

Abstract
Mutations in the gene encoding the transcriptional repressor methyl-CpG binding protein 2 (MeCP2) cause the neurodevelopmental disorder Rett syndrome. Loss of function as well as increased dosage of MECP2 gene cause a host of neuropsychiatric disorders. To explore the molecular mechanism(s) underlying these disorders, we examined gene expression patterns in the hypothalamus of mice that either lack or overexpress MeCP2. In both models, MeCP2 dysfunction induced changes in the expression levels of thousands of genes, but surprisingly the majority of genes (~85%) appeared to be activated by MeCP2. We selected six genes and confirmed that MeCP2 binds to their promoters. Furthermore, we showed that MeCP2 associates with the transcriptional activator CREB1 at the promoter of an activated target but not a repressed target. These studies suggest that MeCP2 regulates the expression of a wide range of genes in the hypothalamus and that it can function as both an activator and repressor of transcription.
Rett syndrome (RTT, MIM 312750) is a progressive neurodevelopmental disorder caused by mutations in the X-linked gene encoding methyl-CpG binding protein 2 (MeCP2) (1). RTT patients appear to develop normally up to the first year of age after which they lose any acquired speech and replace purposeful hand use with stereotypies. MECP2 mutations also result in a host of neurobehavioral abnormalities, ranging from mild learning disabilities to autism, X-linked mental retardation, and infantile encephalopathy. Interestingly, an increase in MECP2 dosage is equally detrimental to the nervous system: duplications spanning the MECP2 locus cause features overlapping with those of RTT and related neurological disorders (2).
MeCP2 aberrations in mice also result in a neurological phenotype. Mecp2-null mice appear normal until 6 weeks (wks) of age when they develop severe neurological dysfunction resulting in premature death by 12 wks of age (3). Mice that overexpress MECP2 under the control of its endogenous promoter (MECP2-Tg) display neurological features similar to the human MECP2 duplication syndrome (4). MeCP2 is believed to function as a transcriptional repressor by binding to methylated CpG dinucleotides and recruiting corepressors and chromatin remodeling proteins (5, 6). Loss of MeCP2 and doubling of MECP2 dosage in mice have opposing effects on excitatory synapse numbers (7), suggesting that similar target genes may be affected in the two mouse models, but in opposite directions. We reasoned that analysis of gene expression profiles in MECP2-Tg and Mecp2-null mice might shed light on the molecular mechanism underlying the MECP2 duplication syndrome, reveal whether this disorder shares downstream molecular pathways with RTT, and possibly distinguish primary versus secondary gene targets of MeCP2. If the MECP2 duplication syndrome is caused by loss or partial loss of normal MeCP2 function (due to sequestration of MeCP2 binding partners because of excess MeCP2), then the gene expression profiles are expected to go in the same direction. However, if the mechanism is due to gain of function, owing to increased MeCP2 activity, then the same primary target genes would be misregulated in opposite directions in MECP2-Tg versus Mecp2-null mice, and the secondary target genes would be specific to each mouse model.
Previous transcriptional profiling studies comparing brain tissue from Mecp2-null and wildtype (WT) mice revealed only subtle differences in gene expression (8). We hypothesized that critical disease-related alterations in gene expression might occur only in specific brain regions or neurons, and therefore would be masked in analyses of whole brain tissue. To circumvent this problem, we investigated the hypothalamus because a number of RTT phenotypes (anxiety, growth deceleration, sleep-wake rhythm disturbance, and autonomic abnormalities) could be attributed to hypothalamic dysfunction (9). We performed microarray analysis using hypothalamic RNA from four Mecp2-null males, four MECP2-Tg males, and their respective WT littermates at 6 wks of age using the Affymetrix Mouse Exon 1.0 ST microarray (10, 11). Given that MeCP2 functions as a transcriptional repressor in vitro, we were surprised to find that 2,184 out of the 2,582 genes misregulated in both mouse models (~85%) were upregulated in the transgenic hypothalami and downregulated in the Mecp2-null hypothalami, suggesting that many of these genes are likely activated by MeCP2. In contrast, only 377 genes were downregulated in the transgenic hypothalami and upregulated in the Mecp2-null hypothalami, suggesting that these are normally repressed by MeCP2 (False discovery rate, FDR-adjusted p-value < 0.05) (Fig. 1A) (tables S1 and S2). The magnitude of the changes in expression levels for both activated and repressed genes was moderate (less than 5-fold). Moreover, only nine genes were upregulated in both mouse models (table S3), and twelve genes were downregulated in both models (table S4) (FDR-adjusted p-value < 0.05). We identified 1,187 genes whose expression was altered only in the transgenic hypothalami, and 369 genes whose expression was altered only in the null hypothalami (tables S5 and S6) (FDR-adjusted p-value < 0.05). Since these changes are specific to each mouse model, they likely represent secondary changes due either to altered expression of primary gene targets of MeCP2 or to disease processes. Interestingly, gene ontology analysis (Fig. S1) revealed that the genes activated and repressed by MeCP2 fall into distinct categories in terms of biological function. For example, neuropeptides were enriched in the list of activated MeCP2 targets, whereas olfactory receptors were enriched in the list of repressed targets. Overall, the genes encoding G-protein coupled receptors (GPCRs) were the most significantly affected by MeCP2 levels; this is expected since most hypothalamic neuropeptide receptors are GPCRs. In addition, we found that the 5kb upstream promoter regions of genes that are upregulated by MeCP2 were significantly enriched in CpG islands, while the downregulated genes were not (Fig. 1B).
