

Abstract
Recurrent hypoglycemia impairs hormonal counterregulatory responses (CRRs) to further bouts of hypoglycemia. The hypothalamus and hindbrain are both critical for sensing hypoglycemia and triggering CRRs. Hypothalamic glucose sensing sites are implicated in the pathogenesis of defective CRRs; however, the contribution of hindbrain glucose sensing has not been elucidated. Using a rat model, we compared the effect of antecedent glucoprivation targeting hindbrain or hypothalamic glucose sensing sites with the effect of antecedent recurrent hypoglycemia on CRR to hypoglycemia induced 24 h later. Recurrent hypoglycemia decreased sympathoadrenal (1,470 ± 325 vs. 3,811 ± 540 pg/ml in controls [t = 60 min], P = 0.001) and glucagon secretion (222 ± 43 vs. 494 ± 56 pg/ml in controls [t = 60]), P = 0.003) in response to hypoglycemia. Antecedent 5-thio-glucose (5TG) injected into the hindbrain did not impair sympathoadrenal (3,806 ± 344 pg/ml [t = 60]) or glucagon (513 ± 56 pg/ml [t = 60]) responses to subsequent hypoglycemia. However, antecedent 5TG delivered into the third ventricle was sufficient to blunt CRRs to hypoglycemia. These results show that hindbrain glucose sensing is not involved in the development of defective CRRs. However, neural substrates surrounding the third ventricle are particularly sensitive to glucoprivic stimulation and may contribute importantly to the development of defective CRRs.
Hypoglycemia is a frequent complication of diabetes therapy that is associated with the use of insulin or glucose-lowering agents (1–3). Recurrent hypoglycemia, prevalent in both type 1 (3) and type 2 (1) diabetes, reduces hormonal counterregulatory responses (CRRs) and symptoms of subsequent bouts of hypoglycemia (4). Collectively, defective CRRs and reduced awareness of hypoglycemic symptoms (hypoglycemia unawareness) are known as the clinical syndrome of hypoglycemia-associated autonomic failure (HAAF). A key defining characteristic of HAAF is that antecedent hypoglycemia shifts the glycemic threshold at which hormonal counterregulation is triggered in response to further bouts of hypoglycemia (4,5). That is, lower plasma glucose levels are required to initiate the CRRs to hypoglycemia. This finding suggests that a reduced or impaired sensitivity of glucose sensing mechanisms may contribute to the development of defective hypoglycemia CRRs.
There is a long history of anatomical and pharmacological evidence supporting the role of both the hypothalamus (6–14) and hindbrain (15–18) in glucose sensing and in the initiation of hormonal CRRs. Although there is evidence supporting the role of the hypothalamus, in particular the ventromedial nucleus, in the development of defective hypoglycemic CRRs (10,19–21), the contribution of hindbrain glucose sensing mechanisms is unknown. Given their anatomical differences and the fact that hindbrain glucoreceptors can function independently from the hypothalamus (16), recurrent hypoglycemic stimulation may differentially affect these two distinct glucose sensing brain regions.
In the present study, we adapted our recurrent hypoglycemia rat model, which results in significant deficits in hormonal CRRs (22), to identify the contribution of a hindbrain glucose sensing site (17) in defective CRRs. We compared the effect of antecedent 5-thio-glucose (5TG), delivered into the hindbrain or third ventricle, with the effect of antecedent recurrent hypoglycemia on hormonal CRRs to subsequent hypoglycemia. We hypothesized that either antecedent hindbrain or third ventricular glucoprivation would impair CRRs to subsequent hypoglycemia. As expected, antecedent third ventricular glucoprivation, similar to recurrent hypoglycemia, blunted hormonal CRRs to subsequent hypoglycemia. In marked contrast, antecedent glucoprivation localized to a caudal hindbrain glucose sensing site did not impair hypoglycemia CRRs. Therefore, the hindbrain does not appear to be vulnerable to the central nervous system's adaptive mechanisms that impair CRRs under conditions of recurrent hypoglycemia.
RESEARCH DESIGN AND METHODS
Adult male Sprague-Dawley rats (Simonsen Laboratories, Gilroy, CA) weighing between 350 and 380 g were used for these experiments. Rats were housed individually and maintained on a 12-h light-dark schedule (lights on at 6:00 a.m., off at 6:00 p.m.) at 22−23°C with ad libitum access to food (Purina rat chow #5001) and water, except where otherwise specified. All procedures were approved by the Animal Studies Subcommittee of the Veterans Affairs Puget Sound Health Care System Research and Development Committee, Seattle Division.
Surgery
All rats were surgically implanted with a Silastic intravenous catheters under ketamine/xylazine (86 mg/kg ketamine; KetaFlo; Abbott Laboratories, Chicago, IL; and 12.9 mg/kg xylazine; Xyla-ject; Phoenix Pharmaceutical, St. Josesph, MO) anesthesia as previously described (22). The catheter was tunneled subcutaneously and exteriorized through a midline incision in the scalp. In addition, some rats received either a 26G stainless steel–guide cannula (Plastics One, Roanoke, VA) aimed at the caudal dorsal medial medulla in the hindbrain using coordinates previously described (17) or the third ventricle using the stereotaxic coordinates (−2.2 bregma; 0 midline; −7.5 dura) according to the atlas of Paxinos and Watson (23). The guide cannula and/or intravenous catheter were held in place with four skull screws (Small Parts, Miami Lakes, FL) and acrylic cement (Lang Dental, Wheeling, IL). Rats received subcutaneous 3 ml lactated Ringer solution (Baxter Pharmaceutical Products, New Providence, NJ) and buprenorphine analgesia (0.05 mg/kg) and were maintained on a circulating water heating pad until recovery from anesthesia. Catheter lines were filled with 60% polyvinylpyrrolidone (PVP10; Sigma, St. Louis, MO)/heparin (1,000 units/ml; Elkins-Sinn, Cherry Hill, NJ) and kept patent by a heparin (10 units/ml) flush every 3 days. All rats regained weight to at least the presurgical level (∼7 days) before the onset of the study.
