

Abstract
The principal source of hydrogen peroxide in mitochondria is thought to be from the dismutation of superoxide via the enzyme manganese superoxide dismutase (MnSOD). However, the nature of the effect of SOD on the cellular production of H2O2 is not widely appreciated. The current paradigm is that the presence of SOD results in a lower level of H2O2 because it would prevent the non-enzymatic reactions of superoxide that form H2O2. The goal of this work was to: a) demonstrate that SOD can increase the flux of H2O2, and b) use kinetic modelling to determine what kinetic and thermodynamic conditions result in SOD increasing the flux of H2O2. We examined two biological sources of superoxide production (xanthine oxidase and coenzyme Q semiquinone, CoQ•−) that have different thermodynamic and kinetic properties. We found that SOD could change the rate of formation of H2O2 in cases where equilibrium-specific reactions form superoxide with an equilibrium constant (K) less than 1. An example is the formation of superoxide in the electron transport chain (ETC) of the mitochondria by the reaction of ubisemiquinone radical with dioxygen. We measured the rate of release of H2O2 into culture medium from cells with differing levels of MnSOD. We found that the higher the level of SOD, the greater the rate of accumulation of H2O2. Results with kinetic modelling were consistent with this observation; the steady-state level of H2O2 increases if K < 1, for example CoQ•− + O2 → CoQ + O2•−. However, when K > 1, e.g. xanthine oxidase forming O2•−, SOD does not affect the steady state-level of H2O2. Thus, the current paradigm that SOD will lower the flux of H2O2 does not hold for the ETC. These observations indicate that MnSOD contributes to the flux of H2O2 in cells and thereby is involved in establishing the cellular redox environment and thus the biological state of the cell.
Keywords: superoxide dismutase, mitochondria, coenzyme Q, hydrogen peroxide, superoxide, free radical, redox environment
Introduction
Superoxide dismutase is an important antioxidant enzyme as it is found in nearly all organisms. In mammals there are at least three forms of SOD: a cytosolic (CuZnSOD), an extracellular (ECSOD), and a mitochondrial form (MnSOD). All SOD enzymes catalyze the dismutation of superoxide, Rxns 2-5 Table 1 [1,2]:
TABLE 1.
The reactions in the kinetic model
| Rate Constant | |||
|---|---|---|---|
| Rxn Nr. | Reaction | M−1 •s−1 or s−1 | Reference/comment |
| 1a | XO-FADH• + O2 → XO-FAD + O2•− + H+ | k1a = 7 × 104 | [30] |
| −1a | XO-FAD + O2•− + H+ → XO-FADH• + O2 | k−1a = 7 | Estimated here using K1b = 104 |
| 1b | CoQ10•− + O2 → CoQ10 + O2•− | k1b = 8 × 103 | [77,95,96, 97] The actual value is uncertain, but an equilibrium constant appears to be ≈ 0.01–0.1. |
| −1b | CoQ10 + O2•− → CoQ10•− + O2 | k−1b = 8 × 105 | [77,95,96] |
| 2 | MnIIISOD + O2•− → MnIIISOD:O2•−a | k2 = 1.5 × 109 | [98, 99] |
| −2 | MnIIISOD:O2•− → MnIIISOD + O2•− | k−2 = 3.5 × 104 | [99] |
| 3 | MnIIISOD: O2•− → MnIISOD + O2 | k3 = 2.5 × 104 | [99] |
| −3 | MnIISOD + O2 → MnIIISOD:O2•−a | k−3 = 0 | [99] |
| 4 | MnIISOD + O2•− → MnIISOD:O2•−b | k4 = 1.5 × 109 | [98, 99] |
| −4 | MnIISOD:O2•− → MnIISOD + O2•− | k−4 = 3.5 × 104 | [99] |
| 5 | MnIISOD:O2•− + 2H+ → MnIIISOD + H2O2 | k5 = 2.5 × 104 | [99] |
| −5 | MnIIISOD + H2O2 → MnIISOD:O2•− + 2H+ | k−5 = 300 | [99] |
| 6 | MnIISOD:O2•− → DEP c | k6 = 650 | [99] |
| −6 | DEP → MnIISOD: O2•− | k−6 = 10 | [99] |
| 7 | 2H+ + 2O2•− → O2 + H2O2 | k7 = 2.4 × 105 | [5] |
| −7 | O2 + H2O2 → 2H+ + 2O2•− | k−7 = 0 | [5] |
| 8 | GPxr + H2O2 + H+ → GPx0 + H2Od | k8 = 2.1 × 107 | [100, 101, 102] |
| −8 | GPx0 + H2O → GPxr + H2O2 | k−8 = 0 | [100, 101] |
| 9 | GPx0 + GSH → GSGPx + H2O | k9 = 4 × 104 | [100, 101] |
| −9 | GSGPx + H2O → GPx0 + GSH | k−9 = 0 | [100, 101] |
| 10 | GSGPx + GSH → GPxr + GSSG + H+ | k10 = 1 × 107 | [100, 101] |
| −10 | GPxr + GSSG + H+ → GSGPx + GSH | k−10 = 0 | [100, 101] |
| 11 | GSSG → 2GSH | ||
| −11 | 2GSH → GSSG | ||
| 12 | CoQ•− + O2•− + 2H+ → CoQ + H2O2 | k12(obs) = 3 × 106 e | Estimated |
MnIIISOD:O2•− is a complex of MnIIISOD and O2•−.
MnIISOD:O2•− is a complex of MnIISOD andO2•−.
DEP is dead end product.
Catalase was not included in the kinetic model because it is primarily a peroxisomal enzyme. We included only GPx1 as a sink for H2O2. Because we kept the capacity of the system for removing H2O2 constant, the inclusion of catalase or peroxiredoxin-III as additional components of the H2O2-removing system would make no difference in the final results of the model.
This is an estimate based on the non-enzymatic dismutation of superoxide. The rate constant will rely on the pH and pKa of these two species. If we assume the maximum rate constant is parallel to that of superoxide, i.e. the reaction of protonated and unprotonated species, then we can assume that the fastest reaction will have either O2•− protonated or CoQ•− protonated. If the effective pKa of CoQH• is 5.9 [103] then at pH 7.4 about 3% of CoQ•− and 0.2% of O2•− will be protonated. With this, an estimate of an observed rate constant would be kobs ≈3 × 106 M−1 s−1. Once superoxide is formed, possible routes to formation of H2O2, as a rate equation, are: +d[H2O2]/dt = {kSOD [SOD] + kCoQ•− [CoQ•−] + kdismut[O2•−] + kother[other]} [O2•−]. Estimating the contributions of each term +d[H2O2]/dt = {2 × 104 s−1 + 3 × 10−1 s−1 + 2.4 × 10−4 s−1 × + kother[other]} × [O2•−], and assuming that [O2•−] is at most on the order of 1 nM, we see that the termination reaction in question is negligible, as well as the chemical dismutation of superoxide. Thus this reaction was not included in the kinetic model. Here, “other” refers to reactions of superoxide with substances such as aconitase, Fe3+cytochrome c, nitric oxide etc. These represent a small fraction of the reactions of superoxide [104, 105].
It has been shown that MnSOD-knockout mice die within 1–18 days after birth, depending on their genetic background [6,7]; thus, MnSOD is an essential enzyme. Changes in SOD levels inside cells can have opposing effects. High overexpression of SOD in E. coli has been found to increase sensitivity to paraquat and to hyperoxia [8], as well as ionizing radiation due to apparent increased production of H2O2 [9]. Human and mouse cell clones that overexpress human CuZnSOD appear to have higher levels of hydrogen peroxide [10,11]. However, transfection of V79 Chinese hamster cells to overexpress CuZnSOD resulted in a decrease in H2O2 in cells [12]. Overexpression of MnSOD in several human cell lines has provided indirect evidence that this increased expression leads to increases in the production of H2O2 [13,14,15,16]. However, the current paradigm in the research community is that SOD decreases the production of H2O2 [17,18,19]: “the proposal that SOD enhances H2O2 by catalyzing the dismutation reaction can be discounted” [17].
The current paradigm
The concentrations of O2•− and H2O2 in a cell are assumed to be in a quasi steady-state. These steady-state concentrations, [O2•−]ss and [H2O2]ss, reflect a balance between the rate of formation and the rate of removal. Thus, the steady-state level can change by either changing the rate of formation and/or the rate of removal. It is widely accepted that changes in levels of cellular SOD will result in:
a change in [O2•−]ss, e.g. an increase in SOD would increase the rate of removal of O2•− and thereby lower [O2•−]ss;
no change in the rate of production of O2•−;
a minor change in the rate of production of H2O2, no more than a factor of two (This assumes that Case 3 described below is not applicable.); this change in the rate of production of H2O2 could result in
a minor change in [H2O2]ss [17].
This paradigm is based on the observation that the rate of production of H2O2 by the enzyme xanthine oxidase (XO) is not affected by SOD. XO is a well-studied enzyme that is widely used as a tool to generate superoxide and hydrogen peroxide in experimental systems [20].
A new paradigm
However, we propose that there are other superoxide-generating systems (e.g. the mitochondrial electron transport chain) that do not behave like the xanthine/xanthine oxidase (X/XO) system. In these systems, changes in levels of cellular SOD can result in:
a change in [O2•−]ss;
a change in the rate of production of O2•−;
a major change in the rate of production of H2O2, more than a factor of two; this could result in
a major change in [H2O2]ss.
How could SOD change the level of H2O2 in cells?
There are various ways that superoxide can be converted to H2O2.
