

Abstract
Diversity measurement is important for understanding community structure and dynamics, but has been particularly challenging for microbes. Microbial community characterization using small subunit ribosomal RNA (SSU rRNA) gene sequences has revealed an extensive, previously unsuspected diversity that we are only now beginning to understand, especially now that advanced sequencing technologies are producing data sets containing hundreds of thousands of sequences from hundreds of samples. Efforts to quantify microbial diversity often use taxon-based methods that ignore the fact that not all species are equally related, which can therefore obscure important patterns in the data. For example, α diversity (diversity within communities) is often estimated as the number of species in a community (species richness), and β diversity (partitioning of diversity among communities) is often based on the number of shared species. Methods for measuring α diversity and β diversity that account for different levels of divergence between individuals have recently been more widely applied. These methods are more powerful than taxon-based methods because microbes in a community differ dramatically in sequence similarity, which also often correlates with phenotypic similarity in key features such as metabolic capabilities. Consequently, divergence-based methods are providing new insights into microbial community structure and function.
Keywords: alpha-diversity, beta-diversity, divergence, phylogeny, microbial communities
Introduction
The explosion of 16S rRNA gene sequence data in the public databases, in conjunction with new high-throughput sequencing technologies such as pyrosequencing (Margulies, et al., 2005), allows us to address a vast range of fundamental questions about microbial communities on an unprecedented scale. The hundreds of thousands of 16S rRNA gene sequences now available allow us to ask key questions such as which environmental factors, such as salinity or temperature, have the greatest influence on how microorganisms are distributed globally (Lozupone & Knight, 2007). The decreased cost of Sanger sequencing, and the ability to produce hundreds of thousands of short sequences in a single pyrosequencing run, may now make it possible to sample communities deeply enough to make accurate estimates of the extent of diversity in even the most complex communities and on a global scale (Huber, et al., 2007). The ability to collect sequence data from dozens of communities simultaneously without cloning using DNA barcoding techniques (Binladen, et al., 2007) will make the comparison of hundreds of microbial community samples, each represented by thousands of sequences, routine. This vastly increased sampling will allow us to address fundamental ecological properties of microbial communities such as whether the distribution of microbial diversity has spatial or temporal patterns, or whether there is a relationship between the amount of diversity and the level of primary productivity in an ecosystem (Prosser, et al., 2007). It will also allow us to address important topics for both human and ecosystem health such as whether the type or amount of microbial diversity differs between healthy and diseased people (Turnbaugh, et al., 2006), or between polluted and pristine environments (Liu, et al., 2007). Understanding the methods for measuring diversity within and between samples, and the assumptions, strengths and weakness of these techniques, is thus critical for the future of microbiology.
Measurements of diversity have historically relied on the species as the fundamental unit of analysis. Diversity within a given community (α diversity) is usually characterized using the total number of species (species richness), the relative abundances of the species (species evenness), or indices that combine these two dimensions (Table 1). Similarly, the partitioning of biological diversity among communities or along an environmental gradient (β diversity) is often characterized using the number of species shared between two communities. Species-based diversity measures have been extensively developed and have been instrumental in understanding fundamental ecology properties of many types of communities. They are the subject of recent reviews both for their general application (Koleff, et al., 2003, Magurran, 2004), and for their specific application to microbial communities (Hughes, et al., 2001, Bohannan & Hughes, 2003, Hill, et al., 2003).
Table 1.
Categories of diversity measurements.
| Measurement of diversity within a single community (α diversity) | Measurement of diversity shared among communities (β diversity) | |
|---|---|---|
| Only presence/absence of taxa considered |
Qualitative α diversity (Richness)
Species-based: Chao 1 ACE Rarefaction Divergence-based: Phylogenetic Diversity (PD) |
Qualitative β diversity
Species-based: Sörensen index Jaccard index Divergence-based: Unweighted UniFrac Taxonomic Similarity (ΔS) |
| Additionally accounts for the number of times that each taxon was observed |
Quantitative α diversity (Richness and/or Evenness)
Species-based: Shannon’s index Simpson’s index Divergence-based: Theta |
Quantitative β diversity
Species-based: Sörensen quantitative index Morisita-Horn measure Divergence-based: Weighted UniFrac FST DPCoA |
Unlike broader taxonomic categories, such as genera or phyla, species are defined based on evolutionary theory and are used as a fundamental unit in ecology (Cohen, 2002, Gevers, et al., 2005). The evolutionary underpinnings of the species concept were first introduced by Ernst Mayr in 1944, who proposed that a species is a group of organisms that remain phenotypically similar because of recombination between them (Mayr, 1944). The species concept has evolved since then, but has been particularly difficult for bacteria and archaea, largely because bacteria and archaea generally reproduce asexually and recombination events can occur between distantly related organisms through horizontal gene transfer (HGT). Species delineations in the bacteria and archaea are usually based on phenotypic traits, such as whether the isolates cause a particular disease. Since the 1970s, species have also been delineated based on the overall relatedness of their genomes, with isolates that have >70% genome DNA-DNA hybridization (DDH) considered to be of the same species (Gevers, et al., 2005). This threshold was chosen because it was consistent with recognized phenotype-based species classifications. To delineate species in molecular surveys that sequence the 16S rRNA gene, a >97% sequence identity threshold is typically applied (Martin, 2002). This threshold was chosen because isolates below this threshold of similarity usually have DDH values below 70% (Stackebrandt & Goebal, 1994). The correlation between 16S rRNA identity and DDH values is highly variable in different lineages, however, and microorganisms with >97% identity in the rRNA gene would often be considered different species with the DDH method and can vary widely in the ecological niches that they can occupy (Fox, et al., 1992). Although a fundamental unit with cohesive properties has been proposed to exist for bacteria, driven by periodic events that select for only the best-adapted bacteria in an ecological niche (Cohen, 2002), this unit cannot be accurately related to a single 16S identity threshold (Cohan & Perry, 2007). Because of these issues, it is questionable what is really being measured when the number of bacterial species in a sample is estimated. For these reasons, many prefer to use the terms Operational Taxonomic Units (OTU) or phylotype, rather than “species” for a cluster of related 16S rRNA sequences.
Recently, there has been considerable interest in divergence-based methods for characterizing diversity within and between microbial communities (Singleton, et al., 2001, Martin, 2002, Eckburg, et al., 2005, Lozupone & Knight, 2005). Divergence-based methods account for the fact that not all species or phylotypes within the sample are equally related to each other, and thus contrast with species-based methods, which all implicitly make this assumption. Divergence-based methods consider a community more diverse if the individuals in it are highly divergent from each other, or are phylogenetically distinct from organisms found elsewhere. Correspondingly, they might consider two communities to be similar if they harbor the same phylogenetic lineages, even if the phylotypes representing those lineages in each of the communities are different.
Divergence based methods are more powerful because similarity in 16S rRNA often correlates with phenotypic similarity in key features such as metabolic capabilities. For instance, since all known representatives of the bacterial division Aquificales exclusively or preferentially use H2 as an energy source, it can be inferred that related 16S rRNA sequences in molecular surveys use this form of energy metabolism (Spear, et al., 2005). Likewise, 16S rRNA data revealed that a division-wide shift from Bacteroidetes towards Firmicutes occurs in obese versus lean mice, indicating that metabolic capabilities and response to an environmental perturbation were consistent for diverse members of deep 16S rRNA evolutionary lineages (Ley, et al., 2005). Although HGT can sometimes allow organisms with very similar 16S rRNA sequences to inhabit very different niches, the genes that tend to be transferred are biased towards particular functions, including cell surface, DNA binding and pathogenicity-related functions (Nakamura, et al., 2004) and the degradation of specific compounds, such as xenobiotics (Top & Springael, 2003), and thus the types of niches that bacteria can expand to from HGT will reflect these biases. In contrast, certain functions are less likely to be transferred, particularly if they involve complex networks of genes encoded on multiple operons (Jain, et al., 1999). Accordingly, certain ecological parameters, such as the ability to live in saline verses non-saline environments, are particularly correlated with 16S rRNA similarity (Lozupone & Knight, 2007), indicating that certain niches cannot easily be occupied by the acquisition of new genes. Divergence-based diversity measures that are based on 16S rRNA may thus be biased towards detection of the functions that are more strongly associated with 16S rRNA similarity, but are still more powerful than species-based methods, in which all of this information is lost.
Many different divergence-based diversity measures have been described and used to understand communities, but these measures were designed with different goals in mind and have different strengths and weaknesses. However, divergence-based measures are generally especially well-suited to the evaluation of microbial communities. Because microbial communities are often evaluated using PCR products that were generated with primers that amplify sequences from a wide taxonomic range, the sequences usually represent microbes that differ dramatically in their degree of relatedness. Also, the availability of sequences from the 16S rRNA gene in most microbial community analyses allows the relatedness between individuals or phylotypes to be estimated easily. Studies of macroorganisms are often much more limited in this regard, because macroscopic communities are often surveyed by visual identification of species for which taxonomic relationships and phylogenetically informative gene sequences may not be known.
Community diversity can be measured in many ways (Table 1), but three main distinctions account for most of the variation between measures. The first distinction is whether the goal is to measure the diversity within a community (α diversity) or to measure the partitioning of diversity among two or more communities (β diversity). The second distinction is whether only presence/absence data for each taxon is used (qualitative measures), or whether the abundance of each taxon is also taken into account (quantitative measures). The third distinction is whether all taxa are treated as equally related to one another (species-based measures), or whether the distance between each pair of taxa is taken into account (divergence-based measures). In general, methods that are identical in all three respects perform similarly, although there are technical and conceptual details that may make one measure or another more suitable for a specific study.
Although all these measures of diversity are in principle applicable to any community, some measures have often only been applied in specific subdisciplines of ecology. For instance, microbial communities have often been characterized using divergence-based measures of β diversity to determine whether two communities are significantly different. These measures include the Phylogenetic test (P test) (Martin, 2002) and LibShuff (Singleton, et al., 2001, Schloss, et al., 2004). In contrast, divergence-based measures of α diversity have been applied only very rarely to microbial communities (Eckburg, et al., 2005, Lozupone & Knight, 2007), but have much more often been developed for and applied to the study of communities of macroorganisms, for applications such as the prioritization nature reserves (Faith, 1992, Virolainen, et al., 1998).
In this review, we describe divergence-based diversity measures and their application to microbial communities. We illustrate which measurements are best-suited to testing specific types of hypotheses, with examples from the literature. We also aim to familiarize microbial ecologists with diversity measurements that have not yet been widely applied in this field, in order to bridge the gap between studies of microscopic and macroscopic communities.
Measurements of divergence between taxa
All divergence-based diversity measures require us to estimate the degree to which pairs of organisms differ. This difference is typically referred to as the divergence between that pair of organisms, and can be measured in several different ways, including sequence distance, phylogenetic distance, topological distance, and taxonomic distance. Sequence distance is measured either as pairwise sequence identity, or using one of many available models such as the Jukes-Cantor model that corrects for the probability that multiple nucleotide changes occurred at the same site and that divergence is thus not linear with evolutionary time (reviewed in (Felsenstein, 2004)). Phylogenetic distance is the sum of the branch lengths that separate two organisms in a phylogenetic tree. Topological distance is the number of nodes separating two organisms in a tree and is equivalent to the phylogenetic distance if all of the branch lengths in a phylogenetic tree are set to 1. Finally, taxonomic distance is the taxonomic level separating the two organisms; for instance, 1 for members of the same species, 2 for genus, 3 for family, etc.