Experimental procedures
Rats were divided into two groups: one receiving only an intravenous catheter and the other an intravenous catheter and a guide cannula implanted into either the caudal dorsal medial medulla hind-brain site or the third ventricle. All experiments began at 9:00 a.m. Nonfasted rats were subjected to a 2-day testing procedure that was conducted in square acrylic testing chambers (∼30 × 30 × 30 cm) to allow for remote blood collection. Before initiating the experiment, rats were familiarized with the testing chambers (4 h/day for 4 days). During this time, rats were extensively handled and mock central and subcutaneous injections administered to mimic the experimental procedure. The concentration of insulin and saline used in all experiments was 2.5 units/kg s.c. (Novolin R, regular humulin insulin, recombinant DNA origin; Novo Nordisk, Princeton, NJ) and 1 ml/kg s.c., respectively. The concentration of 5TG (Sigma) injected into the hindbrain was 24 μg/200 nl (17), and 150 μg/5 μl (24) was injected into the third ventricle. Saline was used as a vehicle control for hindbrain (200 nl) and third ventricle (5 μl) injections.
On the 1st day of testing (day 1), rats surgically fitted with an intravenous catheter subcutaneously received either insulin to induce hypoglycemia (two 2-h bouts of hypoglycemia separated by a 60-min interval) or saline vehicle. Food was available during the 60-min interval. To verify induction of hypoglycemia, blood (0.1 ml) was drawn immediately before each insulin or saline injection and 60 min thereafter for measurement of plasma glucose. On the 2nd day of testing (day 2), all rats were subcutaneously injected with insulin to induce hypoglycemia. Blood was collected (1.5 ml) immediately before insulin injection (t = 0) and 60 and 120 min thereafter for subsequent measurement of plasma glucose, glucagon, epinephrine, norepinephrine, adrenocorticotropic hormone (ACTH), and corticosterone. Blood was immediately replaced with donor blood drawn from unstressed rats before the experiment. Thus, there were two treatment designations: single hypoglycemia (SH) (n = 11; two vehicle injections on day 1 and insulin-induced hypoglycemia on day 2) and recurrent hypoglycemia (RH) (n = 12; two bouts of insulin-induced hypoglycemia on day 1 and one bout of hypoglycemia on day 2).
Rats surgically fitted with an intravenous catheter and a guide cannula directed at the caudal dorsal medial medulla or third ventricle received a single central injection of 5TG on day 1 of testing. Two additional groups of rats received a vehicle control (saline) injection into the hindbrain or third ventricle on day 1. 5TG or saline was delivered using a 33G stainless steel injector (Plastics One) connected to a Hamilton syringe with PE 20 tubing. Injectors extended 0.5 or 1.0 mm beyond the hindbrain or third ventricle guide cannula tip, respectively. Immediately before 5TG or saline injection and 60 min thereafter, blood (0.1 ml) was collected for subsequent measurement of plasma glucose to verify induction of glucoprivation. On day 2, all rats were subjected to a single bout of insulin-induced hypoglycemia. The treatment designations were hindbrain (HB) 5TG on day 1 and hypoglycemia on day 2 (HB 5TG-Hypo; n = 11) and third ventricular 5TG on day 1 and hypoglycemia on day 2 (3v 5TG-Hypo; n = 11). Vehicle control treatment designations were HB saline on day 1 and hypoglycemia on day 2 (HB Sal-Hypo; n = 8) and 3v saline on day 1 and hypoglycemia on day 2 (3v Sal-Hypo; n = 8). Blood (1.5 ml) was collected on day 2 of testing (t = 0, 60, and 120 min) for subsequent measurement of plasma glucose, glucagon, epinephrine, norepinephrine, ACTH, and corticosterone. Blood was immediately replaced with donor blood drawn from unstressed rats before the experiment. Thus, there were two distinct protocols implemented in this study. The recurrent hypoglycemia protocol consisted of two bouts of systemic hypoglycemia on day 1, whereas the central glucoprivation protocol consisted of a single 5TG injection on day 1. In a pilot study, no differences in day 2 hypoglycemia hormonal CRRs were observed in rats that received one versus two central injections of 5TG on day 1 of testing (N.M.S., unpublished data). In addition, since the hyperglycemic response induced by the first injection of 5TG persisted at the onset of the second 5TG injection, we chose the single injection protocol for this study. Positive cannula placements were verified by a hyperglycemic response to hindbrain (17) or third ventricular 5TG stimulation (25) (determined on day 1 of testing). For the vehicle control groups receiving saline on day 1 of testing (HB Sal-Hypo and 3v Sal-Hypo groups), 5TG was administered at the end of the experiment to verify cannula placement. Only rats that exhibited a statistically significant increase in plasma glucose above their baseline values were included in the study. In response to either hindbrain or third ventricular 5TG injection, plasma glucose levels were increased above baseline values by ∼100 mg/dl.
Plasma assays
Blood samples were obtained for the measurement of hormonal CRRs to insulin-induced hypoglycemia and stored at −80°C until assayed. Blood for the catecholamine assays was collected on EGTA glutathione (2.3:1.5 mg/ml; Sigma). Tubes for glucagon assays contained 50 μl of 1 mol/l benzamidine (Sigma) and 1 unit heparin. Blood for glucose, ACTH, and corticosterone assays was collected on EDTA and aprotinin (1.7 tissue inhibitor unit; Sigma). The assays have been previously described (22). Briefly, a radioenzymatic method as described by Evans et al. (26) was used for determination of plasma epinephrine and norepinephrine. A radioimmunoassay procedure was used for plasma corticosterone measurement, as described by Van Dijk et al. (27). Plasma glucose was measured using the Beckman glucose analyzer. Glucagon was assayed by the Linco glucagon radioimmunoassay kit (Linco Research). Measurements of ACTH were made using the Nichols Institute Diagnostics immunoradiometric assay kit (Nichols Institute Diagnostics, San Juan Capistrano, CA).