Case 1: Dismutation reaction
Independent of how O2•− dismutes, i.e. with or without SOD, two O2•− will yield one H2O2, net of Rxns 2-5 of Table 1.
Case 2: Reactions with electron donors
If O2•− reacts with an electron-proton/hydrogen-atom donor, then one O2•− will yield one H2O2.
In addition to cellular reducing agents, the donor could be a transition metal such as Fe2+ yielding Fe3+, for example with the 4Fe-4S cluster of aconitase.
Case 3: Chain reactions that produce superoxide
There are cases where one superoxide can result in the formation of considerably more of both superoxide and hydrogen peroxide, i.e., one O2•− will yield n H2O2, where n can be much greater than one. This scenario can occur in a chain reaction where O2•− is a product of the reaction and is also a chain-carrying radical, initiating a new chain [21,22,23,24].
An example is the oxidation of 6-hydroxydopamine (6-OHDA) to its semiquinone (SQ•−) and quinone (Q):
Here, after initiation, O2•− is formed by electron transfer from the semiquinone radical of 6-OHDA (SQ•−) to O2. Superoxide then serves as a chain-carrying radical to initiate additional oxidation cycles, each forming another O2•− and H2O2. Thus, the formation of one O2•− will lead to n H2O2, where n is essentially the chain-length of this set of reactions. The chain termination reaction of SQ•− + O2•− is probably not of consequence. See Table 1. Iron(III) is shown as the initiating species above, but in higher pH environments metals may not be needed as true autoxidation can occur [25].
Case 4: Reactions with non-hydrogen atom electron acceptors/donors
If superoxide reacts with an electron-acceptor, such as Fe3+cytochrome c, or covalently with an electron-donor, such as nitric oxide, then there will be no H2O2 produced. In this case, SOD would increase the rate of production of H2O2. Although these reactions of superoxide are biologically important, they were not included in this study as they are typically minor sinks for superoxide. In addition, the reactions are not considered reversible, and the focus of this study is on the reversible formation of superoxide.
The current paradigm is that if SOD is present in Cases 2 and 3 above, its reaction with O2•− would dominate reactions of O2•− with donor-H or the chain-initiation by O2•−, thereby converting these processes into Case 1, resulting in less H2O2. In Case 2 the amount of H2O2 would decrease by no more than a factor of two; in Case 3 the decrease would be by a factor of approximately n. In these cases, SOD lowers the level of H2O2. Translating this into a cellular setting would mean that overexpression of SOD in cells would result in lower levels of H2O2. However, experimental data have provided indirect evidence that cells with increased levels of SOD can have increased levels of H2O2 [13–16]. This apparent contradiction can be rationalized if increasing SOD increases the rate of formation of O2•− and consequently H2O2. The paradigm summarized above does not consider the possibility that SOD could change the rate of production of O2•− reversible reactions.
How could SOD change the rate of formation of O2•− and H2O2?
If the rate of production of O2•− in a system is constant and the amount of SOD varies, then as indicated above the rate of production of H2O2 will either remain the same, Case 1, or decrease, Cases 2 and 3. However, if Le Chatelier’s principle1 applies to the reaction forming O2•−, then the rate of production of O2•− could change with changing levels of SOD, resulting in an increase in the rate of production of H2O2.
For example, in the reaction below, if SOD is present it will compete with the reverse reaction and remove some of the O2•− and thereby “pull” the reaction to the right. SOD will have the most influence when the rate constant of the reverse reaction is greater than the rate constant of the forward reaction (kr > kf; K = kf/kr < 1).
Increased levels of SOD will pull the equilibrium of this reversible reaction to the right, and the rate of production of O2•− and subsequently H2O2 will increase. If the reverse reaction is slow (kr < kf; K > 1),
then only a very small fraction of the O2•− molecules will enter the reverse reaction and dismutation will occur with or without SOD. Changes in the amount of SOD would have little impact. In a cellular setting both scenarios can be found. In the case of XO (K > 1) we propose that the current paradigm applies, i.e. increasing SOD will either have no effect, Case 1, or decrease levels of H2O2, because the rate of the reverse reaction is slow compared to the forward reaction that produces O2•−, i.e. Case 2. However, there are reactions in the cell that have K < 1, for example CoQ•− forming O2•−. We propose that when the reverse reaction is comparable to or faster than the forward reaction (K < 1), then Le Chatelier’s principle may apply and the current paradigm no longer holds. Or stated another way, when the equilibrium constant (K) for the reaction forming O2•− is ≫1, then SOD will have little effect on the rate of production of H2O2; but, when K is ≈1 or <1, then SOD can affect the rate of production of H2O2. We propose that the latter is the case in the CoQ•− reaction, but not in the reaction of XO to form O2•−.
A. The xanthine oxidoreductase system, thermodynamics
The metalloenzyme xanthine oxidoreductase (XOR) is a dimer that contains two molybdenum (Mo) atoms and eight irons as four (2Fe-2S) moieties. In addition there are two FAD groups in the enzyme. Thus, each subunit contains, 1 Mo, 2 (2Fe-2S), and 1 FAD [26]. The oxidase form of XOR is a widely used tool to produce superoxide in the laboratory, but it can also be a major source of unchecked superoxide production in vivo [27,28]. The oxidase form (XO) oxidizes xanthine, hypoxanthine or other substrates using oxygen as the preferred electron-acceptor, producing O2•− and H2O2, Figure 1 [29,30,31].
Figure 1. The flow of electrons through xanthine oxidase.

Xanthine makes a complex with XO at the molybdenum site (ka = 6 × 106 M−1 s−1, 37°C [116]); then xanthine is oxidized to urate with reduction of the molybdenum (kb = 15 s−1). This is followed by transfer of electrons to the Fe/S centers (kc = 8.5 × 103 s−1 [117]); then to FAD (kd = 2 × 102 s−1 [117]), and finally production of O2•− from FADH• (k1a = 7 × 104 M−1 s−1). The thermodynamics for the formation of O2•− from the FADH• indicate an equilibrium constant of ≈104. Thus the back reaction would have a rate constant of ≈7 M−1 s−1 [30].
It is the radical of the flavin moiety (XO-FADH•) that interacts with O2 forming O2•− and subsequently H2O2 [32]. (The direct two-electron reduction of oxygen to form H2O2 in general occurs through a fully reduced flavin, FADH2.)
The equilibrium constant for this reaction can be estimated from the reduction potentials of the two redox couples:
Using the fundamental relationship connecting the Gibbs free energy of a reaction to its equilibrium constant, i.e. ΔG°′ = −nFΔE°′ = −RT lnK, the equilibrium constant for the formation of O2•− by the flavin radical in XO is ≫ 1 (K1a = 104). With this equilibrium constant, very little of the O2•− produced will enter into the reverse reaction; in the absence of SOD the O2•− formed will chemically dismute. The addition of SOD would have little effect on the rate of production of H2O2. SOD would convert the chemical dismutation into a faster enzyme-catalyzed dismutation and thereby lower the steady-state level of O2•−. However, it will not have a detectable effect on the production of H2O2 because there is not much superoxide entering into the reverse reaction to draw from.
B. The CoQ system, thermodynamics
The CoQ-system of the mitochondrial electron transport chain has a different equilibrium constant than the X/XO system for forming O2•−. Superoxide is formed by mitochondria [36,37,38]; a major source is thought to be CoQ•−, Rxn 1 (Table 1, Figure 2) [39,40]:
Figure 2. CoQ•− of the ETC can make O2•−, a source of H2O2.

CoQ•− can be formed during electron/proton transfer during the protonmotive Q-cycle, from comproportionation of CoQ and CoQH2, and also by other one-electron oxidation-reduction processes. K1b is the equilibrium constant highlighted in the model. (kc and kd are the rate constants for comproportionation and disproportionation, respectively).
Literature values for this one-electron reduction potential vary considerably due to the difficulty of determining a true reduction potential while in the membrane. Values range from −40 mV to −230 mV [35,41, 42, 43], depending on solvent, environment [44], and whether CoQ is bound. Thus, the one-electron reduction potential for CoQ is estimated as:
Using this range of one-electron reduction potentials, the estimated equilibrium constant for Rxn 1b is K1b ≈10−2 – 10+1, which is much smaller than K1a ≈10+4 for the production of superoxide by the flavin radical in XO. Because of the relatively rapid reverse reaction for Rxn 1b, we propose that the current paradigm, that SOD has no influence on the rate of production, will not apply for the CoQ system. Modulating the level of SOD in the CoQ-system would result in a change in the net rate for the reaction forming O2•−; following Le Chatelier’s principle, an increase in MnSOD would change the rate of production of O2•− and H2O2.
To examine how the value of the equilibrium constant, K1, alters the rate of formation of O2•− and subsequent production of H2O2 and how SOD influences the system, we developed a kinetic model to simulate the production of superoxide and hydrogen peroxide by the mitochondrial CoQ system and the X/XO-system.
Materials and methods
Kinetic Model of the Superoxide-Peroxide System
Antioxidant enzymes are involved in a series of reactions, converting superoxide radical to hydrogen peroxide, and finally to water. Superoxide dismutases convert superoxide radical to hydrogen peroxide. Catalase and glutathione peroxidase on the other hand convert hydrogen peroxide to water. The reactions involved can be separated into three phases:
-
Superoxide Generation
-
Superoxide Dismutation
-
Peroxide removal
When considering the complete process, from the generation of superoxide radicals to the final conversion of hydrogen peroxide to water inside mitochondria, a detailed version can be written as in Table 1. From these reactions a rate expression can be written for time-dependent concentration changes for each species, Table 2. The subsequent sixteen non-linear ordinary differential equations (ODE) are shown in Equations (15) – (30). Evaluating the reaction rate constants, these equations represent a stiff system. Therefore, the modified Rosenbrock numerical method was selected to solve the transient expressions [46].