Although each diversity measure is usually implemented using one particular method for measuring divergence, a different method can often be substituted depending on the type of data available. In general, it is best to apply the method that provides as much reliable information about relatedness between individuals as possible. For example, taxonomic and topological distances have primarily been useful for macroorganisms, where phylogenetic trees with meaningful branch length information are often not available but taxonomic relationships are well defined. For instance, taxonomic distances can be applied when the organisms are described using a “supertree” that compiles phylogenies from multiple studies that are based on either sequence information from different genes or other types of markers. Thus, taxonomic distances avoid the limitation of methods that require data from the same sequence for all taxa. However, if such data are available, substituting sequence or phylogenetic distances can provide more power to the analysis. Likewise, estimates of phylogenetic distance are more often available than sequence distance because they can be generated from non-overlapping sequences. Techniques for combining non-overlapping sequences include measuring homology between metagenomic sequence reads and sequences that are found in organisms that are already described in a phylogeny (Breitbart, et al., 2002) and using parsimony insertion trees, such as those generated in the ARB software package, in which partial sequences are inserted into an existing phylogeny based on their similarity to full-length guide sequences (Ludwig, et al., 2004). Substitution of phylogenetic distances in diversity measures that were originally described using sequence distances can thus broaden their applicability. However, the results may differ because phylogenetic distances approximate distance in evolutionary history, and can be influenced by the models used during tree reconstruction, whereas sequence distances can be affected more by factors such as convergent evolution.
Measures of α diversity
Measuring α diversity is important for comparing the total diversity in different communities. For example, we might want to know whether pristine habitats contain more diverse microbes than human-modified habitats, whether microbial communities associated with disease states are more or less diverse than microbial communities associated with healthy individuals, or whether the availability of less total nutrients or more kinds of nutrients promotes more diverse communities. In this section, we describe divergence-based measurements of α diversity (see Table 2 for a summary), including both qualitative and quantitative measures. In general, these methods follow the intuition that a community that contains taxa that are more divergent from one another should be considered more diverse, all else being equal (Vane-Wright, et al., 1991). In contrast, almost all studies of microbial community α diversity to date have used species-based measures of α diversity. Both qualitative species-based measures, such as Chao 1 (Chao, 1984) or ACE (Chazdon, et al., 1998), and quantitative species-based measures, such as the Shannon (Shannon & Weaver, 1949) or Simpson (Simpson, 1949) indices, have been widely applied. This focus on species-based techniques is surprising because microbial community diversity is usually assessed using molecular data. Consequently, the phylogenetic relationships among the individuals can easily be estimated, and can be exploited to provide a fuller picture of α diversity.
Table 2.
α diversity measures. PR = Phylogenetic Richness, PE = Phylogenetic Evenness, A = Abundance
| Test | Measurement | PR | PE | A | Application | Implementation | Ref |
|---|---|---|---|---|---|---|---|
| Phylogenetic Diversity (PD) | The amount or proportion of branch length in a phylogenetic tree that leads to organisms from a community. | yes | no | no | Determines which communities are the most phylogenetically diverse. (Does the phylogenetic richness of a community decrease with pollution or disease?) | Phylocom: http://www.phylodiversity.net/phylocom/ | (Faith, 1992) |
| Theta | The average divergence between 2 randomly chosen individuals in a community. | no | yes | yes | Determines how phylogenetically distinct individuals in a community are. (Does the phylogenetic evenness of a community decrease with pollution or disease?) | Arlequin: http://lgb.unige.ch/arlequin/ | (Martin, 2002) |
Phylogenetic Diversity: A Qualititative Measure
The most basic question about a community sample, when thinking in a phylogenetic framework, is how much of the tree of life that sample covers. This intuition, that more diverse communities consist of larger numbers of deeper-branching lineages, informs the Phylogenetic Diversity (PD) measure (Faith, 1992). PD is a qualitative divergence-based measure that sums the total branch length in a phylogenetic tree that leads to each member of a community (Fig. 1A). Although PD satisfies the technical requirement to act as a measure of taxon richness in a community (it has set monotonicity, the property that adding a new species always increases the index value), it is highly sensitive to sampling effort because of the implicit assumption that the total diversity in a community has been sampled. However, there is a substantial literature describing how to estimate total species diversity from a limited sample (Soberon & Llorente, 1993, Magurran, 2004), and some of these techniques can be used to estimate the total PD that would be obtained with exhaustive sampling. For example, rarefaction curves plot the cumulative number of species recorded as a function of sampling effort, and can be used to estimate species richness by using curve-fitting methods to predict the asymptote (Hughes, et al., 2001). In the same manner, the total PD of a sample can be extrapolated from the curve of the accumulation of branch length with sampling effort. To illustrate this, we generated PD rarefaction curves using sequences from the bacterial community in the intestine and stool of three healthy individuals that were generated in (Eckburg, et al., 2005) (Figure 1C). These PD rarefaction curves do not level off, indicating that even with this deep level of sequencing, the addition of new sequences still, on average, added more branch length to the tree of sequences from that individual. The bacterial community in Individual B was the most phylogenetically diverse while the community in Individual A was the least.
Figure 1.
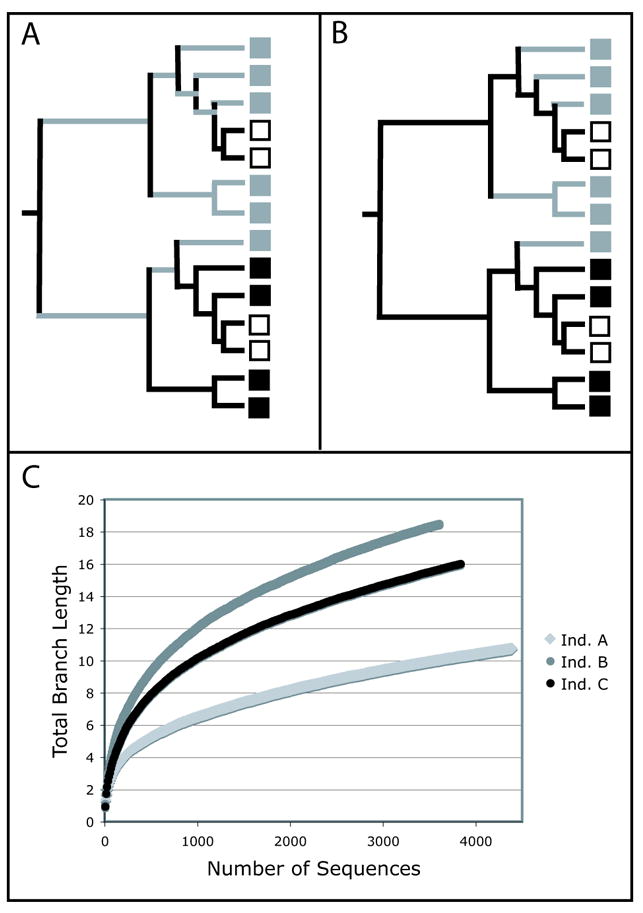
Estimates of Phylogenetic Diversity (PD) and PD Gain (G) for the grey community. The boxes represent taxa from the black, white, and grey communities. (A) PD is the sum of the branches leading to the grey taxa. (B) G is the sum of the branches leading only to the grey taxa. (C) PD rarefaction curves showing the increase in branch length with sampling effort for the intestinal and stool bacteria from three healthy individuals. Aligned16S rRNA sequences from the three individuals were available with the Supplementary Materials in (Eckburg, et al., 2005). The Arb parsimony insertion tool was used to add the sequences to a tree containing over 9,000 sequences (Hugenholtz, 2002) that is available for download at the rRNA Database Project II website (Maidak, et al., 2001). The curves represent the average values for 50 replicate trials.
PD depends on the method used to infer branch lengths on the tree, and are thus potentially sensitive to errors in phylogenetic tree construction. It is thus useful to compare the values from trees generated with different methods or, alternately, to express error estimates by evaluating groups of trees that are equally likely, such as those generated from bootstrapped data. Also, since branch lengths are inconsistent in their meaning with different tree topologies, they should not generally be compared between studies. They are primarily useful for comparing communities in which all individuals are contained within the same tree. For example, we recently used PD to compare the α diversity of different types of physical environments in a global analysis of bacterial 16S rRNA community surveys (Lozupone & Knight, 2007). Using this technique, we made the surprising discovery that soil, which is often described as one of the most diverse types of microbial communities based on species-based diversity measures, has below average phylogenetic diversity in the bacteria. Sediments, on the other hand, have particularly high phylogenetic diversity. This result underscores the importance of describing diversity with divergence based techniques as well as species counts. PD has been implemented in the Phylocom software (Webb, et al., 2006), which is available for download at http://www.phylodiversity.net/phylocom/.
Divergence-based diversity measures differ in the degree to which they are sensitive to whether the data has been de-replicated, for instance by removing sequences that are exact replicates of each other or that are within a per cent identity threshold. PD calculations are insensitive to this factor: adding duplicate sequences to the tree adds no branch length, and adding similar sequences adds only minimal amounts. However, rarefaction analysis with PD requires data that has not been de-replicated, because this analysis measures how the PD changes with the number of individuals that were observed. In this sense, although raw PD values are a qualititative measure, the comparison of rarefaction curves is inherently more quantitative.
Theta: A Quantitative Measure
Measures of α diversity can also be quantitative, accounting for “evenness” as well as divergence between taxa when measuring diversity. Species evenness reflects the distribution of relative species abundances: for example, a community where a few of the species are numerically dominant would have low evenness.
One measure of α diversity that is both quantitative and divergence-based is theta (θ), which is simply the average divergence between two randomly chosen sequences or individuals in a population, and can be calculated using Equation 1:
| Equation 1 |
…where k is the number of individuals sampled, pi is the frequency of the ith sequence, pj is the frequency of the jth sequence, and dij is the divergence between the two sequences. θ is widely applied in molecular evolution and population genetics, but was first described for the analysis of microbial diversity by Martin (Martin, 2002). θ is also identical to the taxonomic diversity index (Δ) described by Clark and Warwick (Warwick & Clarke, 1995, Clarke & Warwick, 1998, Warwick & Clarke, 1998), except that Clark and Warwick use taxonomic distance for dij and Martin uses sequence distance. θ is also equal to ½ the Rao Diversity Coefficient (RaoDIV) (Rao, 1982), which reduces to the Gini-Simpson index when all pairwise distances between species are equal, i.e. if the species follow a star phylogeny (Warwick & Clarke, 1995, Izsak & Papp, 2000, Shimatani, 2001). Because θ is the average divergence of randomly selected pairs of sequences or individuals in the population, it does not increase with sampling effort, and thus does not account for the species richness aspect of a community (Clarke & Warwick, 1998). Instead, θ is a combination of taxonomic distinctness and evenness. For example, a community with equally abundant, divergent lineages would have higher θ than a community in which most of the individuals come from only one of the divergent lineages.
Although θ has not been widely adopted for microbial diversity measurement, it has been used to evaluate both communities of microorganisms and macroorganisms. Eckburg et al. used the related RaoDIV index to compare microbial diversity in multiple mucosal sites in the intestines and the feces of 3 healthy human subjects (Eckburg, et al., 2005). θ has also been used to compare communities of macroorganisms using amino acid diversity (Shimatani, 2001) or taxonomic distances (Izsak & Papp, 1995, Warwick & Clarke, 1995, Shimatani, 2001) for dij. For instance, θ decreased along a gradient of increasing environmental contamination in a marine assemblage (Warwick & Clarke, 1995), and increased with thinning operations to promote species survival in a forest in south-west Michigan (Shimatani, 2001).
Since θ is a quantitative measure, its calculation is sensitive to whether the data has been de-replicated prior to its application. However, if the same sequence is found 5 times in a sample, it does not make a difference whether this sequence is added 5 times to the analysis with a weight of 1, or added once with a weight of 5. It thus does not matter whether OTUs are selected prior to analysis if the abundance of each OTU in the sample is recorded. Since θ is dependent on relative abundance information, however, like all quantitative diversity measures that are described in this review, its value may be affected by bias in DNA extraction efficiency for different bacteria as well as bias in PCR amplification, and cloning efficiency (von Wintzingerode, et al., 1997, Kanagawa, 2003).