Statistical analysis
Data from the plasma assays were analyzed using repeated-measures ANOVA with time as the repeated measure and treatment (SH, RH, HB 5TG-Hypo, 3v 5TG-Hypo, HB Sal-Hypo, and 3v Sal-Hypo groups) as the between-groups factor. In the event of significant effects, Bonferroni's post hoc tests were used to determine significant differences, and t tests were used where appropriate. Significance levels for all tests were taken as P < 0.05.
RESULTS
Day 1 plasma glucose responses
Table 1 presents the plasma glucose data for the experimental groups injected with saline or insulin on day 1 of testing. In the SH group, plasma glucose levels did not change in response to the first or second injection of saline. In response to insulin injection in the RH group, plasma glucose levels were significantly reduced from baseline (t = 0) values (P < 0.05). Table 2 presents the plasma glucose results for the experimental groups receiving a single injection of 5TG into the hindbrain or third ventricle. Both hindbrain and third ventricular delivery of 5TG resulted in a significant increase above baseline plasma glucose at t = 60 (P < 0.05). Vehicle control injection of saline into either the hindbrain (100 ± 4 [t = 0] and 90 ± 5 mg/dl [t = 60]) or third ventricle (92 ± 5 [t = 0] and 102 ± 4 mg/dl [t = 60]) on day 1 did not change plasma glucose levels.
TABLE 1.
Day 1 plasma glucose levels in response to saline or insulin
| First bout |
Second bout |
||||
|---|---|---|---|---|---|
| Treatment group | Drug | t = 0 | t = 60 | t = 0 | t = 60 |
| Single hypoglycemia (SH) | Saline 1 ml/kg, s.c. | 98 ± 5 | 100 ± 4 | 95 ± 3 | 92 ± 8 |
| Recurrent hypoglycemia (RH) | Insulin 2.5 units/kg, s.c. | 107 ± 6 | 34 ± 2* | 83 ± 5 | 39 ± 3* |
Data are means ± SE. Plasma glucose, mg/dl.
P ≤ 0.05 vs. t = 0.
TABLE 2.
Day 1 plasma glucose levels in response to hindbrain or third ventricular 5TG
| Injection site | Drug | t = 0 | t = 60 |
|---|---|---|---|
| Hindbrain | 5TG 24 μg/200 nl | 112 ± 5 | 221 ± 22* |
| Third ventricle | 5TG 150 μg/5 μl | 90 ± 5 | 189 ± 15* |
Data are means ± SE. Plasma glucose, mg/dl.
P ≤ 0.05 vs. t = 0.
Day 2 counterregulatory hormones
Table 3 presents all the raw data for all time points, while Fig. 1 summarizes glucagon, epinephrine, and norepinephrine responses to day 2 hypoglycemia. Baseline (t = 0) values of glucose, glucagon, epinephrine, norepinephrine, ACTH, and corticosterone were not different among the experimental groups (Table 3). In response to insulin-induced hypoglycemia, all experimental groups demonstrated significant decreases in plasma glucose from baseline values (main effect of time: F2,80 = 1398.570, P < 0.001) (Table 3). The declines in plasma glucose levels did not differ among the treatment groups (no interaction between time and treatment: F6,80 = 3.082, P = 0.91).
TABLE 3.
Plasma glucose and hormone responses during day 2 insulin-induced hypoglycemia
| Time point (min) | Glucose (mg/dl) | Glucagon (pg/ml) | Epinephrine (pg/ml) | Norepinephrine (pg/ml) | ACTH (pg/ml) | Corticosterone (ng/ml) |
|---|---|---|---|---|---|---|
| Single hypoglycemia (n = 11) | ||||||
| 0 | 104 ± 7 | 62 ± 13 | 120 ± 25 | 363 ± 30 | 11 ± 3 | 37 ± 6 |
| 60 | 37 ± 3 | 494 ± 56 | 3,811 ± 540 | 925 ± 91 | 226 ± 30 | 223 ± 12 |
| 120 | 25 ± 2 | 428 ± 40 | 3,640 ± 525 | 1,022 ± 85 | 200 ± 19 | 250 ± 21 |
| Recurrent hypoglycemia (n = 12) | ||||||
| 0 | 118 ± 3 | 70 ± 15 | 130 ± 19 | 311 ± 40 | 9 ± 2 | 26 ± 7 |
| 60 | 38 ± 3 | 222 ± 43* | 1,470 ± 325* | 744 ± 75* | 181 ± 19 | 190 ± 20 |
| 120 | 25 ± 1 | 231 ± 34* | 2,500 ± 490 | 1,322 ± 180 | 192 ± 20 | 223 ± 22 |
| Antecedent hindbrain 5TG (n = 11) | ||||||
| 0 | 112 ± 4 | 69 ± 13 | 129 ± 30 | 393 ± 44 | 13 ± 3 | 36 ± 5 |
| 60 | 41 ± 3 | 513 ± 56 | 3,806 ± 344 | 1,154 ± 92 | 243 ± 42 | 208 ± 9 |
| 120 | 27 ± 2 | 403 ± 71 | 4,248 ± 618 | 1,602 ± 170 | 165 ± 17 | 231 ± 10 |
| Antecedent third ventricular 5TG (n = 11) | ||||||
| 0 | 105 ± 7 | 65 ± 6 | 142 ± 18 | 352 ± 41 | 9 ± 3 | 28 ± 7 |
| 60 | 37 ± 3 | 210 ± 21* | 1,162 ± 120* | 723 ± 70* | 220 ± 30 | 180 ± 17 |
| 120 | 28 ± 2 | 290 ± 53 | 3,325 ± 460 | 1,205 ± 180 | 211 ± 35 | 195 ± 19 |
Data are means ± SE; n, no. of rats.
