

Abstract
Context
Genetic inheritance and developmental life stress both contribute to major depressive disorder in adults. Child abuse and trauma alter the endogenous stress response, principally corticotropin-releasing hormone and its downstream effectors, suggesting that a gene × environment interaction at this locus may be important in depression.
Objective
To examine whether the effects of child abuse on adult depressive symptoms are moderated by genetic polymorphisms within the corticotropin-releasing hormone type 1 receptor (CRHR1) gene.
Design
Association study examining gene × environment interactions between genetic polymorphisms at the CRHR1 locus and measures of child abuse on adult depressive symptoms.
Setting
General medical clinics of a large, public, urban hospital and Emory University, Atlanta, Georgia.
Participants
The primary participant population was 97.4% African American, of low socioeconomic status, and with high rates of lifetime trauma (n=422). A supportive independent sample (n=199) was distinct both ethnically (87.7% Caucasian) and socioeconomically (less impoverished).
Main Outcome Measures
Beck Depression Inventory scores and history of major depressive disorder by the Structured Clinical Interview for DSM-IV Axis I Disorders.
Results
Fifteen single-nucleotide polymorphisms spanning 57 kilobases of the CRHR1 gene were examined. We found significant gene × environment interactions with multiple individual single-nucleotide polymorphisms (eg, rs110402, P=.008) as well as with a common haplotype spanning intron 1 (P <.001). Specific CRHR1 polymorphisms appeared to moderate the effect of child abuse on the risk for adult depressive symptoms. These protective effects were supported with similar findings in a second independent sample (n=199).
Conclusions
These data support the corticotropin-releasing hormone hypothesis of depression and suggest that a gene × environment interaction is important for the expression of depressive symptoms in adults with CRHR1 risk or protective alleles who have a history of child abuse.
Although numerous studies suggest that both genetic and environmental influences contribute substantively to vulnerability to major depressive disorder (MDD), the search for direct genetic influences contributing to the risk for depression has been inconclusive so far. Recent studies1,2 examining genetic variants predisposing to MDD suggest that they may only do so via interaction with environmental variables. Several reports have now replicated the finding that the short allele of a functional repeat polymorphism located in the promoter region of the serotonin transporter locus SLC6A43 increases vulnerability to depression only in the presence of significant adverse life events.1,4–8 Although the serotonin transporter is an attractive genetic candidate for moderating the adverse effects of life events, epidemiological studies clearly indicate that the vulnerability to depression is influenced by several genes.9
Genes regulating the effects of stress via the hypothalamic-pituitary-adrenal (HPA) axis have been implicated in the physiological and pathological regulation of stress reactivity.10,11 The physiological stress response is primarily mediated by the release of hypothalamic corticotropin-releasing factor, also known as corticotropin-releasing hormone (CRH), from nerve terminals in the median eminence arising in the paraventricular nucleus, which stimulates adrenocorticotropin release from the anterior pituitary that in turn induces the release of cortisol from the adrenal cortex. Overactivity of the HPA axis is a hallmark of MDD and is at least partly due to a hyperactivity of CRH neurons.12,13
In addition to its effects on the HPA axis, CRH activity at the CRH type 1 receptor (CRHR1) in extrahypothalamic regions is also thought to produce symptoms of anxiety and depression. Preclinical animal models of depression14,15 and clinical studies of patients with depression16,17 have found increased CRH concentrations in the cerebrospinal fluid as well as altered CRHR1 messenger RNA expression and CRH binding in limbic brain regions (eg, hypothalamus, amygdala, and other areas mediating emotion response), lending support to this theory. Furthermore, a nonblinded study demonstrated clinical efficacy of a CRHR1 antagonist in patients with depression.18 Finally, single-nucleotide polymorphisms (SNPs) in the CRHR1 gene (Entrez Gene ID 1394 or RefSeq ID NM_004382) have been associated with MDD in Han Chinese individuals,19 and response to antidepressant medications appears to vary with CRHR1 haplotypes in Mexican American persons.20
Studies suggest that HPA axis hyperactivity related to MDD may be a function of early life stress (ELS).21,22 Given the relationships between ELS, MDD, and the dysregulation of the CRH and CRHR1 system, we hypothesized that genetic polymorphisms altering the function of CRHR1 may moderate the effects of child abuse on adult depression.
METHODS
SAMPLING AND PHENOTYPING METHODS
Sample and Sample Recruitment
The data from this study were collected as part of a larger study investigating the roles of genetic and environmental factors in predicting response to stressful life events in a predominantly African American urban population of low socioeconomic status. The sample for the replication study is discussed later. Research participants were approached, either while waiting for their medical appointments or while waiting with others who were scheduled for medical appointments, in the waiting rooms of the primary care clinic or obstetrical-gynecological clinic of a large, urban, public hospital. Subjects who indicated willingness to participate provided written informed consent, participated in a verbal interview, and provided a salivary sample for DNA extraction (described later). The data presented in this article are from the first 560 subjects screened (we have incomplete genotype or phenotype data for some subjects, so the total number of subjects in the analysis will be fewer than 560, with exact sample sizes stated in the “Results” section). The final genetic analysis was performed on a subset of 422 of these subjects. All of the procedures in this study were approved by the institutional review boards of Emory University School of Medicine and Grady Memorial Hospital, Atlanta, Georgia. Demographic data on sex, self-identified race/ethnicity, education, employment, disability status, and monthly household income are presented in Table 1. The mean (SD) age in the sample was 38.4 (13.3) years (range, 18–81 years).
Table 1.
Primary Sample Demographics
| Characteristic | Participants, No. (%) |
|---|---|
| Sex | |
| Male | 194 (39.0) |
| Female | 303 (61.0) |
| Self-identified race/ethnicity | |
| African American or black | 484 (97.4) |
| Caucasian or white | 4 (0.8) |
| Hispanic or Latino | 2 (0.4) |
| Asian | 1 (0.2) |
| Mixed | 5 (1.0) |
| Other | 3 (0.6) |
| Education | |
| < 12th Grade | 153 (30.8) |
| High school graduate or GED | 217 (43.7) |
| Some college or technical school | 78 (15.7) |
| Technical school graduate | 21 (4.2) |
| College graduate | 21 (4.2) |
| Some graduate school | 9 (1.8) |
| Employment status | |
| Currently unemployed | 338 (67.6) |
| Currently employed | 162 (32.4) |
| Disability status | |
| Not currently receiving disability assistance | 394 (79.3) |
| Currently receiving disability assistance | 103 (20.7) |
| Household monthly income, $ | |
| 0–249 | 158 (31.8) |
| 250–499 | 51 (10.3) |
| 500–999 | 136 (27.4) |
| 1000–1999 | 106 (21.3) |
| > 2000 | 46 (9.3) |
Abbreviation: GED, general equivalency diploma.
Procedure
Participants verbally completed a battery of self-report measures, including the Beck Depression Inventory (BDI) and the brief Childhood Trauma Questionnaire (CTQ), which took 45 to 75 minutes to complete (dependent in large part on the extent of the participant’s trauma history and symptoms). We read instruments to participants to guard against relatively high rates of impaired literacy. Each person was paid $15.00 for participation in this phase of the study.