Measures of β diversity
So far, we have considered measures of α diversity, which is diversity within a single sample. However, many questions require us to inquire about β diversity. β diversity was originally conceived by Whittaker as a measure of change in diversity along transects or across environmental gradients (Whittaker, 1960). In general, β diversity evaluates differences between two or more local assemblages or between local and regional assemblages (Koleff, et al., 2003). β diversity measures that evaluate the extent to which two or more communities differ can be used to evaluate how a microbial community changes over time, or with different disease states. β diversity measures that compare local to regional assemblages can elucidate how much diversity is unique to a local assemblage or which ecological processes, such as habitat filtering or competition, structure the community. β diversity measures allow us to address fundamental ecological questions such as if microbes have biogeography or rather, that because of their small size and high abundance, “Everything is everywhere; the environment selects” (Bass-Becking, 1934). Species-based measures of β diversity have been useful for this question, because they can evaluate whether similar environments contain the same species despite distance and other geographic barriers (Noguez, et al., 2005). Species-based measures are limited, however, because they depend on a poorly defined species concept. In contrast, divergence-based diversity measures can address the same question by determining the spatial distribution of phylogenetic lineages, i.e. whether the microbes in physically separated environments have little unique branch length, indicating little unique evolution endemic to an area.
In this section, we describe several divergence-based measures of β diversity (see Table 3 for a summary), including both qualitative and quantitative measures. In general, these divergence-based measures reflect the intuition that communities should be considered more similar if the taxa they contain are more closely related, even if the taxa are not identical. Whereas some β diversity measures are designed solely to determine whether communities are significantly different, others are measures of distance between pairs of communities that satisfy the requirements of a distance metric, and can thus also be used with multivariate statistical techniques such as clustering and ordination to relate many communities simultaneously. The latter group is related to the Jaccard and Bray-Curtis coefficients for measuring the distance between communities based on the species that they contain.
Table 3.
β diversity measures. S = Significance, C = Clustering, A = Abundance
| Test | Hypothesis tested/Measurement | S | C | A | Unique traits | Implementation | Ref |
|---|---|---|---|---|---|---|---|
| LibShuff | Significance: The sequences in community x are more closely related to each other than they are to the sequences in community y (and vice versa). | yes | no | both | Uses sequence distances exclusively. Can be used to determine whether community differences are due to deep branching or shallow lineages. | LibShuff: http://www.arches.uga.edu/~whitman/libshuff.html
∫-LibShuff: http://www.plantpath.wisc.edu/fac/joh/S-libshuff.html |
(Singleton, et al., 2001, Schloss, et al., 2004) |
| P-test | Significance: There have been fewer transfers of lineages between communities than would be expected by chance. | yes | no | both | Solely uses tree topology. Should be applied when the primary interest is in the relationships between species/individuals and not the extent of divergence of the lineages. | UniFrac: http://bmf.colorado.edu/unifrac
TreeClimber http://www.plantpath.wisc.edu/fac/joh/treeclimber.html |
(Martin, 2002) |
| UniFrac | Significance: More unique evolution has occurred within the communities than expected by chance
Clustering: Similar communities have similar phylogenetic lineages. |
yes | yes | no | Qualitative measure that exclusively uses a phylogenetic tree and accounts for history of shared ancestry between communities. | UniFrac: http://bmf.colorado.edu/unifrac | (Lozupone& Knight, 2005) |
| Weighted UniFrac | Significance: The individuals within the communities are more phylogenetically similar to each other than to those in another community.
Clustering: Similar communities contain more phylogenetically similar individuals. |
yes | yes | yes | Quantitative version of UniFrac. Similar to clustering with FST or DPCoA but exclusively uses a phylogenetic tree. | UniFrac: http://bmf.colorado.edu/unifrac | (Lozupone, et al., 2007) |
| FST | Significance: There is more sequence variation when two communities are pooled than when the two communities are examined individually.
Clustering: Similar communities contain more phylogenetically similar individuals. |
yes | yes | yes | Similar to weighted UniFrac or DPCoA. Uses sequence distances. | Arlequin: http://lgb.unige.ch/arlequin/ | (Martin, 2002) |
| DPCoA | Clustering: Similar communities contain more phylogenetically similar individuals. | no | yes | yes | Similar to clustering with FST or Weighted UniFrac but allows for visualization of the relationships between communities in the same plot as the phylotype relationships. | Requires some programming in the R statistical package. | (Pavoine, et al., 2004) |
| Taxonomic Similarity | Significance: The average distance between the species in two different communities differs from the chance expectation if species were randomly assigned to communities.
Clustering: Similar communities contain more phylogenetically similar species. |
no | yes | no | Qualitative measure that compares communities based on species differences. Requires consistent species definitions. | Phylocom: http://www.phylodiversity.net/phylocom/ | (Izsak & Price, 2001) |
| Gain in Phylogenetic Diversity (G) | The amount or proportion of branch length in a phylogenetic tree that is unique to organisms from a community. | no | no | no | Determines which communities are the most phylogenetically unique. (Is the community in a chemically/physically unique environment different from other characterized environments?) | None | (Faith, 1992) |
| Net Relatedness Index (NRI) | Significance: The average divergence between 2 randomly chosen species in a community are closer or further than would be expected if the species were evenly distributed throughout a region. | yes | no | no | Determines how phylogenetically distinct species in a community are. (Is habitat filtering or interspecies competition more important for structuring the community?) | Phylocom: http://www.phylodiversity.net/phylocom/ | (Webb, 2000) |
| Nearest Taxa Index (NTI) | Significance: The average divergence between each species and its nearest neighbor are closer or further than would be expected if the species were evenly distributed throughout a region. | yes | no | no | Similar to NRI but focuses on lower taxonomic levels and ignores higher-level structure. | Phylocom: http://www.phylodiversity.net/phylocom/ | (Webb, 2000) |
Significance Tests
LibShuff
LibShuff (Singleton, et al., 2001) was among the first divergence-based β diversity measures to be developed. LibShuff uses nucleotide distances to determine whether two communities are significantly different. The library coverage of sequences from community X (CX), defined as the percentage of sequences in X that are not singletons, is calculated after grouping the sequences throughout the range of possible sequence distance levels. CX increases with distance until the maximum distance, where it reaches 1 because all of the sequences are grouped together and there are no singletons (Figure 2). CX is then plotted against sequence distance to produce a homologous coverage curve. The homologous coverage curve is compared to a heterologous coverage curve, which compares the sequences in X to those from another community, Y. In theheterologous case, library coverage (CXY) is calculated as the fraction of sequences that are in X that are also in Y. The Cramer-von Mises statistic (Pettitt, 1982) is used to calculate the distance between the two curves (Equation 2).
Figure 2.
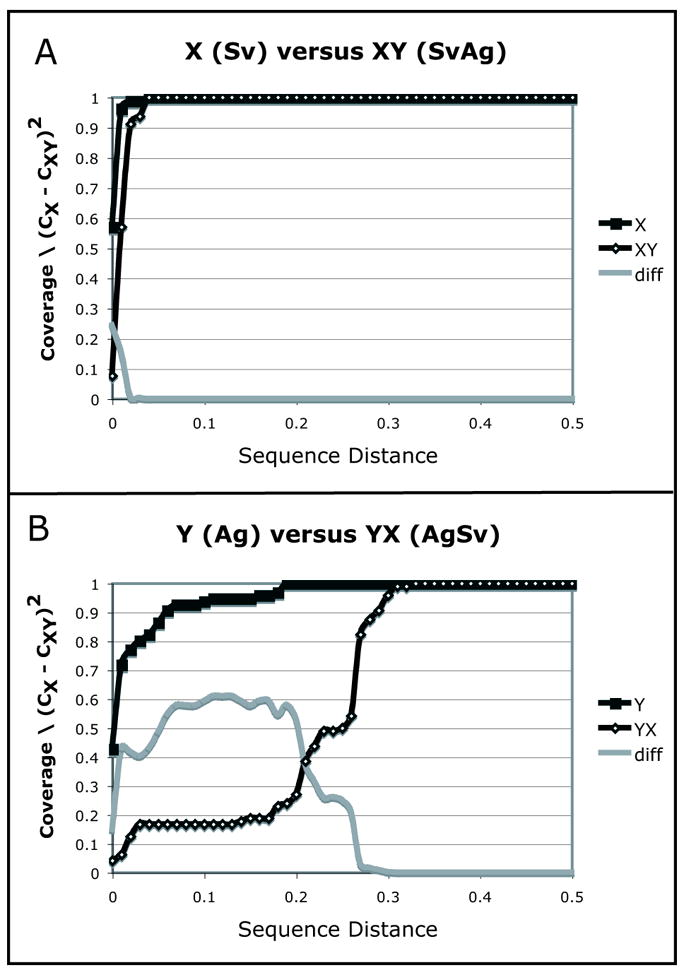
A LibShuff comparison of bacterial 16S rRNA clones from the guts of two wood-boring beetles in the Cerambycidae, S. vestida (X) and A. glabripennis (Y). The sequence data was initially described in (Schloss, et al., 2006). We downloaded the 180 sequences that were deposited in Genbank by the authors, aligned them with the NAST alignment tool (DeSantis, et al., 2006), and used ARB (Ludwig, et al., 2004) to apply a lanemask to exclude hypervariable regions that were not well aligned. We removed 5 short sequences to maximize the region of overlapping sequence reads. A matrix of sequence distances was generated using the Phylip dnadist program with the Jukes-Cantor model of nucleotide substitution. The LibShuff analysis was performed on this distance matrix using the webLIBSHUFF implementation (Henriksen, 2004). The homologous coverage curve (■) represents only S. vestida in panel A and only A. glabripennis in panel B. It shows how the number of groups changes throughout the range of sequence distances. The heterologous coverage curve (◇) shows the percent of groups that the other beetle shares with the first beetle over the range of sequence distances. The solid grey line is the value of (CX − CXY)2 at each level of evolutionary distance. The area under this curve is the raw LibShuff value.
| Equation 2 |
Libshuff uses Monte Carlo simulations to determine whether the two communities are significantly different. Specifically, the sequences are randomly shuffled between the two samples for many replicate trials, and the Cramer-von Mises statistic value is calculated for each replicate. The p-value is the fraction of trials in which the real value is greater than the random values. The shape of the coverage curves can also be evaluated to determine whether community differences are largely due to deep- or shallow-branching lineages.
Note that two comparisons can be made for each pair of environments, CXY and CYX, and that these will not return the same result. For instance, in the comparison of 16S rRNA sequence clones from the guts of two wood-boring beetles illustrated in Figure 2, the results of comparing beetle X (Saperda vestida) to beetle Y (Anoplophora glabripennis) are extremely different. The 16S rRNA gene sequences from S. vestida were all from the γ-Proteobacteria, whereas A. glabripennis had members of the α-β-and γ-Proteobacteria as well as the Firmicutes, Actinobacteria, and Bacteroidetes (Schloss, et al., 2006). Comparison of the homologous coverage curves in Figure 2A and B shows this difference in diversity. All of the sequences from S. vestida bin into just 1 group at a ~0.025 sequence distance, whereas the A. glabripennis sequences do not form one group until ~0.2 sequence distance. The heterologous coverage curve in Figure 2A shows that at ~0.06 sequence distance, all of the sequence groups in S. vestida are also found in A. glabripennis, but the difference in the curves shows that even the γ-Proteobacteria in the two beetles are different from each other, and in fact, the result of this comparison is significant at p < 0.002. The heterologous coverage curve in Figure 2B shows that aside from the similarities at low sequence distances (because of the γ-Proteobacteria), the majority of bacteria in the gut of A. glabripennis are highly divergent from anything in S. vestida. The result of this comparison is also significant at p < 0.002.
