

Abstract
One of the major challenges in transplantation medicine is to control the very strong immune-responses to foreign antigens responsible for graft-rejection. Whereas immunosuppressive drugs efficiently inhibit acute graft rejection, a non-diminishing proportion of patients suffers from chronic rejection which ultimately leads to functional loss of the graft1. Induction of immunological tolerance to transplants would avoid rejection and the need for lifelong treatment with immunosuppressive drugs1,2. Tolerance to self-antigens is ensured naturally by several mechanisms3, one of the major ones relying on the activity of regulatory T lymphocytes4,5. Here we show that in mice treated with clinically acceptable levels of irradiation, regulatory CD4+CD25+Foxp3+ T cells stimulated in vitro with alloantigens induced long-term tolerance to bone marrow and subsequent skin and cardiac allografts. Regulatory T cells specific for directly presented donor antigens prevented only acute rejection, despite hematopoietic chimerism. On the other hand, regulatory T cells specific for directly and indirectly presented alloantigens prevented both acute and chronic rejection. Our findings demonstrate the potential of appropriately stimulated regulatory T cells for future cell-based therapeutic approaches to induce lifelong immunological tolerance to allogeneic transplants.
Keywords: Animals, Bone Marrow Cells, metabolism, CD4-Positive T-Lymphocytes, immunology, Female, Forkhead Transcription Factors, biosynthesis, Graft Rejection, prevention & control, Heart Transplantation, methods, Hematopoietic Stem Cells, metabolism, Immune Tolerance, Interleukin-2 Receptor alpha Subunit, biosynthesis, Isoantigens, chemistry, Male, Mice, Mice, Inbred BALB C, Mice, Inbred C57BL, Skin Transplantation, methods, T-Lymphocytes, Regulatory, immunology, Transplantation, Homologous, methods
Keywords: transplantation, tolerance, regulatory T lymphocyte, acute rejection, chronic rejection, bone marrow, skin, heart, mouse, immunosuppression
CD4+CD25+Foxp3+ regulatory T cells (Treg) play a crucial role in prevention of autoimmune4,5 and immunoinflammatory6 diseases, in regulation of immunity to viral and parasite infections7,8, in maintenance of maternal tolerance to the fetus9, and in inhibition of anti-tumor immunity10. Given their proven physiological role in immune-regulation, it is appealing to attempt to use Treg for induction of immunological tolerance to allografts. We and others opted for a strategy in which Treg are isolated from unmanipulated hosts, cultured in vitro to expand cells with appropriate specificity, and subsequently used to protect allografts in the Mouse. Thus, tolerance to bone marrow11 but not skin allografts12,13 was successfully induced. In this study, we evaluated if immunological tolerance to solid tissue allografts can be induced with a protocol in which mice, preconditioned with clinically acceptable levels of irradiation, were grafted with allogeneic BM, injected with Treg, and subsequently transplanted with donor skin or heart.
We grafted sublethally irradiated BALB/c (H-2d) mice with allogeneic T-cell depleted C57BL/6 (B6, H-2b) bone marrow. Three weeks later the grafted cells had been rejected (Fig. 1a), demonstrating the non-lymphoablative nature of the preconditioning. To prevent rejection of bone marrow allografts, we next injected the preconditioned BALB/c mice with B6 bone marrow and host-type Treg stimulated in vitro with donor strain derived antigen-presenting cells (APC, Supplementary Note 1). The in vitro culture protocol used allowed for expansion of CD4+CD25+Foxp3+ Treg (Fig. 1b). When co-injected with donor bone marrow, these Treg efficiently protected the allograft from rejection (Fig. 1a). Thus, we also induced tolerance to fully allogeneic bone marrow grafts in other donor/host combinations, independently of IL-10 production by Treg (Figs. 1c and S1a,b). Using a modified experimental setup, we showed that allograft-protection required that effector T cells responded to TGF-β (Supplementary Fig. 1b). Finally, we observed no rejection up to 120 days after transplantation (Fig. 1d) and, after engraftment, allogeneic precursors reconstituted all hematopoietic lineages (not shown).
Figure 1.