Table 2.
The Differential Equations of the Model.
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
|
H2O2 measurement from cells
To determine if cells with varying levels of MnSOD produce H2O2 at different rates, we measured the accumulation of H2O2 in their growth medium. The steady-state level of H2O2 in cells has been estimated to be in the range of 10−10 – 10−7 M [47, 48, 49, 50]; the levels arenormally on the order of 10−9 – 10−8 M (1–10 nM), with higher levels associated with pathology [49]. However, intracellular determination of levels of H2O2 is problematic. We relied on the diffusion of H2O2 from cells into culture medium. The diffusion coefficient of H2O2 is nearly the same as H2O, but more importantly, their permeability coefficients to cellular membranes are also nearly the same [47, 51]. The time constant for the exchange of intracellular water for glioma cells in culture is on the order of 50 ms [52]. While the equivalent time constant in the fully functioning brain in vivo is an order of magnitude larger [53], this time constant is still < 1 s. Thus, intracellular H2O2 will rapidly appear in the extra-cellular medium. The rate of appearance will be proportional to the concentration gradient between inside and outside the cell. The levels of H2O2 were measured by the change in fluorescence of Amplex red, 10-acetyl-3,7-dihydroxyphenoxazine (ADHP) [54]], using the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Molecular Probes, Eugene, OR). Amplex red is considered to be a sensitive probe for H2O2, but can have interferences that can overwhelm the H2O2 signal of interest [55]. In the presence of H2O2 and horseradish peroxidase (HRP), ADHP generates a fluorescent oxidation product, resorufin, with an absorption maxima of approximately 563 nm and emission maxima around 587 nm [56]. In the assay exponentially growing cells had their growth medium replaced with a phosphate buffer and the appearance of H2O2 in the buffer was followed with time. The rates of appearance of H2O2, reflecting the intracellular [H2O2]ss, are compared.
Results and discussion
MnSOD overexpression in cells results in an increase in H2O2
There are various cellular sources that can produce H2O2. As a reliable measure of cellular production of H2O2 we have determined the rate of accumulation of extracellular H2O2 from several cell types. To determine if MnSOD influences the formation of H2O2 we used cells that had been transfected with MnSOD cDNA. This transfection provided clones with varying levels of MnSOD. These clones have been well characterized [57, 58, 59] and their antioxidant enzyme profile was verified for these experiments. The relative MnSOD activity for the different clones ranged from the same as wild type to a 19-fold increase in MnSOD activity. CuZnSOD activity in the transfected clones was comparable to parental cells; likewise peroxide-removing ability (catalase and glutathione peroxidase) was similar. We measured the appearance of H2O2 in media from dividing, unstressed cells. The rate of appearance of H2O2 in the media increased as the activity of MnSOD increased, Figure 3. Different MnSOD levels influenced the formation of H2O2 to different degrees. A 3-fold increase in MnSOD activity resulted in a 2-fold increase in the rate of accumulation of H2O2 in the medium, while a 19-fold increase in MnSOD, resulted only in a 3.5-fold increase in the rate of accumulation of H2O2. Thus, in cells with low levels of MnSOD, modest increases in MnSOD will have a greater relative impact on the rate of formation of H2O2 than very large increases in MnSOD.
Figure 3. The rate of accumulation of H2O2 in the media increases with the cellular MnSOD activity.

(♦) The H2O2 released from MCF-7 cells and several clones overexpressing MnSOD (1.1-, 3-, 6-, and 19-fold) in exponential growth was determined at several time points over a period of two hours. The concentrations of H2O2 in the medium were measured using Amplex red. The ordinate represents the change in the [H2O2] in the media versus time compared to wildtype MCF-7 cells (1 on the left ordinate scale represents 2.8 zmol cell−1 s−1). Results are from three independent experiments ± standard deviation.
( ) The mitochondrial steady-state level of H2O2 (nM) from the kinetic model achieved with different levels of MnSOD (0.7–10 μM) as presented in Figure 4D.
Similar measurements were done with wild type prostate cancer cells (PC-3) and an MnSOD-overexpressing clone (8.3-fold increase in MnSOD activity). The rate of appearance of extracellular H2O2 was 1.9-fold higher in the transfected clone compared to wild type PC-3 cells [59]. In a second study with PC-3 cells, we found that a clone with an 8.2-fold increase in MnSOD activity produced a 1.5-fold increase in the rate of appearance of H2O2 in the media compared to wildtype PC-3 cells [58]. Thus, an increase in MnSOD activity increases the rate of appearance of H2O2 in the extracellular medium, consistent with an increase in the rate of production of intracellular H2O2.
Setting up the components of the kinetic model for the formation of H2O2
To understand how increasing the level of SOD increases the rate of production of H2O2 in cells (Figure 3), we set up a kinetic model. Because the experimental results above used cells with varying levels of the mitochondrial form of SOD, MnSOD, we chose as a focus the production of superoxide by the mitochondrial electron transport chain (ETC) and compared this to the formation of O2•− from the X/XO system.
For the initial conditions in the model we started with all the MnSOD in its resting state of MnIIISOD with no H2O2 present, Table 3. To investigate how the rate of formation of H2O2 changes with [MnSOD], we varied the apparent equilibrium constant for the specific reaction that forms superoxide, Rxn 1, by changing the value of the reverse rate constant for Rxn 1, k−1, and thereby changing the apparent value of the equilibrium constant for this reaction, K1 = 0.001 – 1000. For each value of K1 we varied the level of MnSOD from 0.7 μM – 10 μM. We kept the capacity for the removal of peroxide constant. Once the system was allowed to evolve, it took on the order of 10 – 100 s for H2O2 to reach observable levels, Figure 4. After a short time the level of H2O2 reaches a steady-state. The delay in the appearance of H2O2 is because H2O2 is only formed via the reaction of O2•− with MnIISOD, Rxns 4 and 5. Because the initial conditions had MnSOD in the oxidation state of MnIII, it takes some time to convert a significant amount of MnIIISOD to MnIISOD, Rxns 2 and 3. As expected, this lag time increases with increasing initial concentration of MnIIISOD.
Table 3.
The species in the model and their initial concentrations
| Species | [Species]i/M a | Comment/Reference |
|---|---|---|
| CoQ + CoQH2 | 5 × 10−4 | This concentration is based on data on rat liver mitochondria (120,000 total molecules/mitochondrion [78]; volume = 0.42 μm3 [106]), but assumes a homogeneous distribution. If considering only the lipid phase of mitochondria, then [CoQ10] is estimated to be 10 times this concentration (5 mM) in this phase [107] This is comparable to information on heart mitochondria [108, 109, 110] |
| CoQ | 4.5 × 10−4 | 90% of coenzyme Q, ubiquinone, is estimated to be in the oxidized form [78], however this can be expected to vary [111]. |
| CoQH2 | 0.5 × 10−4 | 10% of coenzyme Q is estimated to be in the reduced form [78,112]. |
| CoQ•− | 1 × 10−7 | This estimate assumes that 1 part in 5,000 of CoQ + CoQH2 exists as the radical [77,113]. |
| O2 | 25 × 10−6 | |
| O2•− | 0 | |
| Mn(III)SOD | Varied in the model | The resting state of MnSOD |
| Mn(III)SOD:O2•− | 0 | An intermediate in the reduction of Mn(III)SOD |
| Mn(II)SOD | 0 | The reduced form of MnSOD. The rate production of H2O2 by MnSOD is maximized when [Mn(II)SOD]:Mn(III)SOD is 1:1 |
| Mn(II)SOD:O2•− | 0 | An intermediate in the oxidation of Mn(II)SOD |
| H2O2 | 0 | |
| DEP | 0 | Is a dead end product, i.e. inactivation of MnSOD |
| GPxr | 1 × 10−6 | The reduced form of GPx that reacts with hydroperoxides |
| GPxo | 0 | The oxidized form of GPx that enters into reactions with GSH to recycle back to GPxr |
| H2O2 | 0 | |
| GSH | 1 × 10−3 | Glutathione |
| GSGPx | 0 | An intermediate in the reaction of GSH with GPxo |
| GSSG | 0 | Glutathione disulfide |
This is the initial concentration at time zero.
Figure 4. The steady-state concentration of H2O2 in cells can be a function of the level of MnSOD.