The original Libshuff implementation calculated library coverage at 0.01 increments of nucleotide distance and can be downloaded from http://www.arches.uga.edu/~whitman/libshuff.html or run with the web version at http://libshuff.mib.uga.edu/. An extension of LibShuff, which improves the accuracy of the estimates by using the integral form of the Cramercon Mises statistic, is ∫-LibShuff (Schloss, et al., 2004). This tool is available at http://www.plantpath.wisc.edu/fac/joh/S-libshuff.html. LibShuff has been broadly applied in microbial ecology (Walsh, et al., 2005, Beman & Francis, 2006, Diaz, et al., 2006, Oline, 2006, Santoro, et al., 2006), and can be quantitative or qualitative depending on whether or not duplicate sequences are included in the analysis.
Phylogenetic (P) Test
The Phylogenetic (P) test was introduced by Martin in 2002 as a method to determine whether microbial communities are significantly different by testing whether members of the two communities are randomly distributed over a phylogenetic tree (Martin, 2002). The P test uses the Fitch parsimony algorithm (Felsenstein, 2004) to estimate the minimum number of changes from one community to the other that would be needed to explain the distribution of communities in a phylogenetic tree (Figure 3A). As with LibShuff, statistical significance is determined using Monte Carlo simulations. In this case, the parsimony count is determined for many random trees, and the p-value is the fraction of the trees that have a lower parsimony count than the true tree (Figure 3D). In the initial description of the P test (Martin, 2002) and in its implementation in the TreeClimber software (Schloss & Handelsman, 2006), which is available at http://www.plantpath.wisc.edu/fac/joh/treeclimber.html, the tree topology is randomized by making random joining trees, as described by Maddison and Slatkin (Maddison & Slatkin, 1991). In the P test implementation at the UniFrac website (Lozupone, et al., 2006) http://bmf.colorado.edu/unifrac, the environment labels are randomized over a constant tree topology. Although this results in the testing of a slightly different hypothesis, the results from the two different randomization protocols will most likely be similar, because the randomization methods have been shown to produce similar probability distributions in many circumstances (Maddison & Slatkin, 1991). The P test has also been broadly applied in microbial ecology (Schadt, et al., 2003, Stach, et al., 2003, Breitbart, et al., 2004, Dunfield & King, 2004, Lipson & Schmidt, 2004, Nanba, et al., 2004).
Figure 3.
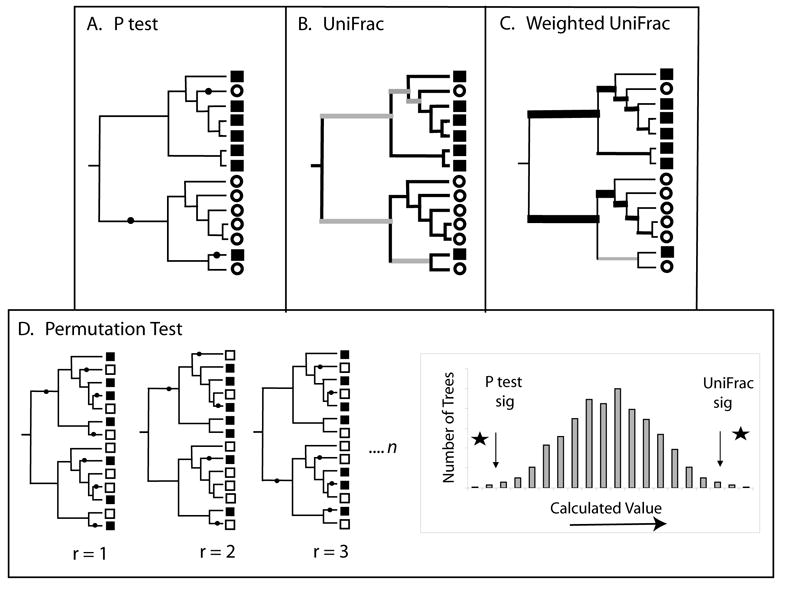
Significance testing with the P test, UniFrac, and weighted UniFrac. The P test and the unweighted and weighted UniFrac significance tests all determine whether two communities are significantly different by comparing a value for the true tree to a collection of random trees and/or trees in which the community labels have been randomly assigned to a constant tree topology. For the P test (A), the calculated value is the minimum number of changes (indicated with black dots) needed to describe the distribution of community labels on a phylogenetic tree (squares and circles denote sequences derived from different communities). For UniFrac (B), the calculated value is the fraction of branch length in the tree that is unique to one community (black branches) verses shared (grey branches). For weighted UniFrac (C), the calculated value is the sum of the branches weighted by the difference in the number of descendants from each community for each branch (represented here by the thickness of the branch). (D) For the P test, the p value is the fraction of the random trees that have a smaller value then the real tree. For both unweighted and weighted UniFrac, the p value is the fraction of the random trees that have a greater value than the real tree.
The P test differs from LibShuff in that it does not explicitly account for the degree of divergence between sequences. It should be applied when the primary interest is in tree topology and not in the extent of divergence of the lineages, for example when rates of evolution are highly heterogeneous. Also, since the P test is dependent on a phylogenetic tree, it has the potential to be affected by errors in inferred tree topology. Although the P test was initially described for the evaluation of a single tree topology, more recently Jones and Martin showed that accounting for uncertainty by using many statistically equivalent phylogenetic trees, such as those obtained with bootstrapping or Bayesian phylogenetic methods, provides a more conservative and robust test (Jones & Martin, 2006).
Whether or not data is de-replicated prior to the application of the P test will have a major impact on the results, because the tree topology over which the randomizations are performed and the sample size will change, and so the expected distribution to which the true number of parsimony changes is compared will differ. If species-level OTUs are chosen before performing the P test, the hypothesis that is being tested is that the species in the communities are more clustered than expected by chance, and the P test is a qualitative measure. If the raw data is used, the hypothesis is that the individuals from each community are more clustered than expected by chance, and the P test is a quantititative measure (see (Lozupone, et al., 2006) for more discussion).
Measures of Community Distance
The measures of β diversity that are described in this section differ from the P Test and LibShuff because they provide measures of distances between pairs of communities that can be used with multivariate statistical techniques such as clustering and ordination to relate many communities simultaneously. However, some of these, including UniFrac, weighted UniFrac, and FST can additionally be used as tests of significance. FST was initially described for this purpose alone (Martin, 2002).
UniFrac: A Qualitative Measure
The Unique Fraction metric, or UniFrac, follows the intuition that communities that differ more should require more unique evolution of the lineages they contain, presumably reflecting adaptation to one community that would be deleterious in the other. UniFrac therefore measures the phylogenetic distance between sets of taxa in a phylogenetic tree as the fraction of branch length of the tree that leads to descendants from either one community or the other but not both (Fig. 3B)(Lozupone & Knight, 2005), and falls within a broader family of divergence-based β diversity measures related to PD that differ in the details of how the unique and shared branches are combined (Faith, 2007). UniFrac is a qualitative β diversity measure, because duplicate sequences contribute no additional branch length to the tree (by definition, the branch length connecting a pair of duplicate sequences is zero, because no substitutions separate them). It assumes that if two communities were similar, few adaptations would be required for an organism to transfer between them. Consequently, most nodes in a phylogenetic tree would have descendants from both communities, and much of the branch length in the tree would be shared. In contrast, if two communities were so distinct that an organism adapted to one could not survive in the other, the lineages in each community would be distinct, and most of the branch length in the tree would lead to descendants from only one of the two communities. UniFrac analyses most often use phylogenetic trees from the 16S rRNA gene, and thus tracks the functional similarities that tend to be associated with this molecule. It does not account for functional differences that may have been acquired by HGT. UniFrac can also, however, be applied to trees made from other genes. For example, UniFrac was used with nitrogenase reductase (nifH) gene sequences to compare nitrogen-fixing communities between soils (Lamarche & Hamelin, 2007).
Like the P test and LibShuff, UniFrac can be used to determine whether two communities differ significantly using Monte Carlo simulations (Fig. 3D). Two communities are considered different if the UniFrac value is greater than would be expected by chance. Randomizations are performed by keeping the tree constant and randomizing the community label that is assigned to each sequence for many replicate trials (typically 100 or 1000). The p-value is the fraction of trees that had higher UniFrac values than the true tree (Fig. 3D). The UniFrac significance test differs from the P test because it accounts for branch lengths in addition to tree topology when comparing diversity. Whereas the P test determines if the sequences are more clustered into monophyletic lineages than would be expected by chance, UniFrac determines whether there has been an excess of unique evolution within each community (more branch length leading to descendants from only one community).
One major advance of UniFrac over LibShuff and the P test is that it can be used with clustering or ordination techniques to compare many communities simultaneously (Fig. 4). These multivariate statistical techniques are applied to a distance matrix describing the pairwise UniFrac distances between the sets of sequences collected from many different microbial communities. For instance, Figure 4 shows the results of using a UniFrac distance matrix to visualize the relationship between the cecal microbial communities of 19 mice in a study of the effects of obesity on gut microbial communities (Ley, et al., 2005). The results of both hierarchical clustering with the Unweighted Pair Group Method with Arithmetic mean (UPGMA) method (Fig. 4B) and ordination with Principal Coordinates Analysis (PCoA) (Fig. 4C) showed that mice with the same mother had similar bacteria in their cecum. We also implemented a sequence jackknifing technique, which can be used to test the robustness of the UPGMA results to sampling effort. In this technique, a subset of the data is selected for many replicate trials and the percent of the time that each node in the hierarchical cluster is recovered is reported (Lozupone & Knight, 2005, Lozupone, et al., 2006, Lozupone, et al., 2007). For instance, for the cecal mouse samples, sub-sampling 200 sequences from each mouse for 100 replicate trials (there were at most 484 sequences per mouse) resulted in strong recovery for many of the nodes in the UPGMA cluster (Fig. 4B).
Figure 4.
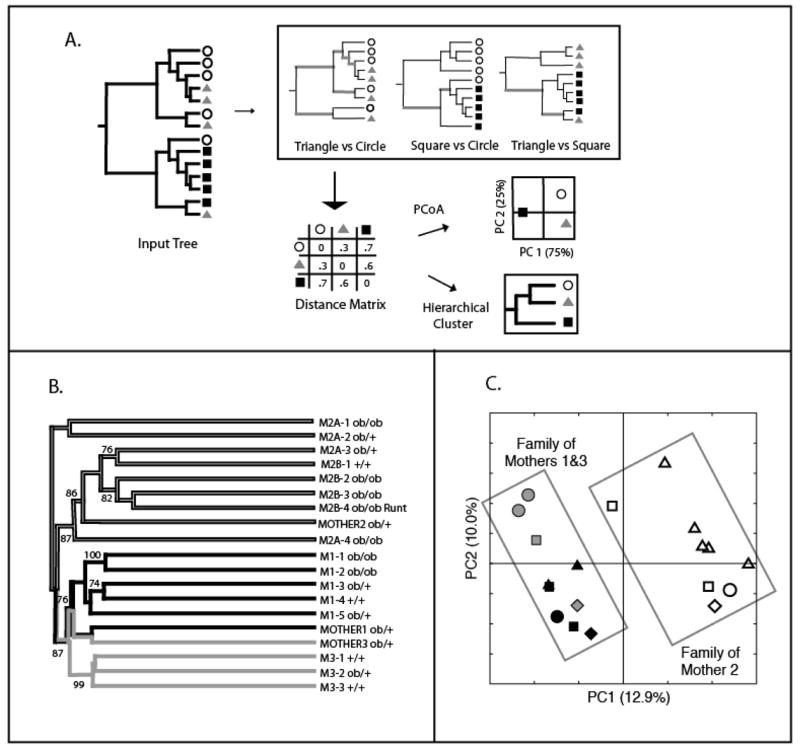
Clustering with UniFrac. (A) Schematic showing how clustering is performed, adapted from (Lozupone & Knight, 2005). The circles, squares, and triangles represent sequences from three different communities. The UniFrac value is calculated for all pairs of communities, and the resulting distance matrix can be used to cluster the samples using Principal Coordinates Analysis (PCoA) or hierarchical clustering. (B) The results of hierarchical clustering and jackknifing of cecal microbial communities from three mother mice (MOTHER1-3) and their offspring (M1-, M2A-, M2B-, and M3) with unweighted UniFrac (Adapted from (Ley, et al., 2005, Lozupone, et al., 2007)). Genotypes are ob/ob for homozygotes for the mutant leptin allele that confers obesity, ob/+ for heterozygotes, and +/+ for wild-types. The percentage support for nodes supported at least 70% of the time with sequence jackknifing with a maximum of 200 sequences from each mouse for 100 replicates is indicated. The main clustering is by mother. (C) Plot of the first 2 principal coordinates axes for PCoA with unweighted UniFrac. Symbols represent individual animals. The rectangles highlight the family of Mother 2 (open symbols), and the families of Mothers 1 and 3 (grey and black symbols), who are sisters.
