Google Scholar]
- Rattiner LM, Davis M, French CT, Ressler KJ. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci. 2004;24:4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Paschall G, Zhou XL, Davis M. Regulation of synaptic plasticity genes during consolidation of fear conditioning. J Neurosci. 2002;22:7892–7902. doi: 10.1523/JNEUROSCI.22-18-07892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routtenberg A, Rekart JL. Post-translational protein modification as the substrate for long-lasting memory. Trends Neurosci. 2005;28:12–19. doi: 10.1016/j.tins.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Sanders SK, Shekhar A. Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacol Biochem Behav. 1995;52:701–706. doi: 10.1016/0091-3057(95)00153-n. [DOI] [PubMed] [Google Scholar]
- Santini E, Muller RU, Quirk GJ. Consolidation of extinction learning involves transfer from NMDA-independent to NMDA-dependent memory. J Neurosci. 2001;21:9009–9017. doi: 10.1523/JNEUROSCI.21-22-09009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz RD, Wess MJ, Labarca R, Skolnick P, Paul SM. Acute stress enhances the activity of the GABA receptor-gated chloride ion channel in brain. Brain Res. 1987;411:151–155. doi: 10.1016/0006-8993(87)90692-5. [DOI] [PubMed] [Google Scholar]
- Serra M, Pisu MG, Littera M, Papi G, Sanna E, Tuveri F, Usala L, Purdy RH, Biggio G. Social isolation-induced decreases in both the abundance of neuroactive steroids and GABAA receptor function in rat brain. J Neurochem. 2000;75:732–740. doi: 10.1046/j.1471-4159.2000.0750732.x. [DOI] [PubMed] [Google Scholar]
- Serra M, Sanna E, Mostallino MC, Biggio G. Social isolation stress and neuroactive steroids. Eur Neuropsychopharmacol. 2007;17:1–11. doi: 10.1016/j.euroneuro.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Shibata S, Yamashita K, Yamamoto E, Ozaki T, Ueki S. Effects of benzodiazepine and GABA antagonists on anticonflict effects of antianxiety drugs injected into the rat amygdala in a water-lick suppression test. Psychopharmacology (Berl) 1989;98:38–44. doi: 10.1007/BF00442003. [DOI] [PubMed] [Google Scholar]
- Smith TA. Type A gamma-aminobutyric acid (GABAA) receptor subunits and benzodiazepine binding: significance to clinical syndromes and their treatment. Br J Biomed Sci. 2001;58:111–121. [PubMed] [Google Scholar]
- Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci. 1998;19:500–505. doi: 10.1016/s0165-6147(98)01270-x. [DOI] [PubMed] [Google Scholar]
- Stork O, Ji FY, Obata K. Reduction of extracellular GABA in the mouse amygdala during and following confrontation with a conditioned fear stimulus. Neurosci Lett. 2002;327:138–142. doi: 10.1016/s0304-3940(02)00387-7. [DOI] [PubMed] [Google Scholar]
- Szapiro G, Vianna MR, McGaugh JL, Medina JH, Izquierdo I. The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus. 2003;13:53–58. doi: 10.1002/hipo.10043. [DOI] [PubMed] [Google Scholar]
- Teuber L, Watjens F, Jensen LH. Ligands for the benzodiazepine binding site – a survey. Curr Pharm Des. 1999;5:317–343. [PubMed] [Google Scholar]
- Tietz EI, Huang X, Chen S, Ferencak WF., III Temporal and regional regulation of [alpha]1, [beta]2 and [beta]3, but not [alpha]2, [alpha]4, [alpha]5, [alpha]6, [beta]1 or [gamma]2 GABAA receptor subunit messenger RNAs following one-week oral flurazepam administration. Neuroscience. 1999;91:327–341. doi: 10.1016/s0306-4522(98)00516-8. [DOI] [PubMed] [Google Scholar]
- Tsunashima K, Schwarzer C, Kirchmair E, Sieghart W, Sperk G. GABAA receptor subunits in the rat hippocampus III: altered messenger RNA expression in kainic acid-induced epilepsy. Neuroscience. 1997;80:1019–1032. doi: 10.1016/s0306-4522(97)00144-9. [DOI] [PubMed] [Google Scholar]
- Vizi S, Palfi A, Hatvani L, Gulya K. Methods for quantification of in situ hybridization signals obtained by film autoradiography and phosphorimaging applied for estimation of regional levels of calmodulin mRNA classes in the rat brain. Brain Res Protocols. 2001;8:32–44. doi: 10.1016/s1385-299x(01)00082-4. [DOI] [PubMed] [Google Scholar]
- Walker DL, Davis M. The role of amygdala glutamate receptors in fear learning, fear-potentiated startle, and extinction. Pharmacol Biochem Behav. 2002;71:379–392. doi: 10.1016/s0091-3057(01)00698-0. [DOI] [PubMed] [Google Scholar]
- Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I Telencephalon, diencephalon, mesencephalon. J Neurosci. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SH, Mao SC, Lin HC, Gean PW. Synaptic expression of glutamate receptor after encoding of fear memory in the rat amygdala. Mol Pharmacol. 2006;69:299–308. doi: 10.1124/mol.105.017194. [DOI] [PubMed] [Google Scholar]
- Zangrossi H, Jr, Viana MB, Graeff FG. Anxiolytic effect of intra-amygdala injection of midazolam and 8-hydroxy-2-(di-n-propylamino) tetralin in the elevated T-maze. Eur J Pharmacol. 1999;369:267–270. doi: 10.1016/s0014-2999(99)00075-8. [DOI] [PubMed] [Google Scholar]
- Zhang L, Rubinow DR, Ma W, Marks JM, Feldman AN, Barker JL, Tathan TA. GABA receptor subunit mRNA expression in brain of conflict, yoked control and control rats. Brain Res Mol Brain Res. 1998;58:16–26. doi: 10.1016/s0169-328x(98)00061-8. [DOI] [PubMed] [Google Scholar]