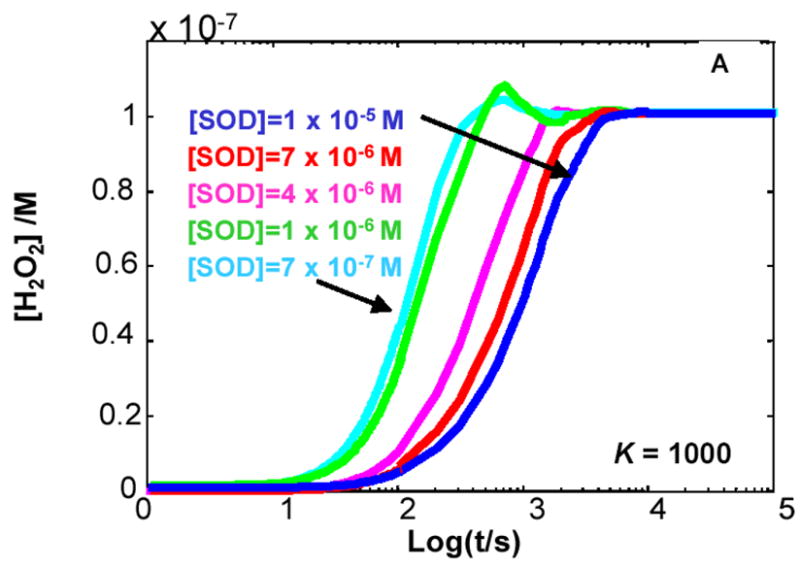
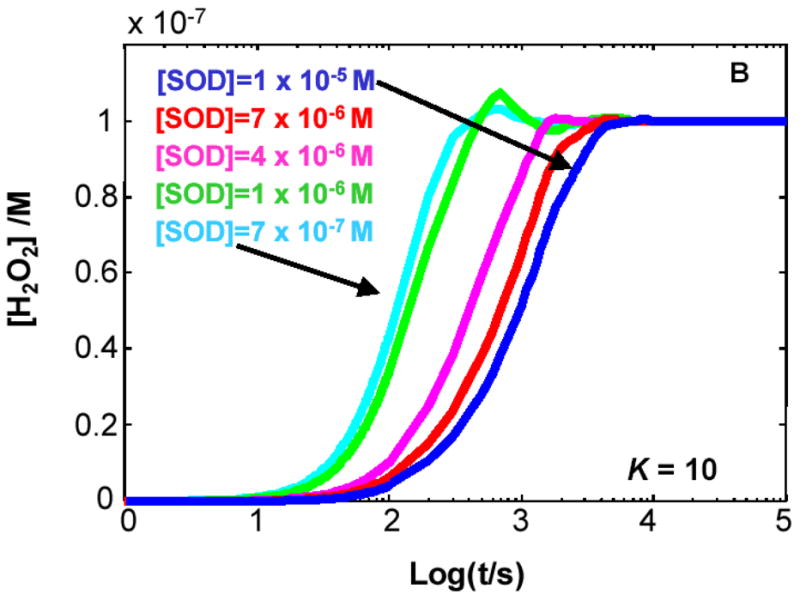
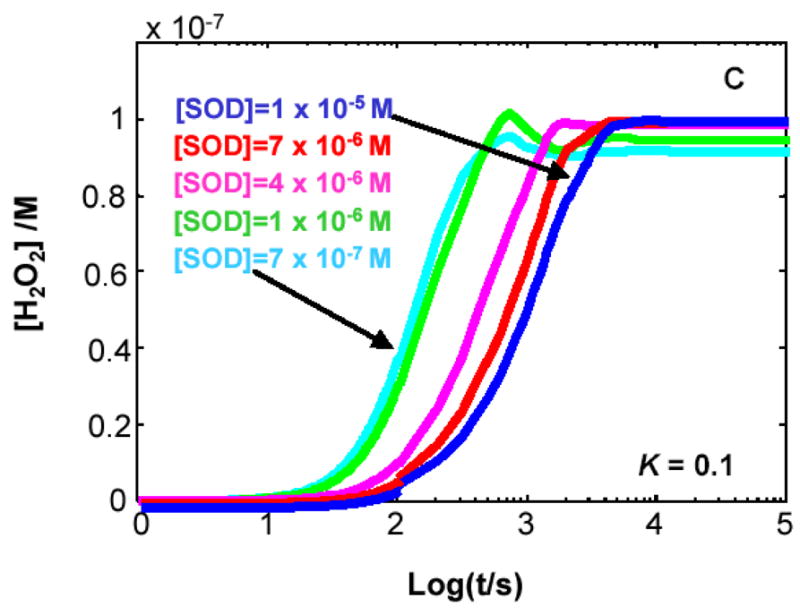
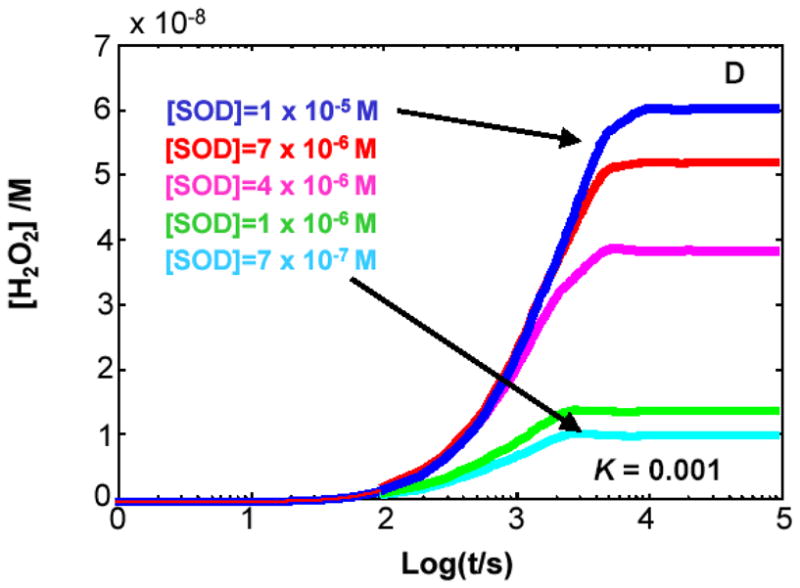
The kinetic model clearly demonstrates that when K < 1 for Rxn 1 the level of SOD will determine the flux of H2O2 as well as its steady-state level. (A) K= 1000; (B) K= 10; (C) K= 0.1; (D) K= 0.001. The different values for the equilibrium constant were achieved by varying the rate constant for the reverse reaction of Rxn 1, keeping the value of the forward rate constant at 8 × 103 M−1 s−1. In the kinetic model, the capacity to remove H2O2 was kept constant.
For our kinetic model we have chosen concentrations of participants that are our best interpretation of the literature, Table 3. However, even if future research suggests concentrations that differ by an order of magnitude or more, the principal findings of this model still hold.
If K1 > 1, e.g. XO, then SOD has no influence on the production of H2O2
When the rate constant for the forward reaction of Rxn 1 is greater than the rate constant for the reverse reaction, i.e. kf > kr or K1 > 1, then [H2O2]ss does not change with varying concentrations of MnSOD, Figures 4A and 4B. When K1 > 1, e.g. K1 = 1000 (Figure 4A), the same steady-state level of H2O2 is achieved for all [MnSOD] examined. This is because little of the O2•− formed enters into the reverse reaction, so essentially all superoxide dismutes to give H2O2, with or without SOD. This scenario applies for the XO-xanthine system.
The kinetics for superoxide production by X/XO is initiated by the formation of a XO-xanthine complex, Figure 1. The actual production of superoxide is accomplished by reaction of the flavin radical of FAD, FADH•, with O2. In the blood plasma of healthy humans typical levels of hypoxanthine are approximately 2 μM with xanthine levels on the order of only 1 μM or less [62]; these values can vary considerably. Using the rate constants of Figure 1, a pseudo first-order rate constant for X/XO complex formation will be about 10 s−1. These electrons are quickly transferred to FAD. Oxygen levels in blood of course vary, but if we assume a typical concentration to be ≈150 μM, then the pseudo first-order rate constant for the oxidation of FADH• by oxygen to form O2•− will be 10 s−1. This estimate shows that the rate of enzyme reduction and subsequent oxidation to form O2•− are about the same. Thus, SOD could have an effect on the rate of production of H2O2 because the oxidation of FADH• is not necessarily rate-limiting. However, the thermodynamics of the FADH• + O2 reaction to form O2•− has K1a = 104; see Introduction. The model predicts that SOD would have little or no effect on the rate of production of superoxide and hydrogen peroxide because of this high value for K1.
When K1 > 1, to change the rate of production of O2•−, and subsequently H2O2, the concentration of the limiting reagents must be modulated. The limit for the rate of formation of O2•− in this setting is the rate of the forward reaction for Rxn 1. Thus, increasing the concentration of the radical source for O2•− (FADH•) or O2 would increase the flux of O2•− and H2O2 as either FADH• or O2, or both, are the limiting reagents. The level of MnSOD will affect [O2•−]ss. In this kinetic model, if [MnSOD] is increased by a factor of two, the steady state level of O2•− decreases by a factor two, i.e. [O2•−]ss will be a direct function of 1/[MnSOD], assuming there are no other significant sinks for O2•−. However, the level of MnSOD would not affect the flux of O2•− and H2O2.
If K1 < 1, e.g. CoQ, then SOD can influence the production of H2O2
When kf < kr, K1 < 1, the steady-state level of H2O2 achieved is a function of the level of MnSOD, the higher [MnSOD] the greater [H2O2]ss, Figures 4C and 4D. The steady-state level of H2O2 will change if either its rate of removal or rate of formation changes. In the model we kept the capacity to remove H2O2 constant; thus, the rate of production must change. The rate of production of H2O2 changes in the presence of SOD because the very fast reverse reaction for Rxn 1 gives little chance for chemical dismutation of O2•− to H2O2 and O2. The very fast reaction of SOD consumes the O2•− before it can enter the reverse reaction of Rxn 1. From an examination of the slopes of the lines at ≈1000 s (log t = 3) in Figure 4D (K1 = 0.001), we can see that indeed the rate of formation of H2O2 increases with increasing MnSOD. Thus, the kinetic model predicts that a change in the level of MnSOD will change the rate of production of O2•− and subsequently the flux of H2O2. This scenario applies to the CoQ-system.