Fig. 1.
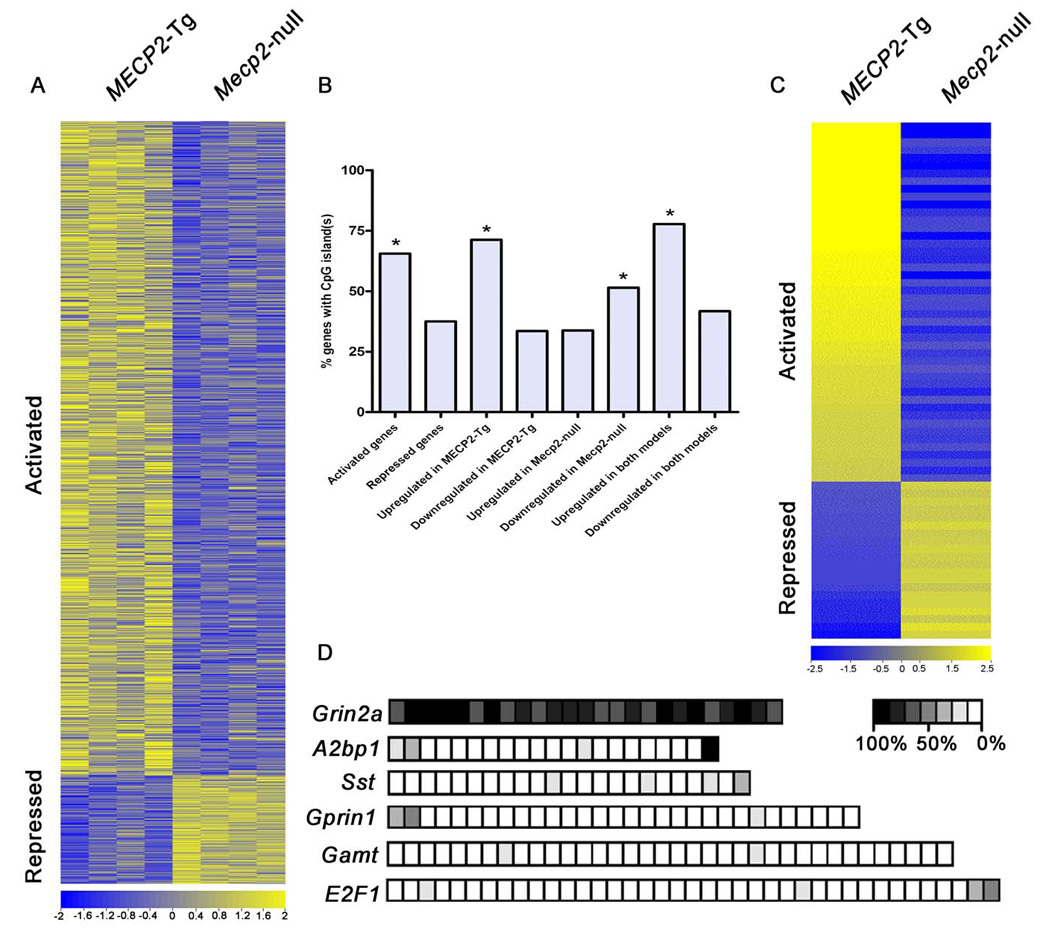
Significant gene expression changes in hypothalami of MeCP2 mouse models. (A) Heatmap showing hypothalamic gene expression profiles in MECP2-Tg and Mecp2-null mice. Yellow and blue colors indicate increased and decreased expression, respectively, relative to WT. Each column represents one RNA sample from each genotype, and each row represents one gene. Expression levels are depicted according to the color scale at the bottom. 2,184 genes are activated in the presence of MeCP2, while 377 genes are repressed (FDR-adjusted p-value < 0.05). (B) Promoter regions of genes upregulated by MeCP2 contain significantly more CpG islands compared to those of genes downregulated by MeCP2 (* p≤0.006). (C) Validation of expression changes for 66 genes by quantitative real-time RT-PCR analysis. Gene expression levels from the microarray were validated in four