P ≤ 0.05 vs. SH.
FIG. 1.
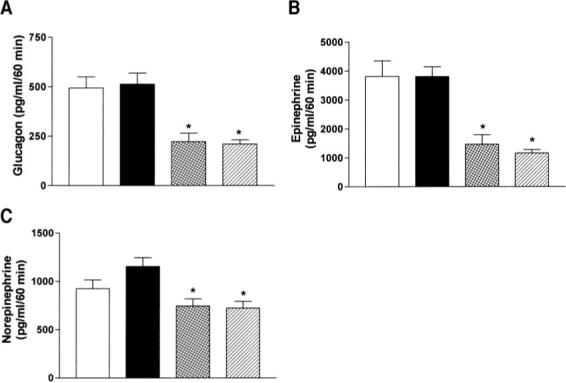
Day 2 plasma glucagon (A), epinephrine (B), and norepinephrine (C) levels (t = 60) in response to insulin-induced hypoglycemia after antecedent (day 1) saline (□), antecedent hindbrain glucoprivation (■), antecedent recurrent hypoglycemia  , or antecedent third ventricular glucoprivation
, or antecedent third ventricular glucoprivation  . Data are expressed as means ± SE. *P < 0.05 vs. SH.
. Data are expressed as means ± SE. *P < 0.05 vs. SH.
There was a significant main effect of treatment on glucagon secretion during day 2 of insulin-induced hypoglycemia (treatment effect: F3,41 = 12.5, P = 0.001; time × treatment interaction: F6,82 = 3.603, P = 0.0032). Recurrent hypoglycemia significantly blunted the glucagon response to day 2 hypoglycemia 60 and 120 min after insulin injection compared with SH-treated rats (P = 0.003 and P = 0.001, respectively) (Table 3 and Fig. 1A). Antecedent third ventricular glucoprivation also significantly suppressed glucagon secretion in response to day 2 hypoglycemia at t = 60 (P = 0.003) (Table 3 and Fig. 1A). In marked contrast, antecedent hindbrain glucoprivation had no effect on glucagon secretion during day 2 of hypoglycemia (Table 3 and Fig. 1A).
There was a significant treatment effect on epinephrine secretion during day 2 of insulin-induced hypoglycemia (treatment effect: F3,35 = 5.7, P = 0.0027; time × treatment interaction: F6,70 = 3.6, P = 0.0036) (Table 3 and Fig. 1B). Compared with the SH group, RH- and 3v 5TG-Hypo–treated rats exhibited a significantly reduced epinephrine response 60 min after insulin injection (P = 0.0001 and P = 0.001, respectively). In contrast, plasma epinephrine levels in HB 5TG-Hypo rats were not different from epinephrine values in the SH group (P = 0.91 and P = 0.478, respectively). There was also a significant treatment effect on the norepinephrine response (treatment effect: F3,34 = 4.15, P = 0.0132; time × treatment interaction F6,68 = 2.74, P = 0.0191). Norepinephrine secretion was reduced in response to hypoglycemia at t = 60 in both RH and 3v 5TG-Hypo groups (P = 0.014 and P = 0.009, respectively) but not in the HB 5TG-Hypo group (Table 3 and Fig. 1C).
All treatment groups exhibited significant increases in plasma ACTH in response to insulin-induced hypoglycemia (main effect of time: F2,58 = 86.9, P = 0.0001) (Table 3). The increases in plasma ACTH levels did not differ among the treatment groups (no interaction between time and treatment: F6,58 = 0.607, P = 0.7235). Plasma corticosterone levels also increased in all experimental groups across the 120-min hypoglycemia session (main effect of time: F2,88 = 86.7, P = 0.0001), with no treatment effect (F3,44 = 2.094, P = 0.1147; no time × treatment interaction: F6,88 = 0.607, P = 0.7235).
Control injections do not blunt counterregulatory responses
Data for hindbrain and third ventricular vehicle control injections are presented in Table 4. As is evident, vehicle injection into the hindbrain or third ventricle on day 1 did not blunt hormonal CRRs as compared with control SH-treated rats (for HB time × treatment: glucagon, F2,42 = 1.491, P = 0.2368; epinephrine, F2,32 = 0.750, P = 0.4803; norepinephrine, F2,34 = 2.099, P = 0.1382; ACTH, F2,30 = 0.951, P = 0.3976; corticosterone, F2,32 = 4.667, P = 0.167; for third ventricle time × treatment: glucagon, F2,42, P = 1.7, P = 0.1962; epinephrine, F2,32 = 2.072, P = 0.1425; norepinephrine, F2,34 = 1.951, P = 0.1577; ACTH, F2,30 = 1.664, P = 0.2063; corticosterone, F2,38 = 0.340, P = 0.7137).
TABLE 4.
Plasma glucose and hormone responses during day 2 insulin-induced hypoglycemia after day 1 hindbrain or third ventricular vehicle injection
| Time point (min) | Glucose (mg/dl) | Glucagon (pg/ml) | Epinephrine (pg/ml) | Norepinephrine (pg/ml) | ACTH (pg/ml) | Corticosterone (ng/ml) |
|---|---|---|---|---|---|---|
| Antecedent hindbrain saline | ||||||
| 0 | 128 ± 2 | 55 ± 17 | 139 ± 29 | 261 ± 16 | 15 ± 4 | 47 ± 10 |
| 60 | 42 ± 3 | 518 ± 70 | 3,666 ± 605 | 852 ± 50 | 203 ± 22 | 186 ± 14 |
| 120 | 34 ± 3 | 540 ± 56 | 4,624 ± 389 | 1,144 ± 97 | 185 ± 10 | 185 ± 13 |
| Antecedent third ventricle saline | ||||||
| 0 | 127 ± 2 | 71 ± 10 | 109 ± 14 | 276 ± 30 | 7 ± 2 | 46 ± 11 |
| 60 | 43 ± 2 | 402 ± 28 | 2,841 ± 298 | 900 ± 91 | 150 ± 10 | 236 ± 21 |
| 120 | 31 ± 3 | 430 ± 45 | 4,468 ± 770 | 1,170 ± 131 | 188 ± 18 | 282 ± 25 |
Data are means ± SE; n = 8 rats in each group.