The semiquinone radical (CoQ•−) of coenzyme Q (ubiquinone) of the ETC is thought to be the primary source of superoxide in most non-phagocytic cells, Figure 2 [63, 64, 65, 66, 67, 68, 69, 70, 71]. A small percent (on the order of 1% or less) of the overall electron-flow through the ETC in normal, healthy cells is thought to result in the production of superoxide [70, 72, 73]. Values for some of the parameters in the kinetic model are sparse. To ensure that the conclusions drawn from the model are reliable, we tested wide ranges of concentrations to determine when significant changes in the outcome occurred. In the kinetic model we assumed a constant level of CoQ•− and dioxygen [74, 75]. We reasoned that the level of CoQ•− would be in a relative steady-state in an unchanging environment because of its formation as an intermediate in the protonmotive Q cycle [71, 76]; in addition CoQ and CoQH2 readily comproportionate to form 2CoQ•− (k ≈250 M−1 s−1; K ≈6 × 10−5 [77]). Thus, any CoQ•− lost to its reaction with O2 to form O2•−, Rxn 1, would be rapidly replaced.
The thermodynamics of the CoQ•− + O2 reaction to form superoxide is considerably different than for the X/XO system. The range of ΔE for Rxn 1 with CoQ•− as the electron source for O2•− yields an equilibrium constant in the range of 10−2 to 101. This much smaller value for K1 allows MnSOD to change the rate of production of superoxide and H2O2, Figure 3.
A pool of electrons is essential
Another difference between the CoQ system and the XO system is the size of the electron pool. Only a very small percentage (≈1% or less) [70,72,73] of the electrons passing through the electron transport chain actually result in superoxide formation. This loss of electrons from the ETC due to the increase in the rate of superoxide formation with increasing levels of SOD is only a small perturbation on the overall electron-flow through the mitochondria. The ETC has at any time more than 40,000 redox active electrons, of which 60% reside in Complexes I, II and CoQH2, Table 4. This large pool of electrons in the ETC coupled with the mobility of CoQ in the mitochondrial inner membrane [78] will instantly re-establish the steady-state level of CoQ•−, because CoQ•− is formed as an intermediate in the protonmotive Q-cycle [71,76] as well as through comproportionation of CoQ and CoQH2. The system is then poised to produce yet another molecule of superoxide. This large pool of electrons is a ready source for the production of O2•− and H2O2 that can be modulated by SOD, Figure 5.
Table 4.
The pool of electrons in the ETC is large.
| Redox component | Molecules per inner membrane[78]a | % reduced/oxidized of redox partners at steady state[78,114] | Redox e− [115] | Number of redox electrons in pool per mitochondrion |
|---|---|---|---|---|
| Complex I | 2,000 | I(5red) → CoQ(90ox)b | 2c; 8d | 800 |
| Complex II | 3,800 | II(5red) →CoQ(90ox) | 2; 4 | 800 |
| CoQ | 120,000 | CoQ(10red) →III(84ox) | 2; 2 | 24,000 |
| Complex III | 5,700 | III(16red) →c(89ox) | 1; 3e | 2,700 |
| Cytochrome c | 17,000 | c(11red) →IV(80ox) | 1; 1 | 1,900 |
| Complex IV | 13,000 | IV(20red) | 4; 4 | 10,600 |
|
Total = 40,800
Total in Complexes I, II and CoQ = 25,000 | ||||
These estimates from [78] have been rounded to two-significant digits.
These estimates are for uncoupled mitochondria from rat liver. The percent of reduced species will vary considerably, being greater for active mitochondria.
number of electrons transferred in turnover in the ETC.
maximum number of redox electrons that could in principal be held in each complex or redox species.
This does not include CoQ.
Figure 5. The ability of SOD to modulate the flux of H2O2 and [H2O2]ss depends on the availability of a sufficient pool of electrons.

The ETC has a large pool of electrons. The vast majority flow through complex IV to O2 forming H2O at the end of the ETC; a very small fraction form O2•−. The CoQ-system has appropriate thermodynamics and access to a large pool of electrons so that MnSOD can modulate the flux of O2•− and subsequently H2O2. XO has unfavorable thermodynamics, which do not allow SOD to modulate the flux of O2•−; in addition there is a very limited pool of electrons to draw from. Although CoQ•−is thought to be the principal source for formation of superoxide in mitochondria, other sites may also be sources of O2•−.
In contrast to CoQ•−, the X/XO system has a very small pool of electrons. Each enzyme molecule can hold six electrons per subunit. X/XO makes both H2O2 and O2. The ratio of products depends on pH, substrate, and substrate concentration [79]. Superoxide is formed from FADH•; this moiety exists only when XO is relatively empty of electrons [80, 81]. The nearer XO is to full capacity, the greater the proportion of H2O2 formed. SOD will have no effect on the direct formation of H2O2 by XO; it will also have no effect on the production of H2O2 via superoxide.
Even if the thermodynamics of a reaction are appropriate and there are no limiting reagents on the reactant side of Rxn 1, the rate of the process must be on the time-scale of the biochemistry of the cell for mass action to be of significance. Or stated another way, the magnitude of the activation energies must be appropriate to allow reactions to proceed on the time scale of the biochemistry of the cell. We evaluated the literature and chose to use a forward rate constant for the CoQ•− system of ≈104 M−1 s−1 for Rxn 1b, Table 1. If this rate constant were lower, e.g. ≈10−2 M−1 s−1, and the equilibrium constant was ≈0.01, as with Rxn 1b, then SOD would still “pull’ the reaction as per Le Chatelier’s principle. However, in a biological setting and biological time scale there would be no detectable change in the flux of H2O2. The rate of formation of H2O2 would be a million times slower and would not be of consequence; The curves of Figure 4 would be shifted from a sub-μM concentration-range to sub-pM, i.e. less than 1 molecule of H2O2 from this source in a typical cell. Thus, rate constants must be appropriate for the time scale of the process; the rate constants for Rxn 1b are appropriate for the biological time scale.
Biological implications: a new view on the function of MnSOD When K < 1, SOD can change [H2O2]ss
The experimental results shown in Figure 3 are consistent with our kinetic model. The MCF-7 wildtype cells have very low MnSOD activity. Increasing this activity modestly results in a large increase in the rate of appearance of H2O2 in the media, but additional increases result in a smaller relative increase in H2O2.
Our kinetic model demonstrates that maximal [H2O2]ss is achieved when K1 > 1. When K1 > 1, the reverse reaction is relatively slow and the vast majority of the superoxide formed dismutes, Figures 4A and 4B, resulting in maximal flux of O2•−/H2O2. Most interesting is that when K1 < 1, the [H2O2]ss achieved is less than the maximum possible [H2O2]ss as most of the superoxide formed is lost in the reverse reaction, Figure 4D. Of special interest in Figure 4D is that even when [MnSOD] = 10 μM, the steady-state level of H2O2 is only 60% of that when K1 > 1, e.g. Figures 4A and 4B. When K1 = 0.001 (Figure 4D), it would take [MnSOD] > 100 μM to approach the maximum achievable [H2O2]ss. These findings on H2O2 are of considerable biological significance as they suggest that for cells with relatively low MnSOD, an increase in enzyme activity will produce a relatively large increase in the rate of formation of H2O2 and [H2O2]ss. But for cells that have robust levels of MnSOD, more MnSOD will have only a minor effect on the rate of formation of H2O2.
These findings establish a basis for a new paradigm for the biological function of SOD. The traditional view is that by controlling the steady-state level of superoxide, SOD protects cells and tissues from potential damage. Thus, SOD is a primary antioxidant enzyme. Here we have shown how SOD can alter the flux of H2O2. Both, superoxide and H2O2 are signalling molecules. Superoxide is known to react with certain Fe-centers in enzymes while hydrogen peroxide is in general changing the sulfhydryl tone of the cell. SOD serves as a rheostat for each of these processes; an increase in MnSOD would lower [O2•−]ss and increase [H2O2]ss; thus, MnSOD could serve as the switching mechanism between one-electron signalling and two-electron signalling processes, Figure 6. Being able to modulate the intracellular H2O2 flux, positions SOD as a major player in establishing the redox environment of a cell. It is thought that the redox environment of the cell is tightly connected to the biological status of the cell. Changes in the redox environment from reducing to more oxidizing can move cells through biological states such as proliferation, differentiation, and cell death [92]. H2O2 has been implicated in bringing about these biological changes. Thus, MnSOD could be a regulator of the cellular redox environment and with this control in part the biological state of the cell. This could explain the role of O2•− and MnSOD in the basic biology of proliferating cells, such as cancer cells, as well as a role for SOD in the treatment of cancer. MnSOD may have an even larger influence in pathologies that affect mitochondria.
Figure 6. MnSOD serves as a rheostat for both superoxide and hydrogen peroxide signalling.

By controlling [O2•−]ss MnSOD influences certain reactive Fe-centers in enzymes and proteins that are sensitive to oxidation/reduction by O2•−. By changing the flux of H2O2, MnSOD will also modulate signalling processes that go through the thiol-system. Thus, MnSOD serves as a rheostat for both one-electron and two-electron signalling pathways. These signalling processes set the redox environment of cells, which in turn establishes the biological state of cells and tissues.
MnSOD has been found to be low in many cancer cells [58,82,83,84,85,86], fetal cells [87,88,89], as well as stem cells [90]. Forced overexpression of MnSOD slows the growth of cancer cells both in vitro and in vivo [58,85,86,91]. These and many other studies have clearly demonstrated that MnSOD suppresses cell growth. This growth suppression could be in part a result of increasing the flux of H2O2 and thereby pushing the redox status of the cell to a more oxidized state. This more oxidized redox environment will no longer support cell proliferation, but rather is associated with differentiation [92,93,94].