Phenotype Measures
Beck Depression Inventory
Depressed mood was assessed with the 21-item BDI,23 a well-validated, commonly used, continuous measure of the level of depressive symptoms. In this sample, the BDI had a standardized α coefficient of.99 (mean [SD] BDI score, 14.43 [13.11]). In addition, we examined validity of the BDI using data collected in a later phase of the study for which the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID-I),24 a validated interview assessment of DSM-IV mood disorders, was administered. In this subsample of participants (n=202), 19.3% met the criteria for current MDD and 40.1% met criteria for lifetime MDD. In addition, 1.1% met criteria for current bipolar I disorder and 2.0% met lifetime criteria for bipolar I disorder; 2.2% met criteria for current bipolar II disorder and 3.0% met lifetime criteria for bipolar II disorder. Based on MDD diagnosis with the SCID-I, the BDI total scores for those with current MDD (mean [SD] BDI total score, 28.26 [15.01]) were significantly different from the BDI total scores for those without a current MDD diagnosis (mean [SD] BDI total score, 13.48 [12.73]) (F1,173=32.9; P <.001). Additionally, because these analyses were performed in subjects identified in a general medical clinic, it is possible that the BDI score is a proxy for medical-related depressive symptoms separate from MDD.
To examine this, we used history of prescribed medications as a proxy variable for the medical illness category. Although we have only limited diagnostic information, we have medical pharmaceutical data for approximately 120 subjects from the full genotype and phenotype data set. Similar to the linear regression analyses with permutations for SNP analyses (described later), we used linear regression with permutation procedures (using 10 000 permutations with BDI as the dependent variable and each of the 13 medical variables as predictors) to establish whether the proxy variables for different types of medical illness predict the BDI score. These permutation procedures randomly assigned the sample BDI scores to subjects (sampled without replacement) while holding each subject’s medical prescription variables fixed. We conducted these analyses using appropriate components of SAS version 9.1 statistical software (SAS Institute, Inc, Cary, North Carolina). There were no significant effects found for any of the 13 available classes of medications examined (all P >.10). This demonstrates that the moderation effects of child abuse and CRHR1 genotype were unlikely affecting medical condition as a possible contributor to depressive symptoms.
Childhood Trauma Questionnaire
The CTQ25 is a self-report inventory assessing 3 types of childhood abuse: sexual, physical, and emotional. Studies have established the internal consistency, stability over time, and criterion validity of both the original 70-item CTQ and the current brief version.26 The CTQ yields a total score and subscale scores for each of the types of child abuse. Our CTQ data demonstrated good internal consistency reliability (α=.99 for physical abuse; α=.94 for sexual abuse; α=.93 for emotional abuse; and α=.98 for the total of these 3 scales). Bernstein and Fink25 established scores for none, mild, moderate, and severe for each type of abuse. The data from the CTQ were used to classify participants into 2 categories for each type of abuse (physical, sexual, and emotional): (1) those with CTQ scale scores in the none to mild range, and (2) those with CTQ scores in the moderate to severe range. We then created a composite variable across all of the 3 types of abuse. Using this composite, we divided participants into 2 groups with respect to the numbers of types of abuse that fell into the moderate to severe range: (1) those with no type of abuse in the moderate to severe range, and (2) those with at least 1 type of abuse in the moderate to severe range.
ADDITIONAL SUPPORTIVE SAMPLE
A sample of 204 women recruited at the Women’s Mental Health Center at Emory University had been assessed for lifetime psychiatric diagnoses using the SCID-I24 and for the presence of child abuse using the CTQ. The severity of child abuse was then scored as none to mild vs moderate to severe. In this sample, the mean (SD) age was 31.9 (5.0) years, and 87.7% were Caucasian or white, 7.0% were African American or black, 3.7% were Native American, and 1.6% were Asian. As compared with the previous sample, subjects in this sample were primarily middle-income Americans. The mean (SD) number of years of education in this sample was 16.0 (2.0) years. Among the subjects, 23.6% reported moderate to severe child abuse. Most subjects (96.4%) fulfilled criteria for at least 1 Axis I DSM-IV diagnosis, with 62.6% of the patients presenting with a principal lifetime diagnosis of MDD. The other reported lifetime diagnoses included bipolar I disorder, bipolar II disorder, panic disorder, generalized anxiety disorder, anxiety disorder not otherwise specified, obsessive-compulsive disorder, specific phobias, dysthymia, depressive disorder not otherwise specified, brief psychotic disorder, schizophrenia, mood disorder due to the general medical condition, cannabis abuse, opioid abuse, post-traumatic stress disorder, adjustment disorder, and binge eating. Women with these latter diagnoses did not fulfill criteria for MDD at any time and represented the group with no MDD.
GENETIC METHODS
DNA Extraction
We extracted DNA from saliva collected into Scope mouthwash (Procter and Gamble, Cincinnati, Ohio) (n=46) or into Oragene saliva kits (DNA Genotek Inc, Ottawa, Ontario, Canada) using the Qiagen M48 system (Qiagen, Hilden, Germany). High-quality DNA was available for 502 individuals.
SNPs and Genotyping
Fifteen SNPs validated in African populations were selected from public databases (dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/; HapMap, http://www.hapmap.org) to span the CRHR1 gene (NM_004382) from the promoter region to the 3′ end of the gene, with an average intermarker distance of 4.1 kilobases (kb) over a total region of 57 kb. Ten of these SNPs had a minor allele frequency greater than 5.0% and were thus included in the analysis (full SNP information is available from us, and identification numbers of 10 selected SNPs are listed in Figure 1A). In our population, we observed moderate linkage disequilibrium (LD) in this gene, especially in the part containing the coding regions with 2 separate blocks of LD (Figure 1A). All of the SNPs were genotyped using a TaqMan allelic discrimination assay27 developed for use on the 7900HT instrument (Applied Biosystems, Foster City, California) in 502 individuals using predesigned and validated TaqMan assay reagent kits containing 1 pair of polymerase chain reaction primers and 1 pair of fluorescently labeled probes (Applied Biosystems). Assay identification numbers for all of the 15 assays are available on request. Polymerase chain reactions were performed in 5-μL reaction volumes in 384-well plates and contained 5 ng of DNA. The standard protocol provided with the kit was followed. Thermal cycler conditions were 95°C for 10 minutes and then 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. The SDS 2.2 software (Applied Biosystems) was used for allelic discrimination. For quality control, 9.0% of the samples were genotyped as duplicates across and within a 384-well plate. Only 2 discordances were recorded with the 6255 unique genotypes (0.03%) and excluded from the analysis. All of the SNP identification numbers, their locations, Hardy-Weinberg equilibrium, and minor allele frequencies are available on request.
Figure 1.