DISCUSSION
This study evaluates the potential contribution of two distinct brain glucose sensing regions to the development of defective hypoglycemia counterregulation. Our findings demonstrate that a hindbrain glucoreceptive site makes little, if any, contribution to impairments in hormonal CRRs that occur following recurrent bouts of hypoglycemia. Thus, we find that antecedent delivery of 5TG into a hindbrain glucose sensing site does not impair the hormonal CRRs to subsequent hypoglycemia. In contrast, antecedent delivery of 5TG into the third ventricle does reproduce the impairments in hormonal CRRs observed in recurrent hypoglycemic rats.
It has long been known that glucoreceptive sites exist within the hindbrain, and it has been suggested that glucoreceptors exist only in the hindbrain (15,16). For example, acute cerebral aqueduct blockade was shown to abolish hyperglycemic and feeding responses to lateral but not fourth ventricular injection of 5TG (15), demonstrating that 5TG must be transported to caudal hindbrain glucoreceptive sites to initiate these responses. More recently, cannula mapping studies have identified the hindbrain sites from which stimulation of food intake and hyperglycemia could be elicited by local glucoprivation (17). In that study, multiple hindbrain sites were identified as glucoreceptive. However, unilateral injection of 5TG into 61 different hypothalamic sites only revealed 1 site that was positive for stimulation of feeding (17). In the present study, we evaluated the role of the caudal dorsomedial hindbrain glucoreceptive site because delivery of 5TG into this site was shown to elicit the largest feeding and sympathoadrenal response of all the hindbrain sites evaluated (17).
Despite the ability of the caudal hindbrain site to detect glucoprivation and elicit CRRs, we find that a single antecedent bout of 5TG-induced glucoprivation localized to this site does not by itself produce defective hormonal CRRs to a subsequent bout of systemic insulin-induced hypoglycemia. In fact, we find that antecedent glucoprivic stimulation of this hindbrain site resulted in hormonal responses to subsequent hypoglycemia that closely matched the magnitude of hormonal responses elicited by a single bout of hypoglycemia. In marked contrast to antecedent hindbrain glucoprivation, antecedent delivery of 5TG into the third ventricle did impair hormonal CRRs to a subsequent bout of hypoglycemia, which mimicked the effects of recurrent systemic hypoglycemia on hormonal CRRs. Similarly, Marin-Spiotta et al. (28) reported that 2-deoxy-D-glucose (2DG) delivered into the third ventricle also attenuated the epinephrine response to a subsequent bout of hypoglycemia. However, in that study there was no deficit in the glucagon response as we found here for third ventricular 5TG. This discrepancy may be due to the greater potency of 5TG in producing glucoprivation compared with 2DG (29).
Although third ventricular delivery of 5TG would be expected to reach the hindbrain, we believe our findings were due to the actions of 5TG within the third ventricle and surrounding hypothalamic nuclei for the following reasons. First, and most importantly, direct glucoprivic stimulation of the caudal hindbrain glucose sensing site did not impair CRRs during subsequent hypoglycemia. Second, 5TG, like glucose, is actively transported into neurons and glia. Thus, it does not seem likely that an adequate concentration of 5TG would remain in the cerebrospinal fluid once it reached the hindbrain to elicit a CRR. Finally, the majority of cerebrospinal fluid exits the ventricular system via the lateral apertures, located at the pontomedullary junction (30), a site rostral to this hindbrain glucoreceptive site, which is located deep within the ventral medulla (17).
The fact that third ventricular glucoprivation blunted CRRs to subsequent hypoglycemia reinforces the idea that hypothalamic sites are critical not only for the expression of CRRs to single hypoglycemia, but also for the blunting of CRRs with recurrent hypoglycemia. For example, bilateral dialysis of 2DG into the ventromedial hypothalamus (VMH), including the ventromedial hypothalamic nucleus (VMN) and arcuate nucleus, stimulates a robust hormonal CRR (8). Conversely, VMH lesions (7) or bilateral VMH glucose perfusion during systemic hypoglycemia (9) effectively block the hormonal CRRs. In addition, glucose sensing neurons exist in the VMN (14), arcuate (31), and lateral hypothalamus (6). These specialized neurons alter their firing rate in response to physiological changes in glucose availability (14) and are hypothesized to initiate CRRs (32). The VMH is also an important brain site in the HAAF pathway: Recurrent hypoglycemia impairs the activation of CRRs induced by bilateral VMH glucoprivation (10). Further, it was recently demonstrated that recurrent hypoglycemia reduces the glucose sensitivity of glucose sensing neurons residing in the VMN (33), a potential mechanism contributing to the development of defective CRRs. Taken together with our current findings, these studies suggest that altered function of neurons surrounding the third ventricle, particularly in the VMH, are primarily responsible for much of the reduced CRRs to hypoglycemia.