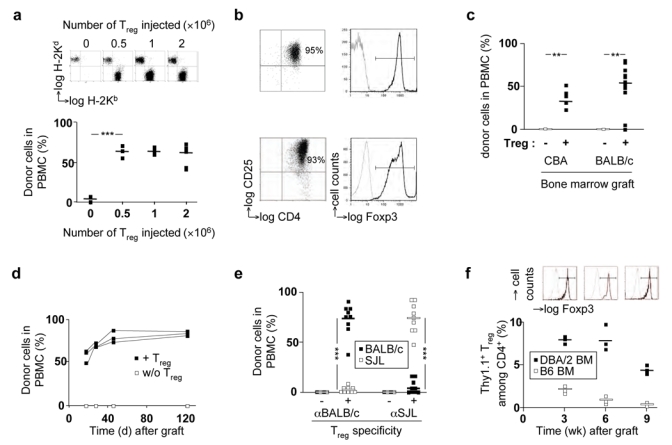
In vitro pre-activated Foxp3+ Treg induce durable tolerance to folly allogeneic bone marrow grafts, (a) BALB/c hosts were grafted with B6 bone marrow and injected with BALB/c Treg pre-activated in vitro with B6 APC. Hematopoietic chimerism was assessed by FACS analysis of peripheral blood mononuclear cells (PBMC) three weeks after bone marrow transplantation. Typical FACS plots of H-2Kb vs. H-2Kd staining are shown in the upper panels. In the lower panels, the percentage of donor (H-2Kb+) cells among PBMC from individual mice is shown, (b) Phenotype of Treg before (upper panels) and after in vitro culture with donor-type APC (lower panels). In right panels, black lines indicate staining with antibody to Foxp3, gray curves staining with isotype-matched control antibodies, (c, d) B6 hosts were grafted with allogeneic donor bone marrow from indicated (c) or BALB/c (d) donors with or without Treg. Hematopoietic reconstitution was assessed at 3 weeks (c) or at indicated time points (d). Values for individual mice are shown, bars indicate means, (e) B6 hosts were grafted with a mixture of BALB/c and SJL bone marrow cells with or without Treg of indicated specificity. Hematopoietic reconstitution by cells of BALB/c (■) and SJL (□) origin was assessed at 3 weeks, (f) B6 hosts were grafted with B6 (□) or (■) DBA/2 bone marrow and injected with B6.Thy1.1 Treg cultured in vitro in presence of DBA/2 APC. At indicated time points, splenocytes were analyzed by FACS. Indicated is the percentage of Thy1.1 Treg among CD4+ splenocytes. The FACS plots indicate Foxp3-staining on Thy1.1+CD4+ splenocytes. Horizontal bars indicate mean values. ***P<0.001, **P<0.01 (Student’s t test).
To be activated, Treg require antigen specific stimulation with MHC/peptide complexes. However, once activated, these cells exert their suppressor effector function in a non antigen-specific-manner in vitro14. It was therefore important to evaluate if Treg induced generalized immunosuppression in our system. We stimulated B6 Treg with BALB/c APC and subsequently injected them into sublethally irradiated B6 recipient with BALB/c and SJL (H-2s) bone marrow. Three weeks later, the target bone marrow had reconstituted the hosts, but the third party SJL bone marrow had been fully rejected (Fig. 1e). Similarly, Treg specific for SJL antigens protected SJL but not BALB/c bone marrow from rejection (Fig. 1e). These results show that the APC used in the in vitro cultures determined the specificity of the Treg in vivo. Moreover, despite the fact that in these mice Treg had clearly been activated (they prevented rejection of target bone marrow), they failed to protect third party grafts. Their suppressor effector function was therefore donor-specific and the Treg had not induced generalized immunosuppression.
Our data show that Treg allow for establishment of hematopoietic chimerism. We next analyzed if this chimeric state in its turn created a favorable environment for persistence of injected Treg. We sublethally irradiated B6 mice, grafted them with allogeneic DBA/2 (H-2d) or syngeneic bone marrow, injected them with Treg previously stimulated with DBA/2 APC in vitro and monitored persistence of injected Treg in these mice (Fig. 1f). We observed that substantially more injected Treg persisted in spleens (but not in blood or lymph nodes, not shown) of mice that had been injected with donor-type bone marrow. Moreover, these cells had maintained their expression of Foxp3 (Fig. 1f). These results show that donor hematopoietic cells and donor-specific Treg mutually favor their in vivo persistence.