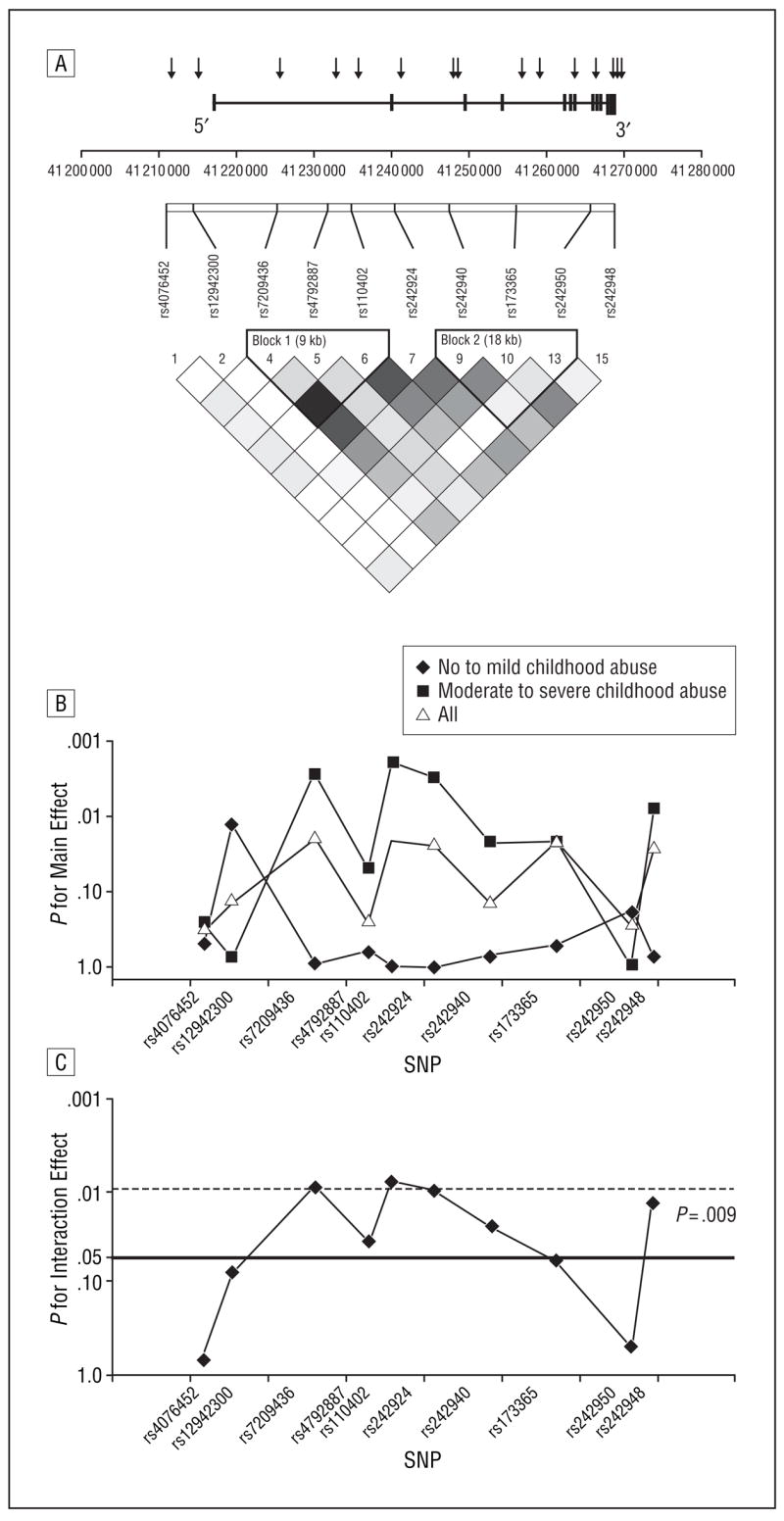
Main and interaction effects of CRHR1 single-nucleotide polymorphisms (SNPs) with child abuse on adult depression. A, CRHR1 linkage disequilibrium (LD) map demonstrating the physical location (arrows) and LD pattern of the selected CRHR1 SNPs. For initial genotyping, we selected 15 SNPs covering the gene from the promoter region to the 3′ end of the gene, with an average intermarker distance of 4.1 kilobases (kb) over a total region of 57 kb. The x-axis shows the position on chromosome 17. Ten of these SNPs had a minor allele frequency greater than 5.0% and were thus included in the analysis. The lower part shows an LD plot generated with Haploview using r 2 as the measure of LD, which ranges from 1 (or complete LD, indicated by black squares) to 0 (or absence of LD, indicated by white squares). In our population, we observed moderate LD in this gene, especially in the part containing the coding regions with 2 separate blocks of LD according to the confidence interval method. B, Main genetic effects. The P values (log scale) for the main genetic effect (for a model containing SNP genotype, Childhood Trauma Questionnaire scores, age, and sex as independent variables and Beck Depression Inventory scores as outcome) for all of the genotyped subjects (n=422), subjects with no to mild child abuse (n=262), and subjects with moderate to severe child abuse (n=160). Concordant with a gene × environment interaction effect, the main genetic effect is carried by individuals with moderate to severe child abuse and is stronger than in the whole sample, whereas no SNPs significant in the whole sample remain significant in the group with no to mild child abuse. C, Interaction effects. The y-axis shows the P value (log scale) for the interaction effect of the 10 SNPs that were present with a minor allele frequency greater than 5.0%. Horizontal lines indicate P=.05 (nominally significant) and P=.0094 (significance level after correction for multiple testing as described in the text).
Ancestry-Informative Markers
Genotyping of ancestry-informative markers (AIMs) and data analysis for estimates of ancestry were performed by DNA Print Genomics, Sarasota, Florida (http://www.dnaprint.com). One hundred thirty-four AIMs were genotyped in 280 individuals from our sample using single-base primer extension and a Beckman SNPstream instrument (Beckman-Coulter, Fullerton, California). Maximum likelihood estimates of individual biogeographical ancestry admixture were determined using methods described previously by others28 and 134 AIMs29–31 chosen from the genome based on their information for a continental, 4-population model. The choice of a 4-population model was based on previously published hypothesis-free cluster analyses of worldwide populations,31,32 and this model lent itself to use of the terms European (genetic ancestry shared among European, Middle Eastern, and, to a lower extent, South Asian populations), sub-Saharan African, East Asian, and indigenous American (genetic ancestry shared among American Indian, Latin and South American Indian, and certain Central Asian populations). The names of these parental populations were chosen to describe extant elements of genetic structure and are arbitrary in that they are reflective of modern population distributions—not necessarily the distributions of the original parental populations 15 000 to 50 000 years ago. Individuals with more than 35 failed AIM genotypes were excluded from the analysis (7.1%) because their biogeographical ancestry admixture could not be estimated with sufficient certainty.
STATISTICAL ANALYSIS
LD Structure
We used Haploview33 to determine the LD structure of the SNPs within the CRHR1 gene and test for Hardy-Weinberg equilibrium (Figure 1A). We also compared the LD structure of a subgroup of CRHR1 SNPs across our study population and 2 populations genotyped in the HapMap project (Yorubans [a sub-Saharan African population] and Centre d’Étude du Polymorphisme Humain [Caucasian population]). We compared a tag SNP selection for the CRHR1 locus based on these HapMap populations covering the same area on chromosome 17 using an r2 cutoff of 0.75 and a minor allele frequency greater than 0.05. We found that across HapMap populations, our SNP sample covers at least 60.0% of the genetic variation described in Hap-Map. In addition, we used 3 SNPs that have not yet been typed in the HapMap project that cover additional, likely overlapping genetic variation of the gene.
Regression and Permutation Analyses for Interaction Effects
We used linear regression to assess whether SNPs in the CRHR1 gene interacted with child abuse to influence BDI scores. For these analyses, a maximum sample size of 422 individuals was informative, but the total sample sizes for the single-SNP analyses vary by a maximum of 5.0% depending on the call rate of each individual SNP. We first considered single-SNP analyses that regressed the BDI outcome (continuous variable) on genotype (coded under an additive model), with child abuse coded into the 2 groups of none to mild and moderate to severe. We further adjusted the regression models for potential confounders including age, sex, and ancestry. We established the significance of genotype–child abuse interaction effects using permutation-based procedures34,35 that randomly assigned the sample BDI scores to subjects (sampled without replacement) while holding each subject’s genotype and environmental variables fixed. This permutation method is robust against nonnormal distribution of the outcome variable as we observed with the BDI scores in this population. For each analysis, the empirical P value was based on 10 000 permutations. We conducted these analyses using appropriate components of SAS version 9.1 statistical software.
After completing the single-SNP interaction analyses, we next considered whether SNP-based haplotypes interacted with child abuse to influence BDI scores. Using linear regression, we modeled the BDI score as a function of the various haplotypes (coded under an additive model), child abuse, and the haplotype–child abuse interaction effects. We pooled all of the haplotypes with less than 10.0% estimated frequency into a rare category. We further adjusted the regression models for potential confounders, such as age and sex. We assessed the significance of haplotype-environment interaction effects using permutation procedures that randomly shuffled BDI scores within the sample while holding genotype and environmental data for each subject fixed. For each interaction analysis, we based inference on 100 000 permutations.