Whether or not antecedent third ventricular glucoprivation specifically impaired hypothalamic glucose sensing mechanisms cannot be determined from our findings. Although impairments in afferent mechanisms are possible, it is also possible that impairments in efferent responses contributed to defective CRRs. Indeed, this has been suggested by previous studies. For example, local administration of lidocaine into key hypothalamic autonomic effector nuclei, the paraventricular nucleus (PVN) (13) or dorsal medial nucleus (DMH) (12), during hypoglycemia partially simulates the impaired hormonal responses observed with recurrent hypoglycemia. Likewise, hindbrain catecholamine neurons, which are required for the expression of glucoprivic feeding and sympathoadrenal responses (18), exhibit reduced activation with repeated glucoprivic episodes (34). Thus, non–glucose sensing hypothalamic and hindbrain sites may also be involved in the pathogenesis of HAAF. Although it is not clear from these findings if antecedent third ventricular glucoprivation targeted afferent or efferent neural substrates, our findings demonstrate that the neural and/or glial substrates surrounding the third ventricle are unique, since antecedent delivery of 5TG into this site, but not the hindbrain, reproduced HAAF.
The results of this study clearly demonstrate differential effects of antecedent third ventricular versus hindbrain glucoprivation on subsequent hypoglycemic CRRs. However, there are some caveats to consider. Glucoprivation was localized to only one of the several reported hindbrain glucoreceptive sites (17). It may be that multiple hindbrain glucoreceptive sites must be affected collectively, as would be expected to occur with systemic hypoglycemia, in order to reveal the full contribution of the hindbrain in defective CRRs. On the other hand, antecedent delivery of 5TG into the third ventricle would likely induce glucoprivation in several brain sites, thus more closely reproducing the effects of systemic hypoglycemia. This is consistent with previous studies indicating that the complete blunted hormonal profile of HAAF involves multiple hypothalamic sites (12,13). Another factor to consider in our study is the lack of insulin administration in the central glucoprivation treatment on day 1. We cannot specifically rule out a role for hyperinsulinemia in defective CRRs; however, our results do not support such a role since both antecedent third ventricular glucoprivation and insulin-induced hypoglycemia impaired subsequent hypoglycemia CRRs. In addition, previous work implementing a similar protocol reported that two antecedent bouts of hyperinsulinemia/euglycemia did not impair subsequent hypoglycemia CRRs (35). However, there are inconsistencies in the literature since a more aggressive protocol of hyperinsuinemia/euglycemia does impair the epinephrine response to subsequent hypoglycemia (36). Lastly, this study induced central glucoprivation using the pharmacological agent 5TG. 5TG induces cellular glucoprivation through the inhibition of phosphoglucomutase, glucose-6-phosphate dehydrogenase, and hexokinase activity (37). Although 5TG has been shown to be toxic, these effects require much greater concentrations than used in the present study and are specific to cultured cells exposed to hypoxic conditions (38).
It is also important to point out that although our findings did not identify a critical role for this hindbrain glucoreceptive site in defective CRRs, it does not negate its importance in hypoglycemia detection or in CRRs. In fact, our findings suggest that this hindbrain glucoreceptive site remains responsive despite antecedent glucoprivation. This is consistent with human (39) and animal (22,40–42) studies demonstrating that even during recurrent hypoglycemia, hormonal responses are still elevated above baseline, demonstrating that some glucose sensing mechanisms must remain intact under recurrent hypoglycemic conditions. The identification of the phenotype of hindbrain glucose sensing neurons and the underlying glucose sensing mechanisms may provide insight into why this glucose sensing site remains responsive under recurrent glucoprivic conditions and why other hypothalamic sites do not.
In summary, we find that hindbrain and hypothalamic glucose sensing sites do not contribute equally to the development of HAAF. Prior glucoprivic stimulation of a hindbrain glucoreceptive site did not impair subsequent hypoglycemic CRRs. Thus, this hindbrain glucoreceptive site may not play a role in the development of HAAF. In marked contrast, antecedent third ventricular glucoprivation impaired CRRs to a subsequent bout of hypoglycemia, thus reproducing the effects of recurrent systemic hypoglycemia. These findings demonstrate that hindbrain glucose sensing sites are not subject to the pathogenic mechanisms that result in defective hypoglycemia CRRs, whereas those surrounding the third ventricle are.
ACKNOWLEDGMENTS
These studies were supported by the Research Services of the VA, National Institutes of Health grants (DK 40963 to D.P.F. and DK 50154 to G.J.T.), Merit Review Program and Research Career Scientist awards (to D.P.F. and G.J.T.), and the American Diabetes Association (Junior Faculty Award to N.M.S.).
We gratefully acknowledge the technical expertise of Aryana Zavosh, Pam Gronbeck, Carl Sikkema, and the Metabolism Lab for excellent technical support with as-says and surgical preparations.