Summary
The results presented here indicate that:
the flux of H2O2 increases in cells as MnSOD increases;
SOD can modulate the flux of O2•− and H2O2 only when the equilibrium constant for the reaction forming superoxide is less than or equal to ≈1, e.g. CoQ10•− + O2;
in addition, for MnSOD to be able to influence the flux of H2O2, a sufficient pool of electrons is needed; the ETC has such a pool;
the rate constant for the reaction of CoQ•−with O2 to form O2•− is of a magnitude to yield a rate of formation of H2O2 appropriate for the time scale of the biochemistry of the cell;
an increase in MnSOD will lower [O2•−]ss in proportion to 1/[MnSOD];
an increase in MnSOD will increase the relative flux of H2O2 and [H2O2]ss in cells with low MnSOD more, compared to those with robust levels of MnSOD.
The results presented here suggest an entirely new function for MnSOD. MnSOD not only controls the levels of O2•−, but it can also be viewed as an enzyme that plays a role in establishing the flux of H2O2 in cells. Because H2O2 is a key to determining the redox environment of cells and tissues, MnSOD should now be viewed not only as an antioxidant enzyme but also as a key enzyme involved in the establishment of the cellular redox environment and thereby the biological status of cells and tissues.
Acknowledgments
This work was supported in part by NIH grants CA66081 and CA81090.
Footnotes
Le Chatelier’s Principle states that if the conditions of a system, initially at equilibrium, are changed, the equilibrium will shift in such a direction as to tend to restore the original conditions.
References
- 1.Fridovich I. Superoxide radical and superoxide dismutases. Annual Review of Biochemistry. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 2.Fridovich I. Superoxide dismutases. An adaptation to a paramagnetic gas. J Biol Chem. 1989;264:7761–7764. [PubMed] [Google Scholar]
- 3.Klug D, Rabani J, Fridovich I. A direct demonstration of the catalytic action of superoxide dismutase through the use of pulse radiolysis. J Biol Chem. 1972;247:4839–4842. [PubMed] [Google Scholar]
- 4.Klug-Roth D, Fridovich I, Rabani J. Pulse radiolytic investigations of superoxide catalyzed disproportionation. Mechanism for bovine superoxide dismutase. J Am Chem Soc. 1973;95:2786–2790. doi: 10.1021/ja00790a007. [DOI] [PubMed] [Google Scholar]
- 5.Bielski BHJ, Cabelli DE, Arudi RL, Ross; AB. Reactivity of hydroperoxyl/superoxide radicals in aqueous solution. J Phys Chem Ref Data. 1985;14:1041–1100. [Google Scholar]
- 6.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 7.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scott MD, Meshnick SR, Eaton JW. Superoxide dismutase-rich bacteria. Paradoxical increase in oxidant toxicity. J Biol Chem. 1987;262:3640–3645. [PubMed] [Google Scholar]
- 9.Scott MD, Meshnick SR, Eaton JW. Superoxide dismutase amplifies organismal sensitivity to ionizing radiation. J Biol Chem. 1989;264:2498–2501. [PubMed] [Google Scholar]
- 10.Elroy-Stein O, Bernstein Y, Groner Y. Overproduction of human Cu/An-superoxide dismutase in transfected cells: extenuation of paraquat-mediated cytotoxicity and enhancement of lipid peroxidation. EMBO J. 1986;5:615–622. doi: 10.1002/j.1460-2075.1986.tb04255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amstad P, Peskin A, Shah G, Mirault M-E, Moret R, Zbinden I, Cerutti P. The balance between Cu,Zn-superoxide dismutase and catalase affects the sensitivity of mouse epidermal cells to oxidative stress. Biochem. 1991;30:9305–9313. doi: 10.1021/bi00102a024. [DOI] [PubMed] [Google Scholar]
- 12.Teixeira HD, Schumacher RI, Meneghini R. Lower intracellular hydrogen peroxide levels in cells overexpressing CuZn-superoxide dismutase. Proc Natl Acad Sci USA. 1998;95:7872–7875. doi: 10.1073/pnas.95.14.7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li S, Yan T, Yang JQ, Oberley TD, Oberley LW. The role of cellular glutathione peroxidase redox regulation in the suppression of tumor cell growth by manganese superoxide dismutase. Cancer Res. 2000;60:3927–3939. [PubMed] [Google Scholar]
- 14.Zhang HJ, Zhao W, Venkataraman S, Robbins ME, Buettner GR, Kregel KC, Oberley LW. Activation of matrix metalloproteinase-2 by overexpression of manganese superoxide dismutase in human breast cancer MCF-7 cells involves reactive oxygen species. J Biol Chem. 2002;277:20919–20922. doi: 10.1074/jbc.M109801200. [DOI] [PubMed] [Google Scholar]
- 15.Kim KH, Rodriguez AM, Carrico PM, Melendez JA. Potential mechanisms for the inhibition of tumor cell growth by manganese superoxide dismutase. Antiox Redox Signal. 2001;3:361–373. doi: 10.1089/15230860152409013. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez AM, Carrico PM, Mazurkiewicz JE, Melendez JA. Mitochondrial or cytosolic catalase reverses the MnSOD-dependent inhibition of proliferation by enhancing respiratory chain activity, net ATP production, and decreasing the steady state levels of H2O2. Free Radic Biol Med. 2000;29:801–813. doi: 10.1016/s0891-5849(00)00362-2. [DOI] [PubMed] [Google Scholar]
- 17.Liochev SI, Fridovich I. The role of O2•− in the production of HO•: In vitro and in vivo. Free Radic Biol Med. 1994;16:29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 18.Gardner R, Salvador A, Moradas-Ferreira P. Why does SOD overexpression sometimes enhance, sometimes decrease, hydrogen peroxide production? A minimalist explanation. Free Radic Biol Med. 2002;32:1351–1357. doi: 10.1016/s0891-5849(02)00861-4. [DOI] [PubMed] [Google Scholar]
- 19.Kowald A, Lehrach H, Klipp E. Alternate pathways as mechanism for the negative effects associated with overexpression of superoxide dismutase. J Theoretical Biol. 2006;238:828–840. doi: 10.1016/j.jtbi.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 20.McCord JM, Fridovch I. Superoxide dismutase: an enzymatic function for eythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 21.Misra HP, Fridovich I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 1972;247:3170–3175. [PubMed] [Google Scholar]
- 22.Bors W, Michel C, Saran M, Lengfelder E. The involvement of oxygen radicals during the autoxidation of adrenalin. Biochim Biophys Acta. 1978;540:162–172. doi: 10.1016/0304-4165(78)90445-2. [DOI] [PubMed] [Google Scholar]
- 23.Heikkila RE, Cohen G. 6-Hydroxydopamine: evidence for superoxide radical as an oxidative intermediate. Science. 1973;181:456–457. doi: 10.1126/science.181.4098.456. [DOI] [PubMed] [Google Scholar]
- 24.Lloyd RV. Mechanism of the manganese-catalyzed autoxidation of dopamine. Chem Res Toxicol. 1995;8:111–116. doi: 10.1021/tx00043a015. [DOI] [PubMed] [Google Scholar]
- 25.Buettner GR. In the absence of catalytic metals, ascorbate does not autoxidize at pH 7: Ascorbate as a test for catalytic metals. J Biochem Biophys Meth. 1988;16:27–40. doi: 10.1016/0165-022x(88)90100-5. [DOI] [PubMed] [Google Scholar]
- 26.Massey V, Brumby PE, Komai H, Palmer G. Studies on milk xanthine oxidase. Some spectral and kinetic properties. J Biol Chem. 1969;244:1682–1691. [PubMed] [Google Scholar]
- 27.Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med. 2002;33:774–97. doi: 10.1016/s0891-5849(02)00956-5. [DOI] [PubMed] [Google Scholar]
- 28.Meneshian A, Bulkley GB. The physiology of endothelial xanthine oxidase: from urate catabolism to reperfusion injury to inflammatory signal transduction. Microcirculation. 2002;9:161–75. doi: 10.1038/sj.mn.7800136. [DOI] [PubMed] [Google Scholar]
- 29.McCord JM, Fridovich I. The reduction of cytochrome c by milk xanthine oxidase. J Biol Chem. 1968;243:5753–5760. [PubMed] [Google Scholar]
- 30.Olson JS, Ballou DP, Palmer G, Massey V. The reaction of xanthine oxidase with molecular oxygen. J Biol Chem. 1974;249:4350–4362. [PubMed] [Google Scholar]
- 31.Porras AG, Olson JS, Palmer G. The reaction of reduced xanthine oxidase with oxygen. Kinetics of peroxide and superoxide formation. J Biol Chem. 1981;256:9096–9103. [PubMed] [Google Scholar]
- 32.Olson JS, David P, Ballou DP, Graham Palmer G, Vincent Massey V. The mechanism of action of xanthine oxidase. J Biol Chem. 1974;249:4363–4382. [PubMed] [Google Scholar]
- 33.Porras AG, Palmer G. The room temperature potentiometry of xanthine oxidase. pH-dependent redox behavior of the flavin, molybdenum, and iron-sulfur centers. J Biol Chem. 1982;257:11617–11626. [PubMed] [Google Scholar]