When UniFrac is used as a distance measure for clustering, it is a qualitative β diversity measure, because duplicate sequences contribute no additional branch length to the tree. Because of this property, it does not matter whether OTUs are selected prior to the analysis. In contrast, as with the P test, significance testing with unweighted UniFrac is sensitive to de-replication, and is only truly a qualitative measure if OTUs are selected prior to the analysis and only one member of each OTU is included in the tree. Although failure to choose OTUs will not change the raw UniFrac value, the significance test results can change because the expected distribution to which the true UniFrac value is compared will be different because the sequences will be randomized over a different tree topology (see (Lozupone, et al., 2006) for more discussion). Performing a UniFrac significance test on a tree that has not been de-replicated is still valid, but it is a quantitative rather than a qualititative measure.
Although UniFrac is strictly dependent on a phylogenetic tree, several lines of evidence show that multivariate analysis with UniFrac is robust to errors in tree construction. First, analysis of the same dataset with trees that were constructed with 6 different methods for inferring the tree yielded almost identical results (Lozupone, et al., 2007). This was true even though the trees differed substantially in both topology and tip to tip distances. Second, although partial sequences have fewer phylogenetically informative positions and are thus less robust for phylogenetic inference, UniFrac reanalysis with extremely short sequences (100–200 base pairs from different regions of the 16S rRNA molecule) from 3 different studies that initially used near full-length sequences, recaptured the major clustering patterns remarkably well, particularly if a moderately conserved region of the 16S rRNA gene was selected (Liu, et al., 2007). As with the P test, significance testing with UniFrac may be more robust if uncertainty in the tree topology is accounted for.
UniFrac has been implemented in both the UniFrac web application (http://bmf.colorado.edu/unifrac) (Lozupone, et al., 2006) and a programming API written in python (available for download at http://bayes.colorado.edu/unifrac), and has been widely applied in microbial ecology (Rawls, et al., 2004, Ley, et al., 2005, Ley, et al., 2006, Ley, et al., 2006, Fraune & Bosch, 2007, Lamarche & Hamelin, 2007, Walker & Pace, 2007, Wallenstein, et al., 2007). The development of UniFrac also made it possible for us to perform a global analysis of bacteria in different physical environments, based on 21,752 sequences from 111 studies (Lozupone & Knight, 2007). In this analysis, we showed that salinity was a key factor for determining the presence or absence of bacterial lineages among environments. The properties that made UniFrac uniquely suited to this application are that 1) it can be used with clustering and ordination techniques to relate many communities simultaneously, 2) it uses a phylogenetic tree as input, and so non-overlapping 16S rRNA sequence reads can be included by using the parsimony insertion tool of ARB (Ludwig, et al., 2004) and 3) it is a qualitative metric, which does not require information on relative sequence abundances, which is not available in GenBank. Although the use of partial non-overlapping sequence reads has the potential to affect the resolution of the phylogenetic tree, these effects were not large enough to obscure the major patterns of variation, because there was no correlation between the sequenced region and UniFrac clustering, and qualitatively similar results were obtained using a smaller dataset that only consisted of overlapping sequences (Lozupone & Knight, 2007).
Quantitative Measures of Community Distance
Many analyses can benefit from quantitative rather than qualitative diversity measures (Lozupone, et al., 2007), because sometimes communities change only in the relative abundance of bacterial lineages. For example, a particular lineage may flourish because a limiting nutrient becomes abundant. In fact, quantitative data for 16S rRNA survey data has often lead to insights that would not have been apparent from presence/absence data alone. For instance, changes of the intestinal community with obesity were only evident in changes in the relative abundance of the Bacteroidetes and Firmicutes, and not in the presence or absence of lineages (Ley, et al., 2005, Ley, et al., 2006). Similarly, the abundance of Aquificales in thermal hotsprings in Yellowstone indicated the importance of hydrogen metabolism, which would not be apparent by their presence alone (Spear, et al., 2005). Finally, a correlation with mineral chemistry of acidic thermal springs was only detectable when relative abundances were taken into account (Lozupone, et al., 2007, Mathur, et al., 2007).
Because quantitative measures are dependent on accurate information on the relative abundance of sequences, they can be affected by biases introduced during the DNA extraction, PCR amplification, and cloning procedures that are known to exist (von Wintzingerode, et al., 1997, Kanagawa, 2003). It is thus always desirable to confirm abundance estimates using other methods such as Florescence In Situ Hybridization (FISH) or quantitative PCR. Furthermore, biases can be introduced by differences in rRNA copy number between bacteria (Weider, et al., 2005). These biases, however, are less important for quantitative β diversity measures than for α diversity measures because all of the samples should be subject to the same biases.
Weighted UniFrac
Weighted UniFrac (Lozupone, et al., 2007) is a variant of the UniFrac algorithm that accounts for changes in relative abundance of lineages between different communities, by weighting the branches in the phylogenetic tree based abundance differences when performing the calculations (Fig. 3C). Weighted UniFrac is a quantitative β diversity measure because it detects changes in how many sequences from each lineage are present, as well as changes in which taxa are present.
The weighted UniFrac value (u) is calculated according to Equation 3:
| Equation 3 |
Here, n is the total number of branches in the tree, bi is the length of branch i, Ai and Bi are the number of sequences that descend from branch i in communities A and B respectively, and AT and BT are the total number of sequences in communities A and B respectively. In order to control for unequal sampling effort, Ai and Bi are divided by AT and BT.
The weighted UniFrac value can be normalized so that it has a value of 0 for identical communities and 1 for non-overlapping communities, facilitating comparisons between different studies. This normalization is accomplished by dividing u by a scaling factor (D), which is the average distance of each sequence from the root (Equation 4).
| Equation 4 |
Here, dj is the distance of sequence j from the root, Aj and Bj are the number of times the sequences were observed in communities A and B respectively, and AT and BT are the total numbers of sequences from communities A and B respectively.
Like UniFrac, weighted UniFrac can be used to perform significance tests (Fig. 3D), to cluster many samples with UPGMA, and to ordinate samples with PCoA (Fig. 4). All of this functionality is also available at the UniFrac web interface (http://bmf.colorado.edu/unifrac). We illustrated with two example datasets that multivariate analysis of the same data with unweighted and weighted UniFrac can lead to different, but equally illuminating, conclusions about the main factors that structure microbial communities (Lozupone, et al., 2007). Specifically, the unweighted UniFrac algorithm, because it only considers the presence or absence of lineages, is better suited to detecting factors that are restrictive for microbial growth, such as temperature or pH. In contrast, weighted UniFrac is ideally suited to revealing community differences that are due to changes in relative taxon abundance, for instance with different availability of mineral nutrients in hotspring sediments or with change in diet in the mouse intestine (Lozupone, et al., 2007). Unlike unweighted UniFrac, significance testing with weighted UniFrac is always quantitative, and it is not valid to perform without abundance information. However, adding a duplicate sequence to the tree 5 times is essentially the same as adding it once with a weight of 5.
The FST test
The FST test was adapted from population genetics, and identifies cases in which more sequence variation exists between two communities than within a single community (Martin, 2002). The intuition behind this test is that combining two different populations into a single, large population will result in increased sequence heterogeneity, but combining two identical populations will not. Sequence variation is calculated using θ, which was described earlier in this review as an estimate of quantitative α diversity. FST is calculated using Equation 5:
| Equation 5 |
…where θT is the value of θ for all samples combined and θW is the average θ within each of the communities being compared. The distance between each pair of taxa is calculated as the number of nucleotide differences in aligned sequences in the θ calculations.
Like weighted UniFrac, FST is a quantitative measure because the number of times each sequence was observed affects the calculation. In fact, we have shown that FST and weighted UniFrac behave similarly in situations where both are applicable (Lozupone, et al., 2007). However, because weighted UniFrac takes a phylogenetic tree as input and FST uses a sequence alignment, weighted UniFrac has the advantage that it can be used to combine data in which different parts of the 16S rRNA were sequenced (e.g. when nonoverlapping sequences can be combined into a single tree using full-length sequences as guides).
Like the P test and Libshuff, FST was introduced as a test to determine whether communities are significantly different using Monte Carlo simulations. As a significance test, FST is similar to LibShuff in that it uses sequence distances to evaluate community distances. FST is less sensitive to multiple comparisons than LibShuff because LibShuff must do two comparisons (calculation of CXY and CYX) for each community comparison. Unlike LibShuff, the FST values satisfy the technical requirements of a distance metric, and can thus be used to compare many communities simultaneously using multivariate statistics. This was illustrated in a recent study of the effects of temperature, mineral chemistry and geography on the bacterial communities in acidic thermal springs in Yellowstone National Park (Mathur, et al., 2007). Mathur et al. used PCoA to cluster their samples based on FST values, and showed that mineral chemistry was the most important factor in describing the differences between their samples. Analysis with FST can be carried out using the computer program Arlequin (Schneider, et al., 1997), which is available for download at http://lgb.unige.ch/arlequin/. As with θ and weighted UniFrac, if a given sequence is found 5 times in a sample, it does not make a difference whether the same sequence is added 5 times to the analysis with a weight of 1, or added once with a weight of 5.
Rao Dissimilarity and Double Principal Coordinates Analysis (DPCoA)
In addition to introducing the RaoDIV index for estimating quantitative α diversity, Rao also described a quantitative β diversity measure, RaoDIS. RaoDIS is a dissimilarity coefficient that estimates the dissimilarity between two communities according to Equation 6:
| Equation 6 |
…where k is the total number of individuals sampled in both communities, ci and cj are the absolute value of the difference in frequency of the ith and jth sequence between the two communities, and dij is the divergence between the two sequences. RaoDIS is conceptually similar to raw FST values in that it accounts for both divergence and relative abundances when comparing diversity between communities.
Double Principal Coordinates Analysis (DPCoA) draws upon the concepts of Rao dissimilarity to cluster communities based on the dissimilarities among the lineages they contain (Pavoine, et al., 2004). In DPCoA, a matrix of species distances is first used to ordinate the species using PCoA. The position of the communities in coordinate space is calculated as the average position of the species that they contain, weighted by relative abundances (See Fig. 5A for an example). The result is that the squared Euclidean distance between the samples is equal to their Rao Dissimilarity. The community relationships can be viewed in the same coordinate space as the phylotype relationships in order to visualize how the phylotypes contribute to the results (Fig. 5A, D). DPCoA was initially used to compare bird communities living in different areas under Mediterranean bioclimates using taxonomic distances, differences in foraging substrates, and differences in morphological traits such as wing length and bill height to measure species dissimilarity (Pavoine, et al., 2004). DPCoA has subsequently been applied in several studies of the human microbial flora including studies of community changes in the human intestine (Fig. 5A, D) (Eckburg, et al., 2005), skin (Paulino, et al., 2006, Gao, et al., 2007), and stomach (Bik, et al., 2006). DPCoA is similar to ordination with FST or weighted UniFrac because they are all quantitative measures (e.g. compare Fig. 5A to 5B), but different from ordination with unweighted UniFrac, a qualitative measure (e.g. compare Fig. 5A to 5C). DPCoA has the advantage over these other methods that phylotypes that contribute to the difference can be directly observed (Fig. 5D). Unfortunately, analysis with DPCoA currently requires some programming in the R statistical language (Eckburg, et al., 2005).