Glossary
- 2DG
2-deoxy-d-glucose
- 5TG
5-thio-glucose
- CRR
counterregulatory response
- HAAF
hypoglycemia-associated autonomic failure
- RH
recurrent hypoglycemia
- SH
single hypoglycemia
- VMH
ventromedial hypothalamus
- VMN
ventromedial hypothalamic nucleus
REFERENCES
- 1.Leese GP, Wang J, Broomhall J, Kelly P, Marsden A, Morrison W, Frier BM, Morris AD. Frequency of severe hypoglycemia requiring emergency treatment in type 1 and type 2 diabetes. Diabetes Care. 2003;26:1176–1180. doi: 10.2337/diacare.26.4.1176. [DOI] [PubMed] [Google Scholar]
- 2.The Diabetes Control and Complications Trial Research Group The effect of intensive treatment of diabetes on the development and progression of long-term complications of insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 3.The Diabetes Control and Complications Trial Research Group Hypoglycemia in the diabetes control and complications trial. Diabetes. 1997;46:271–286. [PubMed] [Google Scholar]
- 4.Dagogo-Jack SE, Craft S, Cryer PE. Hypoglycemia-associated autonomic failure in insulin-dependent diabetes mellitus: recent antecedent hypoglycemia reduces autonomic responses to symptoms of, and defense against subsequent hypoglycemia. J Clin Invest. 1993;91:819–828. doi: 10.1172/JCI116302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Segel SA, Paramore DS, Cryer PE. Hypoglycemia-associated autonomic failure in advanced type 2 diabetes. Diabetes. 2002;51:724–733. doi: 10.2337/diabetes.51.3.724. [DOI] [PubMed] [Google Scholar]
- 6.Oomura Y, Ono T, Ooyama H, Wayner MJ. Glucose and osmosensitive neurons of the rat hypothalamus. Nature. 1969;222:282–284. doi: 10.1038/222282a0. [DOI] [PubMed] [Google Scholar]
- 7.Borg WP, During MJ, Sherwin RS, Borg MA, Brines ML, Shulman GI. Ventromedial hypothalamic lesions in rats suppress counterregulatory responses to hypoglycemia. J Clin Invest. 1994;93:1677–1682. doi: 10.1172/JCI117150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI. Local ventromedial hypothalamic glucopenia triggers counterregulatory hormone release. Diabetes. 1995;44:180–184. doi: 10.2337/diab.44.2.180. [DOI] [PubMed] [Google Scholar]
- 9.Borg MA, Sherwin RS, Borg WP, Tamborlane WV, Shulman GI. Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Invest. 1997;99:361–365. doi: 10.1172/JCI119165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borg WP, Tamborlane WV, Brines ML, Shulman GI, Sherwin RS. Chronic hypoglycemia and diabetes impair counterregulation induced by localized 2-deoxy-glucose perfusion of the ventromedial hypothalamus in rats. Diabetes. 1999;48:584–587. doi: 10.2337/diabetes.48.3.584. [DOI] [PubMed] [Google Scholar]
- 11.de Vries MG, Lawson MA, Beverly JL. Hypoglycemia-induced noradrenergic activation in the VMH is a result of decreased ambient glucose. Am J Physiol. 2005;289:977–981. doi: 10.1152/ajpregu.00403.2005. [DOI] [PubMed] [Google Scholar]
- 12.Evans SB, Wilkinson CW, Gronbeck P, Bennett JL, Zavosh A, Taborsky GJ, Jr, Figlewicz Lattemann DP. Inactivation of the dorsomedial nucleus of the hypothalamus selectively inhibits the ACTH and corticosterone response to hypoglycemia. Am J Physiol Regul Integr Comp Physiol. 2004;286:R123–R128. doi: 10.1152/ajpregu.00328.2003. [DOI] [PubMed] [Google Scholar]
- 13.Evans S, Wilkinson C, Gronbeck P, Bennett J, Taborsky GJ, Jr, Figlewicz D. Inactivation of the PVN during hypoglycemia partially simulates hypoglycemia-associated autonomic failure. Am J Physiol Regul Integr Comp Physiol. 2003;284:R57–R65. doi: 10.1152/ajpregu.00439.2002. [DOI] [PubMed] [Google Scholar]
- 14.Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus (VMN). Diabetes. 2001;50:2673–2681. doi: 10.2337/diabetes.50.12.2673. [DOI] [PubMed] [Google Scholar]
- 15.Ritter RC, Slusser PG, Stone S. Glucoreceptors controlling feeding and blood glucose: location in the hindbrain. Science. 1981;213:451–452. doi: 10.1126/science.6264602. [DOI] [PubMed] [Google Scholar]
- 16.DiRocco RJ, Grill HJ. The forebrain is not essential for sympathoadrenal hyperglycemic response to glucoprivation. Science. 1979;204:1112–1114. doi: 10.1126/science.451558. [DOI] [PubMed] [Google Scholar]
- 17.Ritter S, Dinh TT, Zhang Y. Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res. 2000;856:37–47. doi: 10.1016/s0006-8993(99)02327-6. [DOI] [PubMed] [Google Scholar]
- 18.Ritter S, Bugarith K, Dinh TT. Immunotoxic destruction of distinct catecholamine subgroups produces selective impairment of glucoregulatory responses and neuronal activation. J Comp Neurol. 2001;432:197–216. doi: 10.1002/cne.1097. [DOI] [PubMed] [Google Scholar]
- 19.Sanders NM, Dunn-Meynell AA, Levin BE. Third ventricular alloxan reversibly impairs glucose counterregulatory responses. Diabetes. 2004;53:1230–1236. doi: 10.2337/diabetes.53.5.1230. [DOI] [PubMed] [Google Scholar]
- 20.McCrimmon RJ, Evans ML, Fan X, McNay EC, Chan O, Ding Y, Zhu W, Gram DX, Sherwin RS. Activation of ATP-sensitive K+ channels in the ventromedial hypothalamus amplifies counterregulatory hormone responses to hypoglycemia in normal and recurrent hypoglycemic rats. Diabetes. 2005;54:3169–3174. doi: 10.2337/diabetes.54.11.3169. [DOI] [PubMed] [Google Scholar]