We wanted to know whether Treg could also induce tolerance to solid allografts (Fig. 2). Three weeks after irradiation, the time it took the mice to recover from the preconditioning regimen, B6 animals received allogeneic DBA/2 skin transplants. We injected Treg, in vitro cultured with donor-type APC, immediately after irradiation (DO) or just before grafting the skin (D21). In contrast to the data on bone-marrow transplantation, in this setting skin grafts were rapidly rejected (Fig. 2a). Combined with previously published data12,13, these results suggested that alloantigen-specific Treg alone do not induce immunological tolerance to allogeneic skin grafts, at least not at cell-doses tested. We therefore next tried to induce tolerance to skin allografts by combining Treg transfer with bone marrow transplantation in order to prolong Treg persistence. Moreover, the induced chimeric state would contribute to induction of allograft-tolerance. We reconstituted B6 hosts with DBA/2 bone marrow, injected them with in vitro cultured Treg and, three weeks later, grafted them with DBA/2 skin. Allogeneic skins did not show any macroscopical sign of rejection for the 100-day observation period, but third-party SJL skins were rapidly rejected (Fig. 2a). We obtained similar results using five other host/donor combinations and when using mice with substantially lower levels of hematopoietic chimerism (Figs. S2 and S3a,b). We also assessed if mice in which Treg protected a skin allograft from rejection were generally immunosuppressed. SJL mice received B6 bone marrow grafts, B6-specific Treg, and B6 and third-party DBA/2 skins on opposing flanks. Whereas in these mice B6 skins survived, DBA/2 skins were rapidly rejected (Fig. 2b). Treg-mediated allograft protection was therefore specific and these cells did not induce generalized immunosuppression.
Figure 2.

Treg prevent acute and chronic skin allograft rejection, (a) Left panel: B6 recipient mice were preconditioned with sublethal irradiation only (○, n=4) or combined with injection of Treg pre-activated in vitro with donor-type (DBA/2) APC immediately after irradiation (“D 0”, ●, n=8) or three weeks later, just prior to DBA/2 (or control B6) skin transplantation (“D 21”, ▴, n=10). Control mice were irradiated and received a syngeneic skin graft (□, n=4). Skin allograft survival was monitored daily by assessment of macroscopic signs of rejection. Right panel: B6 hosts were irradiated, injected with donor DBA/2 bone marrow with (●, n=12, █, n=8) or without (▵, n=6) Treg. Three weeks later, DBA/2 (or control SJL) skins were transplanted and their survival monitored, (b) SJL hosts were irradiated, injected with B6 bone marrow and Treg cultured with B6 APC, and grafted, three weeks later, with B6 and SJL skins on opposing flanks (n=4). Survival of “target” B6 (●) and third party DBA/2 (○) skins was monitored, (c) As in (a, right panel), but Treg were pre-cultured with (B6 x DBA/2)F1 APC (▵,n=3; ●, n=8,█, n=8). (d) Scoring of infiltrates of DBA/2 skins transplanted on mice that had received DBA/2 bone marrow and Treg cultured with DBA/2 (n=12) or (B6 x DBA/2)F1 (n=8) APC. (e-h) Representative features of skin histopathology 100 days after transplantation (HE, hematoxylin and eosin; F4/80, immunohistochemistry with an antibody to F4/80; Luna, Luna’s eosinophil stain). Scale bars represent 200 μm (HE), 400 μm (F4/80), or 40 μm (Luna).
At 100 days post-transplantation, we submitted skin grafts to histological analysis. The allografts showed only little signs of rejection (i.e. tissue damage), but we observed substantial infiltration by eosinophils and macrophages (Fig. 2d,g), previously observed in chronically rejected skin allografts15. Whereas this observation indicated that the combined Treg/bone-marrow chimerism approach had not induced full immunological tolerance to allogeneic skins, 250 days post-transplantation allogeneic grafts still survived (Supplementary Fig. 3c). These results demonstrated that the combined Treg/hematopoietic chimerism approach protected allogeneic skin grafts from rejection but did not induce full immunological tolerance to the graft, despite persisting hematopoietic chimerism (Supplementary Fig. 3d). It appears therefore unlikely that this protocol will induce long-term protection of tissue allografts in clinical settings.