Figure 5.
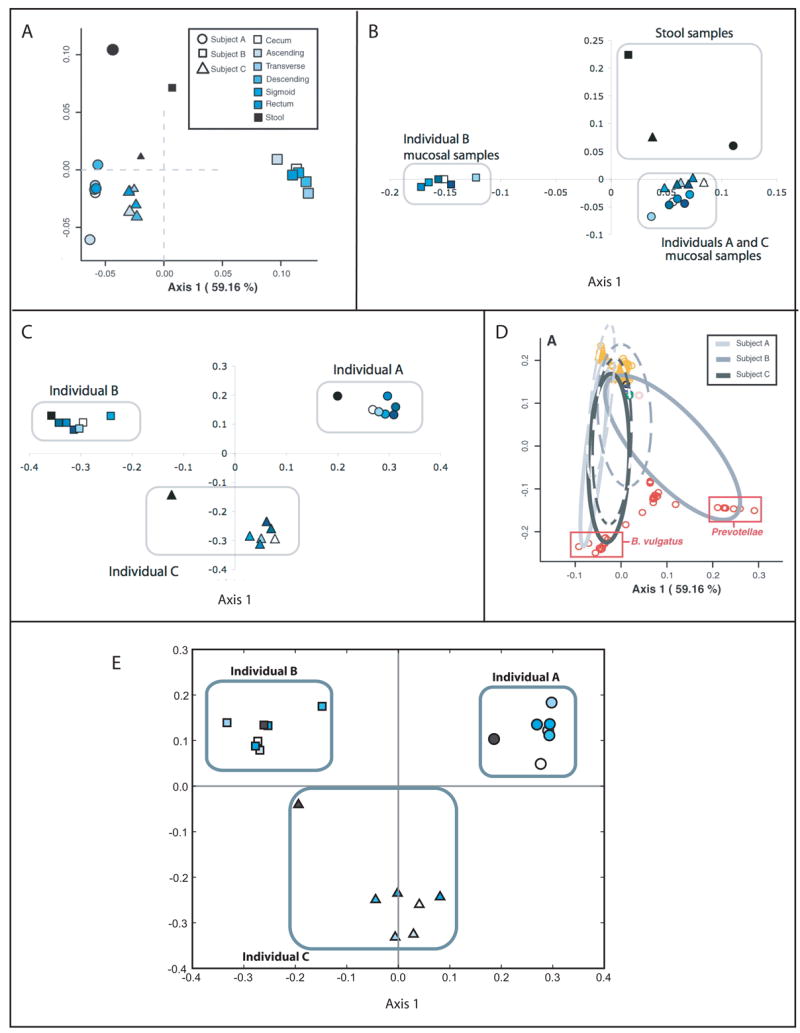
Ordination of human stool and intestinal mucosal samples with DPCoA and UniFrac. (A) Ordination of samples with DPCoA (adapted from (Eckburg, et al., 2005)). Axis 1 separates the mucosal samples of individual B from individuals A and C and Axis 2 separates the stool and mucosal samples. (B) PCoA of weighted UniFrac values (adapted from (Ley, et al., 2005)). The results are almost identical to the DPCoA results from A. (C) PCoA of unweighted UniFrac values (adapted from (Ley, et al., 2005)). Unlike for weighted UniFrac and DPCoA, the stool samples from each individual clusters with the mucosal samples from that individual, indicating that the difference between the stool and mucosal samples is in the relative abundance of lineages rather than which lineages are present. (D) Position of sequences in the same coordinate space used to plot the samples in A. This suggests that the abundance of members of the Prevotellae family in Individual B contributes to the difference with Individuals A and C. (E) PCoA of unweighted UniFrac values calculated from an ARB parsimony insertion tree made with 100 bp sequence regions extending from PCR primer R357 (adapted from (Liu, et al., 2007)). Even with these short sequence reads, UniFrac recaptured the result from the near-full length 16S rRNA sequences.
Taxonomic Similarity
A further β diversity measure is Izsak and Price’s taxonomic similarity (ΔS) (Izsak & Price, 2001). ΔS estimates are derived from the average distance between species from two different communities. Specifically, ΔS measures taxonomic distance between communities (TD) using Equation 7:
| Equation 7 |
…where wiB is the taxonomic distance between species i in community A and all species in community B, wjA is the taxonomic distance between species j in community B and all species in community A, and nA and nB are the number of species in communities A and B respectively. Taxonomic distance between species is 1 for members of the same genus, 2 for family and so on. ΔS is derived from TD with Equation 8, which converts it to a similarity measure rather than a distance and normalizes the raw value to a value between 0 and 1:
| Equation 8 |
L is the number of taxonomic levels used to classify the species. ΔS is a qualitative diversity measure since it only considers presence/absence data.
This measure has not, to our knowledge, been applied to microbial community analysis. Izsak and Price illustrated this measure by comparing the β diversity of echinoderms for 3 regions in the Indo-West Pacific of increasing size. Each of the regions had multiple sampling sites that were further apart for the larger regions. By comparing the average β diversity for pairwise comparisons of sites within each region, Izsak and Price showed that the sites in the smaller regions had more similar echinoderms than in the larger regions.
Although ΔS is exclusively used with taxonomic distances, Webb has implemented a related measure, the comdist function of the Phylocom software package (Webb, et al., 2006) that uses phylogenetic distances. Comdist calculates the distance between two communities, as the mean phylogenetic distance of all possible pairs of taxa in one sample to the taxa in the other. Comdist does not normalize in the same manner as ΔS and it thus is a dissimilarity measure that is not limited to the range 0–1. Comdist returns a distance matrix containing distances between all pairs of communities represented in the tree, so that standard multivariate techniques such as PCoA can be used to compare the samples.
Unlike the other β diversity measures described so far, ΔS is dependent on a species concept, because it determines the degree of similarity of the species that occur in different communities. The successful application of this metric to microbial molecular data, therefore, is dependent on determining what OTU threshold represents a true “species,” which has been difficult for most groups of microorganisms.
Relation of local diversity to regional diversity
So far, we have introduced β diversity measures that can compare two communities, and can compare more than two by making many pairwise comparisons. β diversity measures have also been developed to relate the diversity of a single community to the total diversity in a habitat type or globally. This concept relates to the comparison of within habitat (α) to within landscape (γ) diversity, as introduced by Whittaker (Whittaker, 1972).
Phylogenetic Diversity Gain
The Gain in phylogenetic diversity (G) as described by Faith (Faith, 1992), is related to the α diversity measure PD, but whereas PD tells us about the overall diversity of the lineages within a given community, G is a measurement of which communities contain the most previously unseen diversity (i.e are the most phylogenetically unique). G is defined as the total branch length that a sample adds to a tree that already includes sequences from all of the other samples (Fig. 1B). This measure was developed further by Lewis and Lewis (Lewis & Lewis, 2005), who referred to G as “exclusive molecular phylodiversity”, and used a Bayesian framework to express G and its confidence intervals in terms of the number of nucleotide substitutions that accumulated during the differentiation of each community. Recently, we used G to compare the unique diversity found in different environment types in a global survey of bacterial commmunities (Lozupone & Knight, 2007). By being as comprehensive and representative as possible in the inclusion of 16S sequences from surveyed environments, we were able to determine the types of environments whose exploration was still adding the most branch length to the tree of life. For instance, soils generally added less phylogenetic information per OTU sequenced, in large part because soil is among the more thoroughly sequenced types of environment, and so each new study re-discovered much of the same diversity. In contrast, a hypersaline microbial mat in Guerrero Negro (Ley, et al., 2006), had particularly high G values, indicating that this environment is more distinct relative to the other environments included in this analysis (Lozupone & Knight, 2007).
G measurements have also been used to understand how much unique evolution has occurred in a particular type of environment. For instance, Lewis and Lewis (Lewis & Lewis, 2005) used G to evaluate the diversity of green algae in desert soils, and concluded that a substantial fraction of evolution within the green algae was unique to desert communities. Under sampling is a major issue for G calculations for this application, because using G to estimate the unique evolution in an environment assumes that all of the other environments have been sampled exhaustively. In order to make their initial tree as comprehensive as possible, Lewis and Lewis used BLAST to find the most closely related algal sequences in GenBank. However, this still does not exclude the possibility that the G values were inflated because algae that live in both the desert and other environments had not been sampled. Conversely, if the community being evaluated is undersampled, lineages that are unique to it will be missed, reducing the estimate of G. Assessment of confidence of G estimates to sampling effort needs more exploration.
Measurements of taxonomic distinctness
Taxonomic distinctness is another measure that relates the diversity of a sample to a larger pool, such as comparing diversity in a given habitat to the diversity across a region. Taxonomic distinctness indicates whether the species in a community tend to be closely related (phylogenetically clustered) or distantly related (phylogenetically dispersed), and can thus be used to investigate hypotheses about community structure. For example, phylogenetic clustering could suggest that habitat filtering is important for structuring a community because only similar organisms can live there. Even though HGT and convergent evolution can result in phylogenetically distinct organisms inhabiting the same niche, these processes are not likely to be so widespread that species would not be more phylogenetically clustered than chance expectation if the habitat is filtering out all but the microbes that have particular suites of functions that are rare. Phylogenetic clustering could also indicate biogeographical barriers to dispersion of evolutionary lineages. In contrast, phylogenetic dispersion could suggest that competition restricts all but the most successful species from surviving, and that purifying selection prevents species already in the community from diversifying further.
One measure of taxonomic distinctness is Webb’s Net Relatedness Index (NRI) (Webb, 2000), which is the average taxonomic distance between species in a community. This measure is related to θ, but NRI disregards the species abundance information. For example, to apply NRI to 16S rRNA survey data, species-level OTUs should be chosen, and only one member of each OTU should be considered when calculating k, pi and pj in Equation 1. NRI is essentially the same as Clark and Warwick’s average taxonomic distinctness index (Δ+) (Clarke & Warwick, 1998) and Izsak and Papp’s J index (Izsak & Papp, 2000), differing only in that it is normalized for comparison across trees by dividing the raw value by the maximum value that could be obtained for the same number of taxa in the same phylogeny (Webb, 2000).
A related measure is the Nearest Taxon Index (NTI), which is the average distance between each species and its closest relative in the tree. Like NRI, NTI is normalized by division by the maximum possible value for the specified tree and number of species (Webb, 2000, Webb, et al., 2002, Webb, et al., 2006). NTI focuses on clustering at lower taxonomic levels and is less sensitive to higher level phylogenetic structure than NRI (Webb, 2000). Although originally described using topological distances between species, both NTI and NRI can also use phylogenetic distance measurements (Horner-Devine & Bohannan, 2006), as is implemented in Phylocom (Webb, et al., 2006). NRI and NTI do not account for the relative abundance of species and are not correlated with species richness. As a result, they are relatively robust to incomplete sampling (Clarke & Warwick, 1998).
Monte Carlo simulations can be used to test whether communities are significantly clustered or dispersed phylogenetically, using NRI or NTI. In these simulations, the community assignments in a reference phylogenetic tree are randomized, and the percentage of trials in which the true tree has a more extreme value than the randomized trees is recorded (Clarke & Warwick, 1998, Webb, 2000). The reference tree should include all of the species that are relevant for the comparison, such as all the sequences that are usually found in a given region or habitat type. For example, Webb applied this analysis to trees in the Bornean rain forest to determine whether the phylogenetic distribution of species found in individual plots differed significantly from what would be expected if species were evenly distributed throughout the region. He showed that the species in individual plots were more phylogenetically related than expected by chance, indicating that the co-occurrence of similar species due to variation in habitat among plots played a greater role in structuring the communities than competitive exclusion between similar species (Webb, 2000).