- 34.Barber MJ, Siegel LM. Magnetic Interactions Within Xanthine Oxidase. In: Massey V, Williams CH, editors. Flavins and Flavoproteins. North Holland, New York: Elsevier; 1982. pp. 796–804. [Google Scholar]
- 35.Wardman P. Reduction potentials of one-electron couples involving free radicals in aqueous solution. J Chem Ref Data. 1989;18:1637–1755. [Google Scholar]
- 36.Forman HJ, Kennedy JA. Role of superoxide radical in mitochondrial dehydrogenase reactions. Biochem Biophys Res Commun. 1974;60:1044–1050. doi: 10.1016/0006-291x(74)90418-5. [DOI] [PubMed] [Google Scholar]
- 37.Loschen G, Angelo A, Richter C, Flohé L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974;42:68–72. doi: 10.1016/0014-5793(74)80281-4. [DOI] [PubMed] [Google Scholar]
- 38.Forman HJ, Kennedy J. Superoxide production and electron transport in mitochondrial oxidation of dihydroorotic acid. J Biol Chem. 1975;250:4322–4326. [PubMed] [Google Scholar]
- 39.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 40.Giulivi C, Boveris A, Cadenas E. The steady-state concentrations of oxygen radicals in mitochondria. In: Gilbert DL, Colton CA, editors. Reactive Oxygen Species in Biological Systems: An Interdisciplinary Approach Chapter 3. New York: Kluwer Academic/Plenum Publishers; 1999. pp. 77–102. [Google Scholar]
- 41.DeVries S, Berden JA, Slater EC. Properties of a semiquinone anion located in the QH2:cytochrome c oxidoreductase segment of the mitochondrial respiratory chain. FEBS Lett. 1980;122:143–148. doi: 10.1016/0014-5793(80)80422-4. [DOI] [PubMed] [Google Scholar]
- 42.Sallow AJ. Physical chemistry of semiquinones. In: Trumpower BL, editor. Function of Quinones in Energy Conserving Systems. New York: Academic Press; 1982. pp. 59–72. [Google Scholar]
- 43.Miki T, Yu L, Yu CA. Characterization of ubisemiquinone radicals in succinate-ubiquinone reductase. Arch Biochem Biophys. 1992;293:61–66. doi: 10.1016/0003-9861(92)90365-4. [DOI] [PubMed] [Google Scholar]
- 44.Ohnishi T, Trumpower BL. Differential effects of antimycin on ubisemiquinone bound in different environments in isolated succinate. cytochrome c reductase complex. J Biol Chem. 1980;255:3278–3284. [PubMed] [Google Scholar]
- 45.Sallow AJ. Physical chemistry of semiquinones. In: Trumpower BL, editor. Function of Quinones in Energy Conserving Systems. New York: Academic Press; 1982. pp. 59–72. [Google Scholar]
- 46.Shampine LF, Reichelt MW. The MATLAB ODE suite. SIAM J Sci Computing. 1997;18:1–22. [Google Scholar]
- 47.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 48.Giulivi C, Hochstein P, Davies KJ. Hydrogen peroxide production by red blood cells. Free Radic Biol Med. 1994;16:123–129. doi: 10.1016/0891-5849(94)90249-6. [DOI] [PubMed] [Google Scholar]
- 49.Boveris A, Cadenas E. Cellular sources and steady-state levels of reactive oxygen species. Oxygen, Gene Expression, and Cellular Function. In: Clerch LB, Massaro DJ, editors. Lung Biology in Health and Disease. Vol. 205. New York: Marcell Dekker, Inc; 1997. pp. 1–26. [Google Scholar]
- 50.Johnson RM, Goyette G, Jr, Ravindranath Y, Ho YS. Hemoglobin autoxidation and regulation of endogenous H2O2 levels in erythrocytes. Free Radic Biol Med. 2005;39:1407–1417. doi: 10.1016/j.freeradbiomed.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Dick DA. The permeability coefficient of water in the cell membrane and the diffusion coefficient in the cell interior. J Theoretical Biol. 1964;7:504–531. doi: 10.1016/0022-5193(64)90019-0. [DOI] [PubMed] [Google Scholar]
- 52.Pfeuffer J, Flögel U, Dreher W, Leibfritz D. Restricted diffusion and exchange of intracellular water: theoretical modelling and diffusion time dependence of 1H NMR measurements on perfused glial cells. MNR Biomed. 1998;11:19–31. doi: 10.1002/(sici)1099-1492(199802)11:1<19::aid-nbm499>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 53.Quirk JD, Bretthorst GL, Duong TQ, Snyder AZ, Springer CS, Jr, Ackerman JJ, Neil JJ. Equilibrium water exchange between the intra- and extracellular spaces of mammalian brain. Magnetic Resonance Med. 2003;50:493–499. doi: 10.1002/mrm.10565. [DOI] [PubMed] [Google Scholar]
- 54.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 55.Votyakova TV, Reynolds IJ. Detection of hydrogen peroxide with Amplex Red: interference by NADH and reduced glutathione auto-oxidation. Arch Biochem Biophys. 2004;431:138–144. doi: 10.1016/j.abb.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 56.Gutheil WG, Stefanova ME, Nicholas RA. Fluorescent coupled enzyme assays for D-alanine: application to penicillin-binding protein and vancomycin activity assays. Anal Biochem. 2000;287:196–202. doi: 10.1006/abio.2000.4835. [DOI] [PubMed] [Google Scholar]
- 57.Zhang HJ, Yan T, Oberley TD, Oberley LW. Comparison of effects of two polymorphic variants of manganese superoxide dismutase on human breast MCF-7 cancer cell phenotype. Cancer Res. 1999;59:6276–6283. [PubMed] [Google Scholar]
- 58.Venkataraman S, Jiang X, Weydert CJ, Zhang Y, Zhang HJ, Goswami PC, Ritchie JM, Oberley LW, Buettner GR. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene. 2005;24:77–89. doi: 10.1038/sj.onc.1208145. [DOI] [PubMed] [Google Scholar]
- 59.Venkataraman S, Wagner BA, Jiang X, Wang HP, Schafer FQ, Ritchie JM, Burns CP, Oberley LW, Buettner GR. Overexpression of manganese superoxide dismutase promotes the survival of prostate cancer cells exposed to hyperthermia. Free Rad Res. 2004;38:1119–1132. doi: 10.1080/10715760400010470. [DOI] [PubMed] [Google Scholar]
- 62.Lartigue-Mattei C, Chabard JL, Bargnoux H, Petit J, Berger JA, Ristori JM, Bussiere JL, Catilina P, Catilina MJ. Plasma and blood assay of xanthine and hypoxanthine by gas chromatography-mass spectrometry: physiological variations in humans. J Chrom. 1990;529:93–101. doi: 10.1016/s0378-4347(00)83810-4. [DOI] [PubMed] [Google Scholar]
- 63.Boveris A, Cadenas E, Stoppani AO. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem J. 1976;156:435–444. doi: 10.1042/bj1560435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cadenas E, Boveris A, Ragan CI, Stoppani AO. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys. 1977;180:248–257. doi: 10.1016/0003-9861(77)90035-2. [DOI] [PubMed] [Google Scholar]
- 65.Boveris A. Mitochondrial production of superoxide radical and hydrogen peroxide. Adv Exp Med Biol. 1977;78:67–82. doi: 10.1007/978-1-4615-9035-4_5. [DOI] [PubMed] [Google Scholar]
- 66.Boveris A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Meth Enzymol. 1984;105:429–435. doi: 10.1016/s0076-6879(84)05060-6. [DOI] [PubMed] [Google Scholar]
- 67.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 68.Forman HJ, Kennedy J. Dihydroorotate-dependent superoxide production in rat brain and liver. A function of the primary dehydrogenase. Arch Biochem Biophys. 1976;173:219–224. doi: 10.1016/0003-9861(76)90252-6. [DOI] [PubMed] [Google Scholar]
- 69.James AM, Smith RA, Murphy MP. Antioxidant and prooxidant properties of mitochondrial Coenzyme Q. Arch Biochem Biophys. 2004;423:47–56. doi: 10.1016/j.abb.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 70.St-Pierre J, Buckingham JA, Roebuck SJ, Brand M. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 71.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robiolio M, Rumsey WL, Wilson DF. Oxygen diffusion and mitochondrial respiration in neuroblastoma cells. Am J Physiol. 1989;256:C1207–C1213. doi: 10.1152/ajpcell.1989.256.6.C1207. [DOI] [PubMed] [Google Scholar]
- 75.Jones DP. Intracellular diffusion gradients of O2 and ATP. Am J Physiol. 1986;250:C663–C675. doi: 10.1152/ajpcell.1986.250.5.C663. [DOI] [PubMed] [Google Scholar]
- 76.Trumpower BL. The protonmotive Q cycle. J Biol Chem. 1990;265:11409–11412. [PubMed] [Google Scholar]
- 77.Audi SH, Zhao H, Bongard RD, Hogg N, Kettenhofen NJ, Kalyanaraman B, Dawson CA, Merker MP. Pulmonary arterial endothelial cells affect the redox status of coenzyme Q0. Free Radic Biol Med. 2003;34:892–907. doi: 10.1016/s0891-5849(03)00025-x. [DOI] [PubMed] [Google Scholar]
- 78.Hackenbrock CR. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J Bioenerg Biomembranes. 1986;18:331–368. doi: 10.1007/BF00743010. [DOI] [PubMed] [Google Scholar]
- 79.Fridovich I. Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J Biol Chem. 1970;245:4053–4057. [PubMed] [Google Scholar]
- 80.Nishino T. The conversion of xanthine dehydrogenase to xanthine oxidase and the role of the enzyme in reperfusion injury. J Biochem. 1994;116:1–6. doi: 10.1093/oxfordjournals.jbchem.a124480. [DOI] [PubMed] [Google Scholar]