Haplotypes
We estimated the haplotype frequencies for the 3 SNP haplotypes of LD blocks 1 and 2 (Figure 1A) as well as for the 3 SNP haplotypes consisting of the 3 SNPs with the strongest gene × environment interaction effects (rs7209436 and rs110402 located in block 1 and rs242924 between the 2 blocks). The haplotype block structure was determined using the confidence interval method according to Gabriel et al36 as implemented in the Haploview software. Given that we generated only genotype data, the phase of the underlying haplotypes of a particular subject may be unknown. Therefore, we estimated sample haplotype frequencies using an expectation maximization algorithm37 and used these frequencies to calculate each subject’s posterior haplotype-pair probability given his or her genotype data. This was done using the SNPHAP program (http://slack.ser.man.ac.uk/progs/snphap.html). We then applied a conservative haplotype test that used only those subjects whose posterior haplotype-pair probability exceeded 95.0%. However, because the region has a low degree of haplotype complexity (high LD with a few common haplotypes), this excluded only 5.2% of subjects from interaction analysis.
Correction for Multiple Testing
We applied the program SNPSpD (http://genepi.qimr.edu.au/general/daleN/SNPSpD/) to adjust the nominal significance level for the testing of multiple SNP variants within the CRHR1 gene. The program, based on the work of Nyholt,38 applies spectral decomposition techniques to determine the effective number of independent SNPs in regions where LD exists. The software then implements a Bonferroni correction that divides the nominal significance level of.05 by this effective number of independent SNPs. Using this method, we determined that α =.0094 provides an appropriate threshold to declare significance within the CRHR1 gene after correcting for multiple testing.
Statistical Analyses in the Second Supportive Sample
The second supportive sample was clinical in nature, so we tested the protective effects of the protective alleles and haplotypes against development of MDD as opposed to other types of psychiatric disorders in the presence of moderate to severe child abuse (Table 2). Haplotypes with a posterior haplotype probability greater than 95.0% could only be estimated in 199 women for the block 1 haplotype and 198 women for the 3 best SNP haplotypes. Differences in the SNP genotypes of rs7209436, rs4792887, rs110402, rs242940, and rs242924 and in the copy number frequency of the TCA haplotype, formed by rs7209436, rs4792887, and rs110402, respectively, and of the TAT haplotype, formed by rs7209436, rs110402, and rs242924, respectively, between the groups with no to mild vs moderate to severe child abuse were tested separately for patients with a lifetime diagnosis of MDD and patients with no lifetime diagnosis of MDD using contingency tables and an additive genetic model (linear by linear association). Our specific directional hypotheses were that the protective alleles of these SNPs and both the TCA and TAT haplotypes (which were protective in the African American sample) would also be protective, and thus over-represented, in women with moderate to severe child abuse but no lifetime diagnosis of MDD. Thus, we report 1-tailed P values for the analyses of these directional hypotheses. The frequencies of the SNPs and for the common haplotypes formed by rs7209436, rs4792887, and rs110402 and by rs7209436, rs110402, and rs242924 in the replication sample are reported in Table 2.
Table 2.
Protective Effects of CRHR1 Alleles and Haplotypes in Second Samplea
| No MDD (n = 74)
|
Lifetime MDD (n = 126)
|
|||||
|---|---|---|---|---|---|---|
| CRHR1 Alleles and Haplotypes | No to Mild Abuse, No. (%) (n = 53) | Moderate to Severe Abuse, No. (%) (n = 21) | P Value | No to Mild Abuse, No. (%) (n = 100) | Moderate to Severe Abuse, No. (%) (n = 26) | P Value |
| rs7209436 | ||||||
| CC | 21 (40.4) | 5 (23.8) | ].04 | 37 (37.0) | 8 (30.8) | ].46 |
| CT | 22 (42.3) | 9 (42.9) | 36 (36.0) | 13 (50.0) | ||
| TT | 9 (17.3) | 7 (33.3) | 27 (27.0) | 5 (19.2) | ||
| rs242940 | ||||||
| AA | 18 (34.6) | 3 (14.3) | ].03 | 32 (32.0) | 9 (36.0) | ].20 |
| AG | 24 (46.2) | 11 (52.4) | 35 (35.0) | 10 (40.0) | ||
| GG | 10 (19.3) | 7 (33.3) | 33 (33.0) | 6 (24.0) | ||
| Protective haplotype block 1, TCA copies, No. | ||||||
| 0 | 22 (41.5) | 5 (25.0) | ].04 | 36 (36.0) | 9 (34.6) | ].31 |
| 1 | 22 (41.5) | 8 (40.0) | 36 (36.0) | 12 (46.2) | ||
| 2 | 9 (17.0) | 7 (35.0) | 28 (28.0) | 5 (19.2) | ||
| Protective haplotype best SNPs, TAT copies, No.b | ||||||
| 0 | 23 (43.4) | 5 (25.0) | ].03 | 36 (36.4) | 9 (34.6) | ].31 |
| 1 | 21 (39.6) | 8 (40.0) | 35 (35.3) | 12 (46.2) | ||
| 2 | 9 (17.0) | 7 (35.0) | 28 (28.3) | 5 (19.2) | ||
Abbreviations: MDD, major depressive disorder; NS, nonsignificant; SNPs, single-nucleotide polymorphisms.
Distribution of the estimated copy numbers of the protective alleles (rs7209436 and rs242940) as well as of the protective TCA and TAT haplotypes between women with no to mild child abuse compared with those with moderate to severe child abuse, with or without lifetime diagnosis of MDD as diagnosed with the Structured Clinical Interview for DSM-IV Axis I Disorders. The P values represent the directional 1-tailed P values comparing the respective genotype frequencies between the 2 levels of child abuse for the no MDD and lifetime MDD groups.
The sample size for the TAT haplotype in the lifetime MDD group with no to mild abuse is 99.
RESULTS
CHILD ABUSE AND ADULT DEPRESSION
To examine the interaction of the CRHR1 genotype and ELS, we initially analyzed data from our first sample group of 560 low-income, primarily (97.4%) African American adults living in an urban area. Of the 476 subjects with complete CTQ and BDI phenotypes, we first examined the effects of child abuse on adult depression. As expected based on prior literature, higher CTQ abuse scores predicted higher adult BDI scores (F3,472 = 37; P <.001) (Figure 2A). Additionally, when child abuse is divided into the none to mild and moderate to severe groups, we see a significant difference in BDI scores (none to mild: n=300, mean BDI score, 11.58; moderate to severe: n=176, mean BDI score, 19.74; P <.001).
Figure 2.

Child abuse interacts with the CRHR1 genotype to enhance risk for depression in adults. A, Mean Beck Depression Inventory (BDI) scores (n = 476) are predicted by continuous scores on the Childhood Trauma Questionnaire (CTQ) physical, sexual, and emotional abuse scales divided into quartiles (quartile 1: CTQ score = 15.0–17.5 [n = 117]; quartile 2: CTQ score = 17.5–22.0 [n = 112]; quartile 3: CTQ score = 22.0–31.0 [n = 123]; quartile 4: CTQ score > 31 [n = 124]). The error bars indicate SEM. The overall analysis of variance comparing the 4 groups on the BDI was significant (F3,472 = 37.12; P <.001). Post hoc analysis indicated significant differences (P <.001) between all pairs of groups except for between groups 2 and 3. Effect of rs7209436 (B), rs110402 (C), and rs242924 (D) genotypes and child abuse on adult depression. Error bars indicate SEM. The sample sizes for no to mild child abuse and moderate to severe child abuse are as follows: rs7209436: CC, 110 and 84, respectively; CT, 115 and 61, respectively; TT, 23 and 9, respectively (B); rs110402: GG, 108 and 84, respectively; AG, 114 and 61, respectively; AA, 23 and 9, respectively (C); and rs242924: GG, 112 and 87, respectively; GT, 111 and 60, respectively; TT, 24 and 10, respectively (D). P values of significance (based on conservative linear permutation analyses) for the main effect of genotype in the moderate to severe abuse group are shown.