Clarke and Warwick also described a jackknifing technique for computing the variation in Δ+ (which is essentially equivalent to NRI) by randomly subsampling species without replacement in many replicate trials (Clarke & Warwick, 1998). This procedure provides a statistical test to determine whether phylogenetic distinctness differs significantly between communities. This technique has been most often used in studies of the biodiversity of marine macroorganisms, including benthic nematodes, corals, and coastal fishes (Hall & Greenstreet, 1998, Warwick & Clarke, 1998, Rogers, et al., 1999, Brown, et al., 2002). These studies generally showed that the taxonomic distinctness in degraded locations was significantly lower than in relatively pristine locations.
Although NTI and NRI have been more often applied in the study of macroorganisms than microbes, Horner-Devine and Bohannan (Horner-Devine & Bohannan, 2006) successfully applied these measures to a meta-analysis of four studies of microbial populations, using 16S rRNA data to measure phylogenetic distances. They found that microbial populations were typically more phylogenetically clustered than expected by chance, suggesting potentially an important role for habitat filtering. Horner-Devine and Bohannan also demonstrated how NTI and NRI values could be correlated with environmental variables. For instance, they found a negative correlation between the NRI of β-proteobacteria communities and primary productivity in aquatic mesocosms, indicating that increased stress in a low productivity environment may increase habitat filtering (Horner-Devine & Bohannan, 2006). NTI and NRI calculations and significance tests can be run using the Phylocom software, which is available for download at http://www.phylodiversity.net/phylocom/.
Although divergence-based diversity measures are typically less sensitive to species definitions than are traditional diversity measurements, NRI and NTI are still dependent on a species concept, because they determine whether similar species tend to coexist in a given community, or competitively exclude one another. The successful application of this metric to microbial molecular data, therefore, is dependent on determining what OTU threshold represents a true “species.”
Concluding Remarks
The increase in speed and decrease in cost of obtaining 16S rRNA sequences from microbial community surveys provides a unique opportunity to make fundamental discoveries about microbial ecology and evolution. The diversity measures described here allow many different hypotheses about community structure to be tested using the sequence information that is generated in molecular microbial community surveys. Although no single number can describe all aspects of diversity, the application of these measures in conjunction with commonly used species based diversity measures can elucidate different aspects of diversity.
Divergence-based diversity measures have the potential to elucidate fundamental ecological properties of microbial communities beyond what has been possible with species-based measures alone. For instance, one way to evaluate whether bacteria are spatially distributed is to evaluate a species area curve; a flat curve indicates that an increase in area does not cause in an increase in species that are endemic to particular sights, and that the bacteria are for the most part cosmopolitan (Noguez, et al., 2005). For a flat-species area curve, the α diversity is equal across spatial scales and the β diversity for the comparison of any two sub-plot would be 0, indicating that the same species are present. A divergence-based analog of this analysis would be to use PD measurements to plot a branch length-area curve, to evaluate whether an increase in area increases the total amount of observed phylogenetic lineages. A flat branch length area curve would have the same predictions for divergence-based α and β diversity values, except that it would indicate that the same phylogenetic lineages are present rather than the same species. This technique has the advantage that it is not dependent on a poorly defined species concept and accounts simultaneously for all levels of divergence between taxa.
By comparing the available divergence-based diversity measures to the sequence-based measures, it is evident some of the possible divergence-based diversity measurements remain to be developed. For instance, a whole suite of tools has been developed for the estimation of species richness, including parametric estimators, non-parametric estimators such as ACE and Chao 1, and extrapolation of species accumulation curves (Magurran, 2004). In contrast, very little work has been done to determine the best way to estimate the total phylogenetic richness from a limited sample.
Despite the apparent importance of abundance information for detecting certain types of community changes, limitations in sequence deposition currently make quantitative measures difficult to apply on a global scale. In particular, many large studies deposit only new sequences or omit abundance information, making quantitative metrics impossible to apply correctly. Improvements in community standards for data deposition, especially enforcement of inclusion of abundance information and metadata about the sampling in machine-readable form, will be essential for full exploitation of the community survey data now being obtained at great expense.
The diversity measures described here vary in their degree of accessibility to microbiologists who do not have any programming expertise. Although the majority are now accessible, such as through the UniFrac web interface or the PhyloCom software package, the application of others is still limited. For instance, DPCoA is only implemented as a function in the R statistical package. Although θ and FST values can be calculated with nucleotide distance values using Arlequin (Schneider, et al., 1997), there is no convenient implementation for using phylogenetic distances as may be desirable. Also, although raw PD values can be calculated using Phylocom, PD rarefaction analysis must be performed manually. One of our goals for the UniFrac web interface is to allow direct comparisons among more of these diversity measures by providing them all within a single user-friendly framework.
It is clear that traditional methods of comparing communities, such as using BLAST to find relatives of each of the sequences in a community sample, cannot scale to the demands of cheaper and faster sequencing technologies. Fortunately, the techniques described here provide convenient methods that have been used successfully to compare hundreds of communities and tens of thousands of unique sequences (Lozupone & Knight, 2007). In addition, we recently have shown that UniFrac clustering patterns from near full length sequences can be recaptured using simulated 100–200 bp reads typical of pyrosequencing if the appropriate region of the rRNA molecule is selected (See Fig. 5E) (Liu, et al., 2007), indicating that divergence-based diversity measures will become even more important with the recent advances in sequencing technology. That some errors in the phylogeny will not dramatically impact the results, indicates that the sequence length requirements for divergence based diversity analyses are not as stringent as they are for robust phylogenetic reconstruction. Ultimately, even a phylogeny with some errors is better than the star topology that is inferred with species-based diversity measures, and the benefit from the dramatic increase in the number of short sequences that can be sampled with pyrosequencing appears to outweigh the disadvantage of phylogenetic errors from short sequence reads (Liu, et al., 2007). We expect that the availability of a database of annotated community samples will provide an increasingly valuable framework for understanding diversity in new environments, and for directing new sequencing efforts to uncover new and divergent lineages.
Acknowledgments
We thank Jesse Zaneveld, Eva Top, and two anonymous reviewers for insightful comments on this manuscript. Zongzhi Liu assisted with Figure 5E. C.L. was supported in part by a developmental grant to R.K. by the Colorado Center for AIDS Research, and by NIH/CU Cell Signaling Training Program T32 GM08759.
References
- Bass-Becking LGM. Geobiologie of Inleiding tot de Milieukunde. 1934. W.p van Stockum & Zoon N.V. [Google Scholar]
- Beman JM, Francis CA. Diversity of ammonia-oxidizing archaea and bacteria in the sediments of a hypernutrified subtropical estuary: Bahia del Tobari, Mexico. Appl Environ Microbiol. 2006;72:7767–7777. doi: 10.1128/AEM.00946-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bik EM, Eckburg PB, Gill SR, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binladen J, Gilbert MT, Bollback JP, Panitz F, Bendixen C, Nielsen R, Willerslev E. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS ONE. 2007;2:e197. doi: 10.1371/journal.pone.0000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohannan BJ, Hughes J. New approaches to analyzing microbial biodiversity data. Curr Opin Microbiol. 2003;6:282–287. doi: 10.1016/s1369-5274(03)00055-9. [DOI] [PubMed] [Google Scholar]
- Breitbart M, Felts B, Kelley S, Mahaffy JM, Nulton J, Salamon P, Rohwer F. Diversity and population structure of a near-shore marine-sediment viral community. Proc Biol Sci. 2004;271:565–574. doi: 10.1098/rspb.2003.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, Salamon P, Andresen B, et al. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci USA. 2002;99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown B, Clarke K, Warwick R. Serial patterns of biodiversity change in corals across shallow reef flats in Ko Phuket, Thailand, due to the effects of local (sedimentation) and regional (climactic) perturbations. Mar Biol. 2002;141:21–29. [Google Scholar]
- Chao A. Non-parametric estimation of the number of classes in a population. Scand J Stat. 1984;11:265–270. [Google Scholar]
- Chazdon RL, Colwell RK, Denslow JS, Guariguata MR. Statistical methods for estimating species richness of woody regeneration in primary and secondary rain forests of northeastern Costa Rica. In: Dallmeier F, Comiskey JA, editors. Forest biodiversity research, monitoring and modeling: conceptual background and old world case studies. Parthenon Publishing; Paris: 1998. pp. 285–309. [Google Scholar]
- Clarke K, Warwick R. A taxonomic distinctness index and its statistical properties. J Appl Ecol. 1998;35:523–531. [Google Scholar]
- Cohan FM, Perry EB. A systematics for discovering the fundamental units of bacterial diversity. Curr Biol. 2007;17:R373–386. doi: 10.1016/j.cub.2007.03.032. [DOI] [PubMed] [Google Scholar]
- Cohen FM. What are bacterial species? Annu Rev Microbiol. 2002;56:457–487. doi: 10.1146/annurev.micro.56.012302.160634. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Keller K, et al. NAST: A multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 2006;34:394–399. doi: 10.1093/nar/gkl244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz PI, Chalmers NI, Rickard AH, Kong C, Milburn CL, Palmer RJ, Jr, Kolenbrander PE. Molecular characterization of subject-specific oral microflora during initial colonization of enamel. Appl Environ Microbiol. 2006;72:2837–2848. doi: 10.1128/AEM.72.4.2837-2848.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunfield KE, King GM. Molecular analysis of carbon monoxide-oxidizing bacteria associated with recent Hawaiian volcanic deposits. Appl Environ Microbiol. 2004;70:4242–4248. doi: 10.1128/AEM.70.7.4242-4248.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61:1–10. [Google Scholar]
- Faith DP. Using generalized dissimilarity modelling to analyse and predict patterns of beta diversity in regional biodiversity assessment. Diversity Distrib. 2007;13:252–264. [Google Scholar]
- Felsenstein J. Inferring Phylogenies. Sinauer Associates, Inc; Sunderland, Massachusetts: 2004. [Google Scholar]
- Fox GE, Wisotzkey JD, Jurtshuk P., Jr How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Bacteriol. 1992;42:166–170. doi: 10.1099/00207713-42-1-166. [DOI] [PubMed] [Google Scholar]
- Fraune S, Bosch TC. Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc Natl Acad Sci USA. 2007;104:13146–13151. doi: 10.1073/pnas.0703375104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Tseng CH, Pei Z, Blaser MJ. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci USA. 2007;104:2927–2932. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Cohan FM, Lawrence JG, et al. Opinion: Re-evaluating prokaryotic species. Nat Rev Microbiol. 2005;3:733–739. doi: 10.1038/nrmicro1236. [DOI] [PubMed] [Google Scholar]