- 81.Nishino T, Nishino T, Schopfer LM, Massey V. The reactivity of chicken liver xanthine dehydrogenase with molecular oxygen. J Biol Chem. 1989;264:2518–2527. [PubMed] [Google Scholar]
- 82.Oberley LW, Buettner GR. The role of superoxide dismutase in cancer: A review. Cancer Res. 1979;39:1141–1149. [PubMed] [Google Scholar]
- 83.Yang J, Lam EW, Hammad HM, Oberley TD, Oberley LW. Antioxidant enzyme levels in oral squamous cell carcinoma and normal human oral epithelium. J Oral Pathol Med. 2002;31:71–77. doi: 10.1034/j.1600-0714.2002.310202.x. [DOI] [PubMed] [Google Scholar]
- 84.Allen RG, Balin AK. Effects of oxygen on the antioxidant responses of normal and transformed cells. Exp Cell Res. 2003;289:307–316. doi: 10.1016/s0014-4827(03)00279-9. [DOI] [PubMed] [Google Scholar]
- 85.Weydert C, Roling B, Liu J, Hinkhouse MM, Ritchie JM, Oberley LW, Cullen JJ. Suppression of the malignant phenotype in human pancreatic cancer cells by the overexpression of manganese superoxide dismutase. Molec Cancer Ther. 2003;2:361–369. [PubMed] [Google Scholar]
- 86.Darby-Weydert CJ, Smith BB, Xu L, Kregel KC, Ritchie JM, Davis CS, Oberley LW. Inhibition of oral cancer cell growth by adenovirus MnSOD plus BCNU treatment. Free Radic Biol Med. 2003;34:316–329. doi: 10.1016/s0891-5849(02)01245-5. [DOI] [PubMed] [Google Scholar]
- 87.Allen BP, Keogh RG, Gerhard G, Pignolo R, Horton J, Cristofalo VJ. Expression and regulation of SOD activity in human skin fibroblasts from donors of different ages. J Cell Physiol. 1995;165:576–587. doi: 10.1002/jcp.1041650316. [DOI] [PubMed] [Google Scholar]
- 88.Allen RG, Balin AK. Developmental changes in the superoxide dismutase activity of human skin fibroblasts are maintained in vitro and are not caused by oxygen. J Clin Invest. 1988;82:731–734. doi: 10.1172/JCI113654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Balin AK, Pratt L, Allen RG. Effects of ambient oxygen concentration on the growth and antioxidant defenses of of human cell cultures established from fetal and postnatal skin. Free Radic Biol Med. 2002;32:257–267. doi: 10.1016/s0891-5849(01)00807-3. [DOI] [PubMed] [Google Scholar]
- 90.Oberley TD, Sempf JM, Oberley LW. Immunohistochemical localization of antioxidant enzymes during hamster kidney development. Histochem J. 1995;27:575–586. [PubMed] [Google Scholar]
- 91.Oberley LW. Mechanism of the tumor suppressive effect of MnSOD overexpression. Biomed Pharmacotherapy. 2005;59:143–148. doi: 10.1016/j.biopha.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 92.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 93.Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Meth Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 94.Allen RG, Newton RK, Sohal RS, Shipley GL, Nations C. Alterations in superoxide dismutase, glutathione, and peroxides in the plasmodial slime mold Physarum polycephalum during differentiation. J Cell Physiol. 1985;125:413–419. doi: 10.1002/jcp.1041250308. [DOI] [PubMed] [Google Scholar]
- 95.Patel KB, Willson RL. Semiquinone free radicals and oxygen. Pulse radiolysis study of one electron transfer equilibria. J Chem Soc Faraday Transactions. 1973;69:814–825. [Google Scholar]
- 96.Sugioka K, Nakano M, Totsune-Nakano H, Minakami H, Tero-Kubota S, Ikegami Y. Mechanism of O2− generation in reduction and oxidation cycle of ubiquinones in a model of mitochondrial electron transport systems. Biochim Biophys Acta. 1988;936:377–385. doi: 10.1016/0005-2728(88)90014-x. [DOI] [PubMed] [Google Scholar]
- 97.Sawada Y, Iyanagi T, Yamazaki I. Relation between redox potentials and rate constants in reactions coupled with the system oxygen-superoxide. Biochem. 1975;14:3761–3764. doi: 10.1021/bi00688a007. [DOI] [PubMed] [Google Scholar]
- 98.Pick M, Rabani J, Yost F, Fridovich I. The catalytic mechanism of the manganese-containing superoxide dismutase of Escherichia coli studied by pulse radiolysis. J Am Chem Soc. 1974;96:7329–7333. doi: 10.1021/ja00830a026. [DOI] [PubMed] [Google Scholar]
- 99.Bull C, Niederhoffer EC, Yoshida T, Fee JA. Kinetic Studies of Superoxide Dismutases: Properties of the Manganese-Containing Protein from Thermus thermophilus. J Am Chem Soc. 1991;113:4069–4076. [Google Scholar]
- 100.Flohé L, Loschen G, Gunzler WA, Eichele E. Glutathione peroxidase, V. The kinetic mechanism. Hoppe-Seylers Zeitschrift fur Physiologische Chemie. 1972;353:987–999. doi: 10.1515/bchm2.1972.353.1.987. [DOI] [PubMed] [Google Scholar]
- 101.Flohé L. Glutathione peroxidase: fact and fiction. Ciba Foundation Symposium. 1978;65:95–122. [PubMed] [Google Scholar]
- 102.Salvador A, Antunes F, Pinto RE. Kinetic modelling of in vitro lipid peroxidation experiments - ‘low level’ validation of a model of in vivo lipid peroxidation. Free Rad Res. 1995;23:151–172. doi: 10.3109/10715769509064029. and references therein. [DOI] [PubMed] [Google Scholar]
- 103.Brandt U. Bifurcated ubiquinone oxidation in the cytochrome bc1 complex by proton-gated charge transfer. FEBS Lett. 1996;387:1–6. doi: 10.1016/0014-5793(96)00436-x. [DOI] [PubMed] [Google Scholar]
- 104.Gardner PR, Raineri I, Epstein LB, White CW. Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem. 1995;270:13399–13405. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- 105.Poderoso JJ, Lisdero C, Schopfer F, Riobo N, Carreras MC, Cadenas E, Boveris A. The regulation of mitochondrial oxygen uptake by redox reactions involving nitric oxide and ubiquinol. J Biol Chem. 1999;274:37709–37716. doi: 10.1074/jbc.274.53.37709. [DOI] [PubMed] [Google Scholar]
- 106.Gear ARL, Bednarek JM. Direct counting and sizing of mitochondria in solution. J Cell Biol. 1972;54:325–345. doi: 10.1083/jcb.54.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rich P. A physicochemical model of quinone-cytochrome b-c complex electron transfers. In: Trumpower BL, editor. Function of Quinones in Energy Conserving Systems. Academic Press; New York: 1982. pp. 73–83. [Google Scholar]
- 108.Vendelin M, Kongas O, Saks V. Regulation of mitochondrial respiration in heart cells analyzed by reaction-diffusion model of energy transfer. Am J Physiol Cell Physiol. 2000;278:C747–C764. doi: 10.1152/ajpcell.2000.278.4.C747. [DOI] [PubMed] [Google Scholar]
- 109.Korzeniewski B, Zoladz JA. A model of oxidative phosphorylation in mammalian skeletal muscle. Biophys Chem. 2001;92:17–34. doi: 10.1016/s0301-4622(01)00184-3. [DOI] [PubMed] [Google Scholar]
- 110.Beard DA. A biophysical model of the mitochondrial respiratory system and oxidative phosphorylation. PLOS Computational Biology. 2005;1:e36. doi: 10.1371/journal.pcbi.0010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Galinier A, Carriere A, Fernandez Y, Bessac AM, Caspar-Bauguil S, Periquet B, Comtat M, Thouvenot JP, Casteilla L. Biological validation of coenzyme Q redox state by HPLC-EC measurement: relationship between coenzyme Q redox state and coenzyme Q content in rat tissues. FEBS Lett. 2004;578:53–57. doi: 10.1016/j.febslet.2004.10.067. [DOI] [PubMed] [Google Scholar]
- 112.Wang Q, Lee BL, Ong CN. Automated high-performance liquid chromatographic method with precolumn reduction for the determination of ubiquinol and ubiquinone in human plasma. J Chrom B, Biomed Sci Appl. 1999;726:297–302. doi: 10.1016/s0378-4347(99)00067-5. [DOI] [PubMed] [Google Scholar]
- 113.Backstrom D, Norling B, Ehrenberg A, Ernster L. Electron spin resonance measurement on ubiquinone-depleted and ubiquinone-replenished submitochondrial particles. Biochim Biophys Acta. 1970;197:108–111. doi: 10.1016/0005-2728(70)90018-6. [DOI] [PubMed] [Google Scholar]
- 114.Klingenberg M, Kröger A. On the role of ubiquinone in the respiratory chain. In: Slater EC, Kaniuga Z, Wojtczak L, editors. Biochemictry of Mitochondria. Academic Press; New York: 1967. pp. 11–27. [Google Scholar]
- 115.Tyler DD. The Mitochondrion in Health and Disease. VCH publishers; New York, NY: 1992. [Google Scholar]
- 116.Mondal MS, Mitra S. Kinetics and thermodynamics of the molecular mechanism of the reductive half-reaction of xanthine oxidase. Biochemistry. 1994;33:10305–10312. doi: 10.1021/bi00200a010. [DOI] [PubMed] [Google Scholar]
- 117.Hille R, Anderson RF. Electron transfer in milk xanthine oxidase as studied by pulse radiolysis. J Biol Chem. 1991;266:5608–5615. [PubMed] [Google Scholar]