Graft rejection is initiated when APC present donor antigens from the transplanted tissue to host lymphocytes, which are then activated to attack the grafted organ via direct cytotoxicity, B-cell help, and induction of an inflammatory response16. Donor antigens from a transplant are presented to T cells in two distinct ways, depending on the origin of the APC1. First, donor APC migrate from the graft to secondary lymphoid organs where they activate host T cells, which therefore recognize donor antigens presented by donor MHC molecules. In contrast to this ‘direct’ pathway, donor antigens can also be picked up in the transplanted tissue by host APC, processed, and then presented to T cells on self MHC molecules (‘indirect allorecognition’). Whereas acute organ graft rejection has been attributed mainly to direct antigen presentation, chronic rejection is thought to be mediated mostly by T cells specific for indirectly presented donor antigens1. In our in vitro culture protocol, we activated host Treg with donor-type APC. The injected Treg population was therefore enriched in cells specific for directly presented alloantigens17. This may explain why acute skin allograft rejection was efficiently inhibited while we still observed chronic rejection. Treg specific for indirectly presented alloantigens may be able to prevent the chronic immune-response to the graft (Supplementary Note 2).
To test this hypothesis, we expanded Treg with (host × donor)F1 APC. B6 hosts were transplanted with DBA/2 bone marrow and simultaneously injected with Treg beforehand expanded in vitro with (B6 x DBA/2)F1 APC. Three weeks later, mice were grafted with DBA/2 (or third party SJL) skin and allograft-survival was monitored. Whereas third-party skins were rejected, ‘target’ skin grafts survived the whole 100-day monitoring period (Fig. 2c). We obtained similar results with the inverse host/donor combination (Supplementary Fig. 3e). When, at 100 days post grafting, we took the DBA/2 skins for histological analysis, we observed healthy skin without eosinophil or macrophage infiltration (Fig. 2d,h). The fundamental difference between our results with Treg specific for directly vs. directly and indirectly presented alloantigens was obtained despite similar hematopoietic chimerism in the two experimental conditions (Supplementary Fig. 3d). These results show that, in combination with hematopoietic chimerism, Treg specific for directly and indirectly presented alloantigens protected skin-allografts from acute and chronic rejection.
We wanted to know whether appropriately in vitro cultured Treg would also induce tolerance to cardiac allografts (Fig. 3). Irradiated host mice received allogeneic bone marrow grafts, in vitro cultured Treg, and, three to eight weeks later, allogeneic heart transplants. Whereas in the control groups hearts were rejected, in thus treated mice the grafted hearts continued beating for more than 100 days after transplantation (Figs. 3a, b, and S4a). Prevention from rejection was partially dependent on IL-10 (Supplementary Fig. 1c). At 100 days post transplantation the beating hearts were removed for histological analysis. In hearts grafted into mice that had received Treg specific for directly presented alloantigens only, we observed large and diffuse infiltrates of mononuclear cells and eosinophils, destruction of cardiac muscle fibers, intima-thickening, arteriosclerosis, and extended areas of fibrosis that had replaced contractile tissue (Figs. 3e and S4b), all typical signs of chronic cardiac allograft rejection. All hearts thus analyzed revealed moderate to severe chronic rejection (Figs. 3c and S4c). In contrast, hearts grafted into mice that had received Treg specific for directly and indirectly presented alloantigens showed little or no signs of rejection (Fig. 3b,c,f). The substantial difference between our results with Treg specific for directly vs. directly and indirectly presented alloantigens was obtained despite similar hematopoietic chimerism (Supplementary Fig. 4d). These data show that Treg specific for directly and indirectly presented donor antigens, in combination with mixed hematopoietic chimerism, prevented both acute and chronic rejection of heart allografts.
Figure 3.

Treg prevent acute and chronic cardiac allograft rejection (a) B6 recipient mice were preconditioned with sublethal irradiation only (○, n=2) or with irradiation and injection of Treg pre-activated in vitro with donor-type (DBA/2) APC immediately after irradiation (“D 0”,▴, n=5) or three weeks later (“D 21”, █, n=5). Three to eight weeks later, recipient mice were transplanted with donor DBA/2 hearts. Control mice were irradiated and received a syngeneic B6 heart graft (□, n=4). Cardiac allograft survival was monitored daily (for 100 days) by abdominal palpation, (b) B6 hosts were irradiated, injected with donor DBA/2 bone marrow with (●, n=12, □, n=9) or without (▵n=2) Treg cultured in vitro with DBA/2 (●) or (B6 x DBA/2)F1 (D) APC. Three to eight weeks later, recipient mice were transplanted with donor DBA/2 hearts. Cardiac allograft survival was monitored daily, (c) Clinical score of DBA/2 cardiac allograft rejection 100 days after transplantation into sublethally irradiated hosts grafted with DBA/2 bone marrow and injected with Treg pre-activated in vitro with DBA/2 (n=12) or (B6xDBA/2)F1 (n=9) APC, as indicated. *** P< 0.001 (Student’s t test). (d,e,f) Representative features of cardiac histopathology 100 days after transplantation of B6 (d) or DBA/2 (e,f) hearts in B6 hosts. Specificity of injected Treg is indicated in the figure. Scale bar represents 200 μm in left panels and 50 μ in right panels.