CRHR1 POLYMORPHISMS AND LD STRUCTURE
The gene encoding CRHR1 is located on chromosome 17q21.31 and contains 13 exons spanning 51 kb. Of the 502 participants in the study from whom we obtained DNA, 422 were genotyped for 15 SNPs located in a 57-kb region spanning the 5′ promoter region to the 3′ end of the CRHR1 gene, with an average intermarker distance of 4.1 kb (Figure 1A). We identified 2 blocks of LD: one within intron 1 spanning at least 8 kb (block 1), and the second from introns 2 through 9 spanning at least 18 kb (block 2). Ten of the SNPs showed a minor allele frequency greater than 5.0% and were thus included in the gene × environment regression analysis described later.
CRHR1 POLYMORPHISMS INTERACT WITH CHILD ABUSE TO PREDICT ADULT DEPRESSION
To test the primary hypothesis that genetic polymorphisms in the CRHR1 gene moderate the effects of child abuse on adult depression, we used linear regression–based methods. We first performed single-SNP analyses in which we regressed, using permutation analyses, continuous BDI scores on genotype (coded under an additive model), child abuse (none to mild vs moderate to severe), and the interaction between genotype and child abuse, adjusting for potential confounders such as age and sex. Statistical significance of main effects and interactions were evaluated using a permutation-based analysis (see the “Methods” section). Prior to adjusting for multiple comparisons, 7 of the 10 tested SNPs showed a significant interaction (lowest P=.008 for rs110402) with child abuse for the prediction of adult depression (Figure 1B and C).
INTERACTION OF INDIVIDUAL CRHR1 POLYMORPHISMS, CHILD ABUSE, AND DEPRESSION
To correct for multiple comparisons, we used spectral decomposition techniques to determine the effective number of independent SNPs in regions where LD exists. Using this method, we determined that α=.0094 provides an appropriate threshold to declare significance within the CRHR1 gene. This α level is derived from 5.3 independent tests accounting for the moderate extent of LD across the 10 tested SNPs.
The interactions between child abuse and 2 of the CRHR1 SNPs—rs110402 and rs7209436—remained significant after correction for multiple testing (Figure 2B and C), and 5 others showed significant interaction effects prior to correction (Figure 2D and Figure 3). The rs110402 and rs7209436 SNPs both reside in the first LD block of CRHR1 that spans intron 1 of the gene and are in high LD (r2=0.93). For both SNPs, no significant genotype effect on depression was detected in the no to mild abuse group, with the mean BDI scores from these subjects ranging from 10.91 to 12.67 across genotypes (Figure 2B and C). Among individuals in the moderate to severe abuse groups, the rare allele of both SNPs had a protective effect on the severity of adult depressive symptoms. While individuals homozygous for the common alleles (CC for rs7209436 and GG for rs110402) had average BDI scores of 22.33 to 22.49 (reflective of moderate depressive symptoms), heterozygous individuals had significantly lower BDI scores (mean, 16.31–16.39, reflective of mild depressive symptoms) and individuals homozygous for the rare alleles (TT or AA, respectively) had a mean BDI score of 10.22 (reflective of low to absent levels of depressive symptoms) despite the presence of at least 1 type of moderate to severe child abuse. The data suggest an additive protective genetic effect of the rare allele of these 2 SNPs in those exposed to child abuse. We also note that similar interactive gene dosage patterns were seen across the other SNPs that were significant prior to correction, including rs242924 (Figure 2D), rs4792887, rs242940, rs173365, and rs242948 (Figure 3).
Figure 3.

Effect of rs242940 (A), rs173365 (B), rs4792887 (C), and rs242948 (D) genotypes and child abuse on adult depression. Error bars indicate SEM. The sample sizes for no to mild child abuse and moderate to severe child abuse are as follows: rs242940: AA, 98 and 74, respectively; AG, 113 and 69, respectively; GG, 31 and 13 respectively (A); rs173365: GG, 79 and 56, respectively; AG, 114 and 78, respectively; AA, 51 and 21, respectively (B); rs4792887: CC, 107 and 58, respectively; CT, 105 and 70, respectively; TT, 29 and 26, respectively (C); and rs242948: GG, 84 and 57, respectively; GT, 107 and 79, respectively; TT, 64 and 22, respectively (D). P values of significance for the main effect of genotype in the moderate to severe abuse group are shown.
CRHR1 HAPLOTYPES SHOW SIMILAR GENE × ENVIRONMENT INTERACTION
A similar permutation-based linear regression approach was applied to investigate gene × environment interactions involving common CRHR1 haplotypes (haplotype frequency >10.0%) in haplotype blocks 1 and 2 (Figure 1A) as well as a haplotype defined by the 3 SNPs carrying the strongest gene × environment interactions in the individual SNP analysis. In block 1, we observed 3 common haplotypes of approximately equal frequency (30.4%–34.1%) that summed to account for approximately 98.5% of the observed haplotypes (Figure 4). Haplotype interaction analysis showed a significant interaction effect of child abuse and haplotypes within block 1. This association was accounted for by the haplotype formed of alleles T (rs7209436), C (rs4792887), and A (rs110402), which appears to be protective in a dose-dependent manner (P <.001) (Figure 4A and C). The other 2 common haplotypes within block 1 and those within block 2 did not show a significant interaction effect.
Figure 4.

Effect of estimated CRHR1 haplotypes and child abuse on adult depressive symptoms. The frequencies of the estimated individual haplotypes within the first linkage disequilibrium block (A) and of the 3 most significant single-nucleotide polymorphisms (SNPs) (B). C, The effects of the estimated copy numbers of specific haplotypes on adult depression in the presence or absence of moderate to severe child abuse. Error bars indicate SEM. Only haplotypes with an estimation likelihood greater than 95.0% were included in the analysis. The sample sizes for no to mild child abuse and moderate to severe child abuse were as follows: TCA haplotype: 0 copies, 107 and 82, respectively; 1 copy, 109 and 60, respectively; 2 copies, 21 and 9, respectively; TAT haplotype: 0 copies, 118 and 91, respectively; 1 copy, 106 and 54, respectively; 2 copies, 19 and 9, respectively. P values of significance for the interaction effect of haplotype in the moderate to severe abuse group are shown. D, A graphical representation of the number of copies of the TAT haplotype in a smaller but ethnically and socioeconomically separate sample. In this separate sample, the best SNP haplotype frequencies were 41.6% for TAT and 56.0% for CGG. The frequencies of TAT haplotypes are shown within child abuse groups for those with no history of major depressive disorder (MDD) and those with a lifetime history of MDD. The protective alleles appear to be present at a higher rate in those with moderate to severe child abuse who have no lifetime MDD compared with those with a lifetime history of MDD.
We then investigated the haplotypes formed by the 3 most significant SNPs in the single-SNP analysis, of which 2 are located in block 1 (rs7209436 and rs110402) and 1 is between blocks 1 and 2 (rs242924). These 3 SNPs form 2 common “yin and yang” haplotypes in our sample accounting for more than 95.0% of the observed haplotypes (CGG=66.5% and TAT=28.8%) (Figure 4B and C). The haplotypes formed by these 3 SNPs showed a significant interaction with child abuse on adult BDI scores (P=.003). The presence of the rarer haplotype appears to decrease the risk of adult depressive symptoms in an additive manner in those with a history of child abuse.