- Hall S, Greenstreet S. Taxonomic distinctness and diversity measures: responses in marine fish communities. Marine Ecology Progress Series. 1998;166:227–229. [Google Scholar]
- Henriksen JR. webLIBSHUFF. 2004 http://libshuff.mib.uga.edu/
- Hill TCJ, Walsh KA, Harris JA, Moffett BF. Using ecological diversity measures with bacterial communities. FEMS Microbiol Ecol. 2003;43:1–11. doi: 10.1111/j.1574-6941.2003.tb01040.x. [DOI] [PubMed] [Google Scholar]
- Horner-Devine MC, Bohannan BJ. Phylogenetic clustering and overdispersion in bacterial communities. Ecology. 2006;87:S100–108. doi: 10.1890/0012-9658(2006)87[100:pcaoib]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Huber JA, Welch DB, Morrison HG, Huse SM, Neal PR, Butterfield DA, Sogin ML. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- Hugenholtz P. Exploring prokaryotic diversity in the genomic era. Genome Biol. 2002;3:1–8. doi: 10.1186/gb-2002-3-2-reviews0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJ. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol. 2001;67:4399–4406. doi: 10.1128/AEM.67.10.4399-4406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izsak C, Price ARG. Measuring β-diversity using a taxonomic similarity index, and its relation to spatial scale. Marine Ecology Progress Series. 2001;215:69–77. [Google Scholar]
- Izsak J, Papp L. Application of the quadratic entropy indices for diversity studies of drosophilid assemblages. Environ Ecol Stat. 1995;2:213–224. [Google Scholar]
- Izsak J, Papp L. A link between ecological diversity indices and measures of biodiversity. Ecol Model. 2000;130:151–156. [Google Scholar]
- Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci USA. 1999;96:3801–3806. doi: 10.1073/pnas.96.7.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RT, Martin AP. Testing for differentiation of microbial communities using phylogenetic methods: accounting for uncertainty of phylogenetic inference and character state mapping. Microb Ecol. 2006;52:408–417. doi: 10.1007/s00248-006-9002-7. [DOI] [PubMed] [Google Scholar]
- Kanagawa T. Bias and artifacts in multitemplate polymerase chain reactions (PCR) J Biosci Bioeng. 2003;96:317–323. doi: 10.1016/S1389-1723(03)90130-7. [DOI] [PubMed] [Google Scholar]
- Koleff P, Gaston KJ, Lennon JL. Measuring beta diversity for presence-absence data. J Anim Ecol. 2003;72:367–382. [Google Scholar]
- Lamarche J, Hamelin RC. No evidence of an impact on the rhizosphere diazotroph community by the expression of Bacillus thuringiensis Cry1Ab toxin by Bt white spruce. Appl Environ Microbiol. 2007;73:6577–6583. doi: 10.1128/AEM.00812-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LA, Lewis PO. Unearthing the molecular phylodiversity of desert soil green algae (Chlorophyta) Syst Biol. 2005;54:936–947. doi: 10.1080/10635150500354852. [DOI] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Harris JK, Wilcox J, et al. Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl Environ Microbiol. 2006;72:3685–3695. doi: 10.1128/AEM.72.5.3685-3695.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson DA, Schmidt SK. Seasonal changes in an alpine soil bacterial community in the Colorado Rocky Mountains. Appl Environ Microbiol. 2004;70:2867–2879. doi: 10.1128/AEM.70.5.2867-2879.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007;35:e120. doi: 10.1093/nar/gkm541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Hamady M, Knight R. UniFrac--an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics. 2006;7:371. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Knight R. Global patterns in bacterial diversity. Proc Natl Acad Sci U S A. 2007;104:11436–11440. doi: 10.1073/pnas.0611525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and Qualitative {beta} Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Appl Environ Microbiol. 2007;73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig W, Strunk O, Westram R, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison WP, Slatkin M. Null Models for the number of evolutionary steps in a character on a phylogenetic tree. Evolution. 1991;45:1184–1197. doi: 10.1111/j.1558-5646.1991.tb04385.x. [DOI] [PubMed] [Google Scholar]
- Magurran AE. Measuring Biological Diversity. Blackwell; Oxford: 2004. [DOI] [PubMed] [Google Scholar]
- Maidak BL, Cole JR, Lilburn TG, et al. The RDP-II (Ribosomal Database Project) Nucleic Acids Res. 2001;29:173–174. doi: 10.1093/nar/29.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AP. Phylogenetic approaches for describing and comparing the diversity of microbial communities. Appl Environ Microbiol. 2002;68:3673–3682. doi: 10.1128/AEM.68.8.3673-3682.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur J, Bizzoco RW, Ellis DG, Lipson DA, Poole AW, Levine R, Kelley ST. Effects of abiotic factors on the phylogenetic diversity of bacterial communities in acidic thermal springs. Appl Environ Microbiol. 2007;73:2612–2623. doi: 10.1128/AEM.02567-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr E. Systematics and the origin of species from the viewpoint of a zoologist. Columbia Univ. Press; New York: 1944. [Google Scholar]
- Nakamura Y, Itoh T, Matsuda H, Gojobori T. Biased biological functions of horizontally transferred genes in prokaryotic genomes. Nat Genet. 2004;36:760–766. doi: 10.1038/ng1381. [DOI] [PubMed] [Google Scholar]
- Nanba K, King GM, Dunfield K. Analysis of facultative lithotroph distribution and diversity on volcanic deposits by use of the large subunit of ribulose 1,5-bisphosphate carboxylase/oxygenase. Appl Environ Microbiol. 2004;70:2245–2253. doi: 10.1128/AEM.70.4.2245-2253.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguez AM, Arita HT, Escalante AE, Forney LJ, Garcia-Oliva F, Souza V. Microbial macroecology: highly structured prokaryotic soil assemblages in a tropical deciduous forest. Global Ecol Biogeogr. 2005;14:241–248. [Google Scholar]
- Oline DK. Phylogenetic comparisons of bacterial communities from serpentine and nonserpentine soils. Appl Environ Microbiol. 2006;72:6965–6971. doi: 10.1128/AEM.00690-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulino LC, Tseng CH, Strober BE, Blaser MJ. Molecular analysis of fungal microbiota in samples from healthy human skin and psoriatic lesions. J Clin Microbiol. 2006;44:2933–2941. doi: 10.1128/JCM.00785-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavoine S, Dufour AB, Chessel D. From dissimilarities among species to dissimilarities among communities: a double principal coordinate analysis. J Theor Biol. 2004;228:523–537. doi: 10.1016/j.jtbi.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Pettitt AN. Encyclopedia of statistical sciences. Wiley-Interscience; New York, N.Y: 1982. Cramer-von Mises statistic; pp. 220–221. [Google Scholar]
- Prosser JI, Bohannan BJ, Curtis TP, et al. The role of ecological theory in microbial ecology. Nat Rev Microbiol. 2007;5:384–392. doi: 10.1038/nrmicro1643. [DOI] [PubMed] [Google Scholar]
- Rao C. Diversity and dissimilarity coefficients: a unified approach. Theor Pop Biol. 1982;21:24–43. [Google Scholar]
- Rawls JF, Samuel BS, Gordon JI. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc Natl Acad Sci USA. 2004;101:4596–4601. doi: 10.1073/pnas.0400706101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers S, Clarke K, Reynolds J. The taxonomic distinctness of coastal bottom-dwelling fish communities of the North-ewast Atlantic. J Anim Ecol. 1999;68:769–782. [Google Scholar]
- Santoro AE, Boehm AB, Francis CA. Denitrifier community composition along a nitrate and salinity gradient in a coastal aquifer. Appl Environ Microbiol. 2006;72:2102–2109. doi: 10.1128/AEM.72.3.2102-2109.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt CW, Martin AP, Lipson DA, Schmidt SK. Seasonal dynamics of previously unknown fungal lineages in tundra soils. Science. 2003;301:1359–1361. doi: 10.1126/science.1086940. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Handelsman J. Introducing TreeClimber, a test to compare microbial community structures. Appl Environ Microbiol. 2006;72:2379–2384. doi: 10.1128/AEM.72.4.2379-2384.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Larget BR, Handelsman J. Integration of microbial ecology and statistics: a test to compare gene libraries. Appl Environ Microbiol. 2004;70:5485–5492. doi: 10.1128/AEM.70.9.5485-5492.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Delalibera I, Handelsman J, Raffa KF. Bacteria associated with the guts of two wood-boring beetles: Anoplophora glabripennis and Saperda vestita (Cerambycidae) Physiol Ecol. 2006;35:625–629. [Google Scholar]
- Schneider S, Kueffer J, Roessli D, Excoffier L. Arlequin ver. 1.1; a software for population genetic data analysis Genetics and Biometry Lab. University of Geneva; 1997. [Google Scholar]
- Shannon CE, Weaver W. The mathematical theory of communication. University of Illinois Press; Urbana, Il: 1949. [Google Scholar]
- Shimatani K. On the measurement of species diversity incorporating species differences. Oikos. 2001;93:135–147. [Google Scholar]
- Simpson EH. Measurement of diversity. Nature. 1949;163:688. [Google Scholar]
- Singleton DR, Furlong MA, Rathbun SL, Whitman WB. Quantitative comparisons of 16S rRNA gene sequence libraries from environmental samples. Appl Environ Microbiol. 2001;67:4374–4376. doi: 10.1128/AEM.67.9.4374-4376.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soberon J, Llorente J. The use of species accumulation functions for the prediction of species richness. Conserv Biol. 1993;7:480–488. [Google Scholar]
- Spear JR, Walker JJ, McCollom TM, Pace NR. Hydrogen and bioenergetics in the Yellowstone geothermal ecosystem. Proc Natl Acad Sci USA. 2005;102:2555–2560. doi: 10.1073/pnas.0409574102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stach JE, Maldonado LA, Masson DG, Ward AC, Goodfellow M, Bull AT. Statistical approaches for estimating actinobacterial diversity in marine sediments. Appl Environ Microbiol. 2003;69:6189–6200. doi: 10.1128/AEM.69.10.6189-6200.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackebrandt E, Goebal BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int Syst Bacteriol. 1994;44:846–849. [Google Scholar]
- Top EM, Springael D. The role of mobile genetic elements in bacterial adaptation to xenobiotic organic compounds. Curr Opin Biotechnol. 2003;14:262–269. doi: 10.1016/s0958-1669(03)00066-1. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Vane-Wright R, Humphries C, Williams P. What to protect: systematics and the agony of choice. Biol Conserv. 1991;55:235–254. [Google Scholar]
- Virolainen K, Suomi T, Suhonen J, Kuitunen M. Conservation of vascular plants in single large and several small mires: species richness, rarity, and taxonomic diversity. J Appl Ecol. 1998;35:700–707. [Google Scholar]
- von Wintzingerode F, Gobel UB, Stackebrandt E. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev. 1997;21:213–229. doi: 10.1111/j.1574-6976.1997.tb00351.x. [DOI] [PubMed] [Google Scholar]
- Walker JJ, Pace NR. Phylogenetic composition of Rocky Mountain endolithic microbial ecosystems. Appl Environ Microbiol. 2007;73:3497–3504. doi: 10.1128/AEM.02656-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallenstein MD, McMahon S, Schimel J. Bacterial and fungal community structure in Arctic tundra tussock and shrub soils. FEMS Microbiol Ecol. 2007;59:428–435. doi: 10.1111/j.1574-6941.2006.00260.x. [DOI] [PubMed] [Google Scholar]
- Walsh DA, Papke RT, Doolittle WF. Archaeal diversity along a soil salinity gradient prone to disturbance. Environ Microbiol. 2005;7:1655–1666. doi: 10.1111/j.1462-2920.2005.00864.x. [DOI] [PubMed] [Google Scholar]
- Warwick R, Clarke K. Taxonomic distinctness and environmental assesment. J Appl Ecol. 1998;35:532–543. [Google Scholar]
- Warwick RM, Clarke KR. New ‘biodiversity’ measures reveal a decrease in taxonomic distinctness with increasing stress. Marine Ecology Progress Series. 1995;129:301–305. [Google Scholar]
- Webb C, Ackerly D, Kembel S. Phylocom: software for the analysis of community phylogenetic structure and character evolution Version 3.40. 2006. [DOI] [PubMed] [Google Scholar]
- Webb CO. Exploring the phylogenetic structure of ecological communities. Am Nat. 2000;156:145–155. doi: 10.1086/303378. [DOI] [PubMed] [Google Scholar]
- Webb CO, Ackerley DD, McPeek MA, Donoghue MJ. Phylogenies and Community Ecology. Annu Rev Ecol Syst. 2002;33:475–505. [Google Scholar]
- Weider LJ, Elser JJ, Crease TJ, Mateos M, Cotner JB, Markow TA. The functional significance of ribosomal (r)DNA variation: impacts on the evolutionary ecology of organisms. Annu Rev Ecol Evol Syst. 2005;36:219–242. [Google Scholar]
- Whittaker RH. Vegetation of the Siskiyou Mountains, Oregon and California. Ecol Monogr. 1960;30:279–338. [Google Scholar]
- Whittaker RH. Evolution and measurement of species diversity. Taxon. 1972;21:213–251. [Google Scholar]