Our findings show that host CD4+CD25+Foxp3+ Treg, when appropriately stimulated in vitro, can be used to induce immunological tolerance to bone marrow and subsequent skin or cardiac allografts in hosts submitted to non-lymphoablative γ-irradiation, preventing both acute and chronic rejection (Supplementary Note 3). Suppression of rejection is most likely due to two interdependent mechanisms. First, Treg suppress host lymphocytes and thus directly contribute to acceptance of the allograft. Second, immunosuppression by Treg also helps establishing a chimeric hematopoietic state, allowing for persistence of injected Treg and contributing to induction of central and peripheral immunological tolerance to the allografts18.
The data reported here demonstrate that mixed hematopoietic chimerism did not induce immunological tolerance to skin and cardiac allografts. This important conclusion is consistent with data on transplantation in mixed hematopoietic chimeras19–22. Transplantation-protocols exclusively based on induction of hematopoietic chimerism will therefore most probably not yield permanent tolerance to, and survival of, allografts.
In conclusion, we have demonstrated that adequately pre-stimulated Treg can be used to protect skin and cardiac allografts from acute and chronic rejection. The preconditioning regimen used in our study has a level of toxicity that may be acceptable in clinical settings . However, other protocols aimed at induction of hematopoietic chimerism are currently tested in clinical trials24–26 and could be used, in combination with injection of in vitro activated Treg, to replace the one used in this report. Moreover, human Treg with indirect specificity can be expanded in vitro27. Induction of tolerance to organs or tissues to be taken from live donors should therefore be feasible using our protocol or a modified version thereof. We can also predict that it could, after adaptation, be used in the future to induce tolerance to transplants taken from cadaveric donors.
METHODS
Mice
Sex-matched mice between 6 and 10 weeks of age were used. Mice were purchased from the Centre de Recherche et d’Elevage Janvier. Thy1.1 and IL-10 deficient B6 mice were purchased from Charles River. DnTβRII-transgenic B6 mice28 were bred in our SPF animal facility. All experiments involving animals, performed in compliance with relevant laws (authorization # 31–13) and institutional guidelines (Institut National de Santé et de la Recherche Médicale, Inserm), were approved by the local ethics committee (Midi-Pyrénées, France; ref MP/01/31/10/03).
Antibodies
Antibodies with the following specificities were used for analyses and purification of Treg: H-2Kb (AF6-88.5), H-2Kd (SF1-1.1), H-2KS (5KH49), H-2Kk (36-7-5) (BD PharMingen); CD4 (GK1.5), CD8 (53.6.7), Thy1.1, CD25 (PC61), Foxp3 (FJK-16s, eBioscience); F4/80 (CI:A3-1, Serotec). Hybridoma supernatants of antibodies recognizing FcγRII/III (2.4G2), CD8 (53.6.7), MHC class II (M5/114.15.2) and Thy1.2 (AT83) were produced in our laboratory.
Purification and in vitro culture of CD4+CD25+ T cells
CD4+CD25+ splenic T cells were purified and co-cultured with γ-irradiated splenocytes as previously described17.
Bone marrow allografts
Bone marrow cells from femurs and tibias were prepared as previously described17. 107 cells were injected intravenously into γ-irradiated mice (5Gy, 137Cs source).
Flow cytometry
Hematopoietic reconstitution and Treg persistence were determined by analyzing PBMC or splenocytes at indicated time points. Erythrocyte-depleted cells were resuspended in 2.4G2 hybridoma supernatant and saturating concentrations of indicated antibodies were added. Acquisition was performed on a FACSCalibur or an LSRII cytometer and data analyzed using CellQuest (BD Biosciences) or FlowJo (Tree Star) software. Foxp3 analysis was performed according to instructions of the manufacturer.