- 21.McCrimmon RJ, Song Z, Cheng H, McNay EC, Weikart-Yeckel C, Fan X, Routh VH, Sherwin RS. Corticotrophin-releasing factor receptors within the ventromedial hypothalamus regulate hypoglycemia-induced hormonal counterregulation. J Clin Invest. 2006;116:1723–1730. doi: 10.1172/JCI27775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans S, Wilkinson CW, Bentson K, Gronbeck P, Zavosh A, Figlewicz D. PVN activation is suppressed by repeated hypoglycemia but not antecedent corticosterone in the rat. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1426–R1436. doi: 10.1152/ajpregu.2001.281.5.R1426. [DOI] [PubMed] [Google Scholar]
- 23.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; New York: 1986. [Google Scholar]
- 24.Tordoff MG, Flynn FW, Grill HJ, Friedman MI. Contribution of fat metabolism to ‘glucoprivic’ feeding produced by fourth ventricular 5-thio-D-glucose. Brain Res. 1988;445:216–222. doi: 10.1016/0006-8993(88)91181-x. [DOI] [PubMed] [Google Scholar]
- 25.Slusser PG, Ritter RC. Increased feeding and hyperglycemia elicited by intracerebroventricular 5-thioglucose. Brain Res. 1980;202:474–478. doi: 10.1016/0006-8993(80)90158-4. [DOI] [PubMed] [Google Scholar]
- 26.Evans MI, Halter JB, Porte D. Comparison of double- and single-isotope enzymatic derivative methods for measuring catecholamines in human plasma. Clin Chem. 1978;24:567–570. [PubMed] [Google Scholar]
- 27.Van Dijk G, Donahey JC, Thiele TE, Scheurink AJ, Steffens A, Wilkinson CW, Tenenbaum R, Campfield LA, Burn P, Seeley RJ, Woods SC. Central leptin stimulates corticosterone secretion at the onset of the dark phase. Diabetes. 1997;46:1911–1914. doi: 10.2337/diabetes.46.11.1911. [DOI] [PubMed] [Google Scholar]
- 28.Marin-Spiotta A, Levin BE, Tkacs NC. A single episode of central glucoprivation reduces the adrenomedullary response to subsequent hypoglycemia in rats. Neurosci Lett. 2004;360:81–84. doi: 10.1016/j.neulet.2004.02.044. [DOI] [PubMed] [Google Scholar]
- 29.Ritter RC, Slusser PG. 5-thio-D-glucose causes increased feeding and hyperglycemia in the rat. Am J Physiol. 1980;238:E141–E144. doi: 10.1152/ajpendo.1980.238.2.E141. [DOI] [PubMed] [Google Scholar]
- 30.King AS. Physiological and Clinical Anatomy of the Domestic Mammals. Oxford Univ. Press; New York: 1987. pp. 13–23. [Google Scholar]
- 31.Wang R, Cruciani-Guglielmacci C, Migrenne S, Magnan C, Cotero VE, Routh VH. Effects of oleic acid on distinct populations of neurons in the hypothalamic arcuate nucleus are dependent on extracellular glucose levels. J Neurophysiol. 2006;95:1491–1498. doi: 10.1152/jn.00697.2005. [DOI] [PubMed] [Google Scholar]
- 32.Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell A. Neuronal glucosensing: what do we know after 50 years? Diabetes. 2004;53:2521–2528. doi: 10.2337/diabetes.53.10.2521. [DOI] [PubMed] [Google Scholar]
- 33.Song Z, Routh VH. Recurrent hypoglycemia reduces the glucose sensitivity of glucose inhibited neurons in the ventromedial hypothalamus nucleus (VMN). Am J Physiol Regul Integr Comp Physiol. 2006;291:R1283–R1287. doi: 10.1152/ajpregu.00148.2006. [DOI] [PubMed] [Google Scholar]
- 34.Sanders NM, Ritter S. Repeated 2-deoxy-D-glucose-induced glucoprivation attenuates Fos expression and glucoregulatory responses during subsequent glucoprivation. Diabetes. 2000;49:1865–1874. doi: 10.2337/diabetes.49.11.1865. [DOI] [PubMed] [Google Scholar]
- 35.Sandoval DA, Ping L, Neill RA, Gong B, Walsh K, Davis SN. Brain region-dependent effects of dexamethasone on counterregulatory responses to hypoglycemia in conscious rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R413–R419. doi: 10.1152/ajpregu.00674.2003. [DOI] [PubMed] [Google Scholar]
- 36.Shum K, Inouye K, Chan O, Mathoo J, Bilinski D, Matthews SG, Vranic M. Effects of antecedent hypoglycemia, hyperinsulinemia, and excess corticosterone on hypoglycemic counterregulation. Am J Physiol Endocrinol Metab. 2001;281:E455–E465. doi: 10.1152/ajpendo.2001.281.3.E455. [DOI] [PubMed] [Google Scholar]
- 37.Chen M, Whistler RL. Action of 5-thio-D-glucose and its 1-phosphate with hexokinase and phosphoglucomutase. Arch Biochem Biophys. 1975;169:392–396. doi: 10.1016/0003-9861(75)90180-0. [DOI] [PubMed] [Google Scholar]
- 38.Song CW, Clement JJ, Levitt SH. Preferential cytotoxicity of 5-thio-D-glucose against hypoxic tumor cells. J Natl Cancer Inst. 1976;57:603–605. doi: 10.1093/jnci/57.3.603. [DOI] [PubMed] [Google Scholar]
- 39.Davis SN, Shavers C, Davis B, Costa F. Prevention of an increase in plasma cortisol during hypoglycemia preserves subsequent counterregulatory responses. J Clin Invest. 1997;100:429–438. doi: 10.1172/JCI119550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tkacs NC, Dunn-Meynell AA, Levin BE. Presumed apoptosis and reduced arcuate nucleus neuropeptide Y and pro-opiomelanocortin mRNA in non-coma hypoglycemia. Diabetes. 2000;49:820–826. doi: 10.2337/diabetes.49.5.820. [DOI] [PubMed] [Google Scholar]
- 41.De Vries MG, Arseneau LM, Lawson ME, Beverly JL. Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes. 2003;52:2767–2773. doi: 10.2337/diabetes.52.11.2767. [DOI] [PubMed] [Google Scholar]
- 42.Flanagan DE, Keshavarz T, Evans ML, Flanagan S, Fan X, Jacob RJ, Sherwin RS. Role of corticotrophin-releasing hormone in the impairment of counterregulatory responses to hypoglycemia. Diabetes. 2003;52:605–613. doi: 10.2337/diabetes.52.3.605. [DOI] [PubMed] [Google Scholar]