GENETIC ADMIXTURE AND POPULATION STRATIFICATION
Varying degrees of genetic admixture of sub-Saharan African, European, and Native American ancestry have been reported in African American individuals31,32 and could lead to population stratification and thus spurious association results. To control for potential differences in genetic ancestry, we genotyped 134 AIMs.31,39 Differences in the extent of admixture in our sample do not account for the observed results because genetic ancestry was correlated with neither the extent of child abuse (Pearson correlations of genetic ancestry with CTQ total abuse scores were nonsignificant, ranging from −0.033 to 0.048 for the 4 different tested populations) nor adult BDI scores (Pearson correlations of genetic ancestry with BDI total scores were nonsignificant, ranging from −0.094 to 0.099). To confirm that admixture was not an important confounder in our analysis, we reinvestigated the interaction effect of rs110402, the SNP with the lowest P value, by using linear regression and adding the percentages of sub-Saharan and European ancestry as independent variables. In this model, the interaction effect between the rs110402 genotype and child abuse on BDI scores was less than P=.05 (P=.04 in a smaller sample of 210 individuals). Percentages of sub-Saharan and European ancestry were not significant predictors in this model, with P values of.38 and.80, respectively. Because the β values for the interaction term were not significantly different with and without ancestry in the model (β [SE], 6.56 [2.70] and 5.59 [2.01], respectively), we also concluded that ancestry is not a confounder in our analysis. Overall, the interaction term remained at P ≤.05 for 5 of the 10 CRHR1 SNPs when adjusting for admixture.
SUPPORTIVE EVIDENCE FROM AN INDEPENDENT SAMPLE
These data suggest that certain alleles of the CRHR1 gene may provide a protective effect on the risk for depression in adults with a history of child abuse. To confirm this hypothesis, we genotyped rs7209436, rs110402, rs4792887, rs242924, and rs242940 in an independent sample that was ethnically (87.7% Caucasian) and socioeconomically (less impoverished) distinct from the original population described earlier. We analyzed the allele distribution of the previously identified protective SNP alleles and haplotypes TCA (rs7209436, rs110402, and rs4792887) (Figure 4A and C) and TAT (rs7209436, rs110402, and rs242924) (Figure 4B and C) in women with and without a lifetime diagnosis of MDD and with and without child abuse (n=199). For haplotype block 1, we found the following frequencies: TCA, 42.1%; CGG, 43.0%; and CTG, 13.1%. For the best SNP haplotype, we found frequencies of 41.6% for TAT and 56.0% for CGG.
We hypothesized that we would find an overrepresentation of 2 copies of the protective haplotypes in women with a history of moderate to severe child abuse but who had not developed MDD within their lifetimes. Indeed, we found a significant overrepresentation of the protective alleles of the tested SNPs (most significant in replication rs7209436 and rs242940) and of 2 protective copies of both the TCA and TAT haplotypes in this group (Table 2). In the group with child abuse but no MDD, 35.0% carried 2 copies of these haplotypes; however, in the group with no child abuse and no MDD, only 17.0% had 2 copies (Table 2). In contrast, in the group with a lifetime diagnosis of MDD, there were no significant differences in the distribution of haplotype copy numbers regardless of child abuse history.
The group with a history of child abuse but no MDD appears to be protected from depression despite the increased psychosocial risk. Notably, this group also has a relative increase in the copy numbers of protective CRHR1 alleles compared with those with a history of lifetime depression (Figure 4D), underscoring the potential protective effects of the identified genotypes in 2 independent and ethnically different samples.
COMMENT
Our results demonstrate that genetic variants in CRHR1 are moderators of the effects of child abuse on adult depressive symptoms in 2 independent, ethnically different populations. A haplotype formed by 3 SNPs located in intron 1 of the CRHR1 gene is most significantly associated with diminished effects of child abuse on adult depressive symptoms. Similar effects were seen on a single-SNP basis and with haplotypes formed by the 3 most significant SNPs. These genotypes and haplotypes potentially serve as predictors of both risk and resilience for adult depression in men and women with a history of child abuse. The protective effect was seen in a predominantly African American sample as well as in a sample that was predominantly Caucasian, and the effect was observed with both current depressive symptoms and lifetime diagnosis of MDD.
It is unlikely that any of these SNPs are actually functional variants in the CRHR1 gene; instead, they are presumably in LD with an as-yet untyped, potentially functional variant. Given the haplotype block structure of the gene, the pattern of association (Figure 1), and the fact that a haplotype in the first LD block showed the most significant interaction effect (Figure 4), it is likely that the causal variant is located in the 5′ region of the gene (ranging from the first 2 kb of the promoter to intron 2). Increasing evidence suggests that much of the regulation of gene function is within the noncoding regions.40,41 Intron 1 of CRHR1 contains 3 highly conserved regions that may have regulatory functions (according to the UCSC Genome Browser database, http://genome.ucsc.edu/). Functional intronic regulatory elements have been reported for several genes42,43; therefore, these CRHR1 intronic regions could affect transcriptional modulation of gene function. Denser fine mapping of this region combined with resequencing followed by in vitro and in vivo studies may allow for the identification of potentially functional variants.
One potential limitation to this study is the use of BDI scores as a continuous variable to measure current depression symptoms, as our genotype effects measure the interactions with child abuse on adult depression symptoms rather than MDD per se. However, this limitation could also be seen as a strength of the study. As pointed out in the “Methods” section, a significant percentage of our sample received a SCID-I–based DSM diagnosis of MDD, and these data strongly validate our BDI measures of depression. Additionally, current research on the assessment of psychopathology points to a number of advantages of a continuous or dimensional assessment of psychopathology, including depressive symptoms. Other potential limitations are the limited size and different phenotype measure (presence or absence of lifetime MDD) in the second supportive study sample. Despite the limitations of this additional sample, we believe that both having a supporting result within a different socioeconomically based sample and finding the apparent protective effects of the CRHR1 polymorphisms using the SCID-I diagnoses support the main findings from the larger primary sample. In the future, we would also like to know the differential effects of age, sex, and type of abuse among other factors that may mediate these effects.
Several studies suggest that depression-related HPA axis hyperactivity may be related to ELS, which may form the physiological basis for the finding that abuse during early life substantially elevates adult risk for depression.21,22 For example, plasma adrenocorticotropin and cerebrospinal fluid CRH concentrations appear to correlate with perceived ELS more strongly than with current depression severity.44,45 Preclinical studies indicate that the persistent hyperactivity of the HPA axis associated with ELS is mediated by hyperactive CRH neurons, with chronic activation of CRHR1 in limbic brain regions.46,47 It is reasonable to hypothesize that alterations in CRHR1 responsiveness during these early emotional critical periods could alter later risk for HPA axis overactivity and depression.
Our genetic results support the hypothesis that the CRH and CRHR1 system moderates the effects of ELS on adult psychopathology. Our data also point out that genetic association and linkage studies that fail to take the environment into account may miss important genetic variants involved in the etiology of complex diseases. Given the clinical and preclinical data12,13,16,21,45,48 that overactivity of CRH-ergic neurotransmission is associated with depressive and anxiety-like symptoms, it is reasonable to expect that the protective polymorphisms identified here associate with either decreasedsensitivityoftheCRHR1orincreasednegativefeed-back regulation of its functioning. These results suggest the possibility that heritable differences in CRH-mediated neurotransmission exacerbate or dampen the effects of child abuse on the stress hormone system, potentially modulating HPA axis sensitivity, extrahypothalamic CRH-ergic circuits, and risk for depressive symptoms in adulthood.