Skin and cardiac transplantation
Skin graft was performed as previously described29. Skins were considered rejected if ≥ 70% of the surface was necrotic. Heterotopic heart transplantation was performed in the surgery section of the Institut Fédératif de Recherche 31 animal facility according to the method of Corry et al. with some modifications30. Functionality of the transplanted heart was monitored daily by abdominal palpation. Clinical rejection was defined by cessation of palpable heartbeats and confirmed by autopsy. Loss of graft function within 48h of transplantation was considered as a technical failure (<5%) and these animals were omitted from analysis.
Histological analysis
Skin biopsies and hearts were fixed in 10% buffered formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin, Masson’s trichrome, Luna’s eosinophil stain, or using antibodies specific for F4/80. Heart rejection was graded from 0 (none) to 4 (severe): 0, no rejection; 1, slight perivascular mononuclear cell infiltration; 2, intense and/or interstitial mononuclear cell infiltration; 3, intense interstitial mononuclear cell infiltration associated with myocyte loss and slight fibrosis; 4, interstitial mononuclear cell infiltration associated with myocardial necrosis and massive fibrosis.
Statistics
Statistical significance was determined using Student’s t-test.
Supplementary Material
Acknowledgments
The authors would like to thank M.-C. Cuturi, J. Cohen, and C. Reis e Sousa for valuable advice and critical comments on the manuscript, J.-C. Guéry for stimulating discussions, F. Powrie (University of Oxford, UK) and R. Flavell (Yale University) for transgenic mice, the personnel of the Institut Fédératif de Recherche 30, Institut Fédératif de Recherche 31, and Institut de Pharmacologie et de Biologie Structurale animal facilities for expert animal husbandry, F. Capilla for preparation of histological specimens, the personnel of the Institut Fédératif de Recherche 30 flow-cytometry facility for technical assistance, and C. Joffre for her permanent support. This work was supported in part by grants from the Région Midi Pyrénées (#01008776 & 03011999), the Etablissement Francais des Greffes (2003), the Roche Organ Transplantation Research Foundation (ROTRF #133456773), and the Ligue Nationale contre le Cancer (#GL/VP-4825 to OJ).
Footnotes
Olivier Joffre present address: Cancer Research UK London Research Institute, Immunobiology Laboratory, London WC2A 3PX, United Kingdom
Denis Hudrisier present address: Centre National de la Recherche Scientifique, UMR5089, Institut de Pharmacologie et de Biologie Structurale, Toulouse, F-31400 France
References
- 1.Lechler RI, Sykes M, Thomson AW, Turka LA. Organ transplantation--how much of the promise has been realized? Nat Med. 2005;11:605–613. doi: 10.1038/nm1251. [DOI] [PubMed] [Google Scholar]
- 2.Waldmann H, Cobbold S. Exploiting tolerance processes in transplantation. Science. 2004;305:209–212. doi: 10.1126/science.1099538. [DOI] [PubMed] [Google Scholar]
- 3.Stockinger B. T lymphocyte tolerance: from thymic deletion to peripheral control mechanisms. Adv Immunol. 1999;71:229–265. doi: 10.1016/s0065-2776(08)60404-6. [DOI] [PubMed] [Google Scholar]
- 4.Shevach EM, et al. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60–73. doi: 10.1111/j.0105-2896.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 5.Sakaguchi S, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 6.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol Rev. 2006;212:256–271. doi: 10.1111/j.0105-2896.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 7.Belkaid Y, Blank RB, Suffia I. Natural regulatory T cells and parasites: a common quest for host homeostasis. Immunol Rev. 2006;212:287–300. doi: 10.1111/j.0105-2896.2006.00409.x. [DOI] [PubMed] [Google Scholar]
- 8.Rouse BT, Sarangi PP, Suvas S. Regulatory T cells in virus infections. Immunol Rev. 2006;212:272–286. doi: 10.1111/j.0105-2896.2006.00412.x. [DOI] [PubMed] [Google Scholar]
- 9.Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol. 2004;5:266–271. doi: 10.1038/ni1037. [DOI] [PubMed] [Google Scholar]