Acknowledgments
Funding/Support: This work was primarily supported by grant MH071537 from the National Institute of Mental Health. This work was also supported by grant P50-MH 68036 from the Women’s Mental Health Program at Emory University; the Emory and Grady Memorial Hospital General Clinical Research Center; grant M01 RR00039 from the National Centers for Research Resources, National Institutes of Health; grants MH-42088 (Dr Nemeroff), MH-58922 (Dr Nemeroff), and MH069884 (Dr Ressler) from the National Institute of Mental Health; grant DA015766 from the National Institute of Drug Abuse (Dr Cubells); and the Burroughs Wellcome Fund (Dr Ressler).
Footnotes
Author Contributions: Drs Bradley and Binder contributed equally to this article. Drs Cubells and Ressler are co-senior authors.
Financial Disclosure: Dr Binder receives grant support or has received awards from Pfizer, GlaxoSmithKline, the National Alliance for Research on Schizophrenia and Depression, and the National Institute of Mental Health and has received speaker’s honoraria from AstraZeneca. Dr Gillespie has received funding from the American Psychiatric Institute for Research and Education/Wyeth and the National Institute on Drug Abuse. Dr Newport has received research support from Eli Lilly, GlaxoSmithKline, Janssen, the National Alliance for Research on Schizophrenia and Depression, the National Institutes of Health, and Wyeth and has served on speakers’ bureaus and/or received honoraria from AstraZeneca, Eli Lilly, GlaxoSmithKline, Pfizer, and Wyeth; he has not served on advisory boards or as a consultant to any biomedical or pharmaceutical corporations, and neither he nor his family members hold equity positions in biomedical or pharmaceutical corporations. Dr Stowe has received grants for clinical research and educational activities, has served as an advisor or consultant to GlaxoSmithKline, Wyeth, and Pfizer, has served as an advisor or consultant for Bristol-Myers Squibb, has served on speakers’ bureaus for GlaxoSmithKline, Wyeth, Pfizer, and Eli Lilly, and receives grants from the National Institutes of Health for his clinical research. In the past, Dr Nemeroff consulted to, served on the speakers’ bureau and/or board of directors of, has been a grant recipient from, and/or owned equity in 1 or more of the following: Abbott Laboratories, Acadia Pharmaceuticals, the American Foundation for Suicide Prevention, the American Psychiatric Institute for Research and Education, AstraZeneca, BMC-JR LLC, Bristol-Myers-Squibb, CeNeRx, Corcept, Cypress Biosciences, Cyberonics, Eli Lilly, Entrepreneur’s Fund, Forest Laboratories, George West Mental Health Foundation, GlaxoSmithKline, i3 DLN, Janssen Pharmaceutica, Lundbeck, the National Alliance for Research on Schizophrenia and Depression, Neuronetics, the National Institute of Mental Health, the National Foundation for Mental Health, NovaDel Pharma, Otsuka, Pfizer Pharmaceuticals, Quintiles, Reevax, UCB Pharma, and Wyeth-Ayerst. Currently, Dr Nemeroff serves on the scientific advisory boards for AstraZeneca, Johnson & Johnson, Pharma Neuroboost, Forest Laboratories, Quintiles, and the National Alliance for Research on Schizophrenia and Depression, is a grant recipient from the National Institutes of Health, the National Alliance for Research on Schizophrenia and Depression, and the American Foundation for Suicide Prevention, serves on the board of directors of the American Foundation for Suicide Prevention, the American Psychiatric Institute for Research and Education, NovaDel Pharmaceuticals, Mount Cook Pharma, Inc, and the George West Mental Health Foundation, owns equity in CeNeRx and Reevax, and owns stock or stock options in Corcept and NovaDel. Dr Ressler has received awards and/or funding support from Lundbeck, the Burroughs Wellcome Fund, Pfizer, the National Alliance for Research on Schizophrenia and Depression, the National Institute of Mental Health, and the National Institute on Drug Abuse and has a consulting agreement with Tikvah Therapeutics for N-methyl-D-aspartate–based therapeutics.
Additional Contributions: Jason Mercer, MS, Taira Everett, BA, Rafael Outland, BA, Abby Powers, BA, Allen Graham, BA, Mark Gapen, BA, and Rachel Hershenberg, BA, provided technical support.
References
- 1.Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301(5631):386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 2.Kendler KS, Kuhn JW, Vittum J, Prescott CA, Riley B. The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression: a replication. Arch Gen Psychiatry. 2005;62(5):529–535. doi: 10.1001/archpsyc.62.5.529. [DOI] [PubMed] [Google Scholar]
- 3.Heils A, Teufel A, Petri S, Stöber G, Riederer P, Bengel D, Lesch KP. Allelic variation of human serotonin transporter gene expression. J Neurochem. 1996;66(6):2621–2624. doi: 10.1046/j.1471-4159.1996.66062621.x. [DOI] [PubMed] [Google Scholar]
- 4.Kaufman J, Plotsky PM, Nemeroff CB, Charney DS. Effects of early adverse experiences on brain structure and function: clinical implications. Biol Psychiatry. 2000;48(8):778–790. doi: 10.1016/s0006-3223(00)00998-7. [DOI] [PubMed] [Google Scholar]
- 5.Wilhelm K, Mitchell PB, Niven H, Finch A, Wedgwood L, Scimone A, Blair IP, Parker G, Schofield PR. Life events, first depression onset and the serotonin transporter gene. Br J Psychiatry. 2006;188:210–215. doi: 10.1192/bjp.bp.105.009522. [DOI] [PubMed] [Google Scholar]
- 6.Sjöberg RL, Nilsson KW, Nordquist N, Ohrvik J, Leppert J, Lindström L, Oreland L. Development of depression: sex and the interaction between environment and a promoter polymorphism of the serotonin transporter gene. Int J Neuropsychopharmacol. 2006;9(4):443–449. doi: 10.1017/S1461145705005936. [DOI] [PubMed] [Google Scholar]
- 7.Grabe HJ, Lange M, Wolff B, Völzke H, Lucht M, Freyberger HJ, John U, Cascorbi I. Mental and physical distress is modulated by a polymorphism in the 5-HT transporter gene interacting with social stressors and chronic disease burden. Mol Psychiatry. 2005;10(2):220–224. doi: 10.1038/sj.mp.4001555. [DOI] [PubMed] [Google Scholar]
- 8.Eley TC, Sugden K, Corsico A, Gregory AM, Sham P, McGuffin P, Plomin R, Craig IW. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908–915. doi: 10.1038/sj.mp.4001546. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157(10):1552–1562. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- 10.De Kloet ER. Hormones and the stressed brain. Ann N Y Acad Sci. 2004;1018:1–15. doi: 10.1196/annals.1296.001. [DOI] [PubMed] [Google Scholar]
- 11.Heinrichs SC, Koob GF. Corticotropin-releasing factor in brain: a role in activation, arousal, and affect regulation. J Pharmacol Exp Ther. 2004;311(2):427–440. doi: 10.1124/jpet.103.052092. [DOI] [PubMed] [Google Scholar]
- 12.Nemeroff CB, Widerlöv E, Bissette G, Walléus H, Karlsson I, Eklund K, Kilts CD, Loosen PT, Vale W. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226(4680):1342–1344. doi: 10.1126/science.6334362. [DOI] [PubMed] [Google Scholar]
- 13.Reul JM, Holsboer F. Corticotropin-releasing factor receptors 1 and 2 in anxiety and depression. Curr Opin Pharmacol. 2002;2(1):23–33. doi: 10.1016/s1471-4892(01)00117-5. [DOI] [PubMed] [Google Scholar]
- 14.Stenzel-Poore MP, Heinrichs SC, Rivest S, Koob GF, Vale WW. Overproduction of corticotropin-releasing factor in transgenic mice: a genetic model of anxiogenic behavior. J Neurosci. 1994;14(5 pt 1):2579–2584. doi: 10.1523/JNEUROSCI.14-05-02579.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holsboer F. The rationale for corticotropin-releasing hormone receptor (CRH-R) antagonists to treat depression and anxiety. J Psychiatr Res. 1999;33(3):181–214. doi: 10.1016/s0022-3956(98)90056-5. [DOI] [PubMed] [Google Scholar]
- 16.Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M. Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry. 1988;45(6):577–579. doi: 10.1001/archpsyc.1988.01800300075009. [DOI] [PubMed] [Google Scholar]
- 17.Merali Z, Du L, Hrdina P, Palkovits M, Faludi G, Poulter MO, Anisman H. Dys-regulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J Neurosci. 2004;24(6):1478–1485. doi: 10.1523/JNEUROSCI.4734-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zobel AW, Nickel T, Künzel HE, Ackl N, Sonntag A, Ising M, Holsboer F. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res. 2000;34(3):171–181. doi: 10.1016/s0022-3956(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 19.Liu Z, Zhu F, Wang G, Xiao Z, Wang H, Tang J, Wang X, Qiu D, Liu W, Cao Z, Li W. Association of corticotropin-releasing hormone receptor1 gene SNP and haplotype with major depression. Neurosci Lett. 2006;404(3):358–362. doi: 10.1016/j.neulet.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 20.Licinio J, O’Kirwan F, Irizarry K, Merriman B, Thakur S, Jepson R, Lake S, Tantisira KG, Weiss ST, Wong ML. Association of a corticotropin-releasing hormone receptor 1 haplotype and antidepressant treatment response in Mexican-Americans. Mol Psychiatry. 2004;9(12):1075–1082. doi: 10.1038/sj.mp.4001587. [DOI] [PubMed] [Google Scholar]
- 21.Gillespie CF, Nemeroff CB. Corticotropin-releasing factor and the psychobiology of early-life stress. Curr Dir Psychol Sci. 2007;16(2):85–89. [Google Scholar]
- 22.Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry. 2001;49(12):1023–1039. doi: 10.1016/s0006-3223(01)01157-x. [DOI] [PubMed] [Google Scholar]
- 23.Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–571. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- 24.First MB, Spitzer RL, Gibbon M, Williams JB. Structured Clinical Interview for DSM-IV Axis I Disorders, Research Version. New York: Biometrics Research Dept, New York State Psychiatric Institute; 1996. [Google Scholar]
- 25.Bernstein DP, Fink L. Childhood Trauma Questionnaire Manual. San Antonio, TX: Psychological Corp; 1998. [Google Scholar]
- 26.Bernstein DP, Stein JA, Newcomb MD, Walker E, Pogge D, Ahluvalia T, Stokes J, Handelsman L, Medrano M, Desmond D, Zule W. Development and validation of a brief screening version of the Childhood Trauma Questionnaire. Child Abuse Negl. 2003;27(2):169–190. doi: 10.1016/s0145-2134(02)00541-0. [DOI] [PubMed] [Google Scholar]
- 27.Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14(5–6):143–149. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 28.Chakraborty R, Weiss KM. Frequencies of complex diseases in hybrid populations. Am J Phys Anthropol. 1986;70(4):489–503. doi: 10.1002/ajpa.1330700408. [DOI] [PubMed] [Google Scholar]
- 29.Frudakis T, Venkateswarlu K, Thomas MJ, Gaskin Z, Ginjupalli S, Gunturi S, Ponnuswamy V, Natarajan S, Nachimuthu PK. A classifier for the SNP-based inference of ancestry. J Forensic Sci. 2003;48(4):771–782. [PubMed] [Google Scholar]
- 30.Shriver MD, Kittles RA. Genetic ancestry and the search for personalized genetic histories. Nat Rev Genet. 2004;5(8):611–618. doi: 10.1038/nrg1405. [DOI] [PubMed] [Google Scholar]
- 31.Shriver MD, Mei R, Parra EJ, Sonpar V, Halder I, Tishkoff SA, Schurr TG, Zhadanov SI, Osipova LP, Brutsaert TD, Friedlaender J, Jorde LB, Watkins WS, Bamshad MJ, Gutierrez G, Loi H, Matsuzaki H, Kittles RA, Argyropoulos G, Fernandez JR, Akey JM, Jones KW. Large-scale SNP analysis reveals clustered and continuous patterns of human genetic variation. Hum Genomics. 2005;2(2):81–89. doi: 10.1186/1479-7364-2-2-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW. Genetic structure of human populations. Science. 2002;298(5602):2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 33.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 34.Epstein MP, Satten GA. Inference on haplotype effects in case-control studies using unphased genotype data. Am J Hum Genet. 2003;73(6):1316–1329. doi: 10.1086/380204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao JH, Curtis D, Sham PC. Model-free analysis and permutation tests for allelic associations. Hum Hered. 2000;50(2):133–139. doi: 10.1159/000022901. [DOI] [PubMed] [Google Scholar]
- 36.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 37.Excoffier L, Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol. 1995;12(5):921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- 38.Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004;74(4):765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halder I, Shriver MD. Measuring and using admixture to study the genetics of complex diseases. Hum Genomics. 2003;1(1):52–62. doi: 10.1186/1479-7364-1-1-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306(5696):636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 41.ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hubler TR, Scammell JG. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones. 2004;9(3):243–252. doi: 10.1379/CSC-32R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finkbeiner S. New roles for introns: sites of combinatorial regulation of Ca2+− and cyclic AMP-dependent gene transcription. Sci STKE 2001. 2001;(94):PE1. doi: 10.1126/stke.2001.94.pe1. [DOI] [PubMed] [Google Scholar]
- 44.Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, Miller AH, Nemeroff CB. Pituitaryadrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA. 2000;284(5):592–597. doi: 10.1001/jama.284.5.592. [DOI] [PubMed] [Google Scholar]
- 45.Carpenter LL, Tyrka AR, McDougle CJ, Malison RT, Owens MJ, Nemeroff CB, Price LH. Cerebrospinal fluid corticotropin-releasing factor and perceived early-life stress in depressed patients and healthy control subjects. Neuropsychopharmacology. 2004;29(4):777–784. doi: 10.1038/sj.npp.1300375. [DOI] [PubMed] [Google Scholar]
- 46.Ladd CO, Huot RL, Thrivikraman KV, Nemeroff CB, Meaney MJ, Plotsky PM. Long-term behavioral and neuroendocrine adaptations to adverse early experience. Prog Brain Res. 2000;122:81–103. doi: 10.1016/s0079-6123(08)62132-9. [DOI] [PubMed] [Google Scholar]
- 47.Plotsky PM, Thrivikraman KV, Nemeroff CB, Caldji C, Sharma S, Meaney MJ. Long-term consequences of neonatal rearing on central corticotropin-releasing factor systems in adult male rat offspring. Neuropsychopharmacology. 2005;30(12):2192–2204. doi: 10.1038/sj.npp.1300769. [DOI] [PubMed] [Google Scholar]
- 48.Heim C, Owens MJ, Plotsky PM, Nemeroff CB. Persistent changes in corticotropin-releasing factor systems due to early life stress: relationship to the pathophysiology of major depression and post-traumatic stress disorder. Psychopharmacol Bull. 1997;33(2):185–192. [PubMed] [Google Scholar]