- 10.Beyer M, Schultze JL. Regulatory T cells in cancer. Blood. 2006;108:804–811. doi: 10.1182/blood-2006-02-002774. [DOI] [PubMed] [Google Scholar]
- 11.Joffre O, van Meerwijk JPM. CD4+CD25+ regulatory T lymphocytes in bone marrow transplantation. Sem Immunol. 2006;18:128–135. doi: 10.1016/j.smim.2006.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishimura E, Sakihama T, Setoguchi R, Tanaka K, Sakaguchi S. Induction of antigen-specific immunologic tolerance by in vivo and in vitro antigen-specific expansion of naturally arising Foxp3+CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:1189–1201. doi: 10.1093/intimm/dxh122. [DOI] [PubMed] [Google Scholar]
- 13.Golshayan D, et al. In vitro-expanded donor alloantigen-specific CD4+CD25+ regulatory T cells promote experimental transplantation tolerance. Blood. 2007;109:827–835. doi: 10.1182/blood-2006-05-025460. [DOI] [PubMed] [Google Scholar]
- 14.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 15.Le Moine A, et al. Critical roles for IL-4, IL-5, and eosinophils in chronic skin allograft rejection. J Clin Invest. 1999;103:1659–1667. doi: 10.1172/JCI5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rocha PN, Plumb TJ, Crowley SD, Coffman TM. Effector mechanisms in transplant rejection. Immunol Rev. 2003;196:51–64. doi: 10.1046/j.1600-065x.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 17.Joffre O, Gorsse N, Romagnoli P, Hudrisier D, van Meerwijk JPM. Induction of antigen-specific tolerance to bone marrow allografts with CD4+CD25+ T lymphocytes. Blood. 2004;103:4216–4221. doi: 10.1182/blood-2004-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sykes M. Mixed chimerism and transplant tolerance. Immunity. 2001;14:417–424. doi: 10.1016/s1074-7613(01)00122-4. [DOI] [PubMed] [Google Scholar]
- 19.Boyse EA, Lance EM, Carswell EA, Cooper S, Old LJ. Rejection of skin allografts by radiation chimaeras: selective gene action in the specification of cell surface structure. Nature. 1970;227:901–903. doi: 10.1038/227901a0. [DOI] [PubMed] [Google Scholar]
- 20.Ildstad ST, Wren SM, Bluestone JA, Barbieri SA, Sachs DH. Characterization of mixed allogeneic chimeras. Immunocompetence, in vitro reactivity, and genetic specificity of tolerance. J Exp Med. 1985;162:231–244. doi: 10.1084/jem.162.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharabi Y, Sachs DH. Mixed chimerism and permanent specific transplantation tolerance induced by a nonlethal preparative regimen. J Exp Med. 1989;169:493–502. doi: 10.1084/jem.169.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo B, Chan WF, Shapiro AM, Anderson CC. Non-myeloablative mixed chimerism approaches and tolerance, a split decision. Eur J Immunol. 2007;37:1233–1242. doi: 10.1002/eji.200636938. [DOI] [PubMed] [Google Scholar]
- 23.Vriesendorp HM. Aims of conditioning. Exp Hematol. 2003;31:844–854. doi: 10.1016/s0301-472x(03)00229-7. [DOI] [PubMed] [Google Scholar]
- 24.Cosimi AB, Sachs DH. Mixed chimerism and transplantation tolerance. Transplantation. 2004;77:943–946. doi: 10.1097/01.tp.0000117779.23431.3f. [DOI] [PubMed] [Google Scholar]
- 25.Fudaba Y, et al. Myeloma responses and tolerance following combined kidney and nonmyeloablative marrow transplantation: in vivo and in vitro analyses. Am J Transplant. 2006;6:2121–2133. doi: 10.1111/j.1600-6143.2006.01434.x. [DOI] [PubMed] [Google Scholar]
- 26.Salama AD, Womer KL, Sayegh MH. Clinical transplantation tolerance: many rivers to cross. J Immunol. 2007;178:5419–5423. doi: 10.4049/jimmunol.178.9.5419. [DOI] [PubMed] [Google Scholar]
- 27.Jiang S, Camara N, Lombardi G, Lechler RI. Induction of allopeptide-specific human CD4+CD25+ regulatory T cells ex vivo. Blood. 2003;102:2180–2186. doi: 10.1182/blood-2003-04-1164. [DOI] [PubMed] [Google Scholar]
- 28.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 29.Coudert JD, Coureau C, Guery JC. Preventing NK cell activation by donor dendritic cells enhances allospecific CD4 T cell priming and promotes Th type 2 responses to transplantation antigens. J Immunol. 2002;169:2979–2987. doi: 10.4049/jimmunol.169.6.2979. [DOI] [PubMed] [Google Scholar]
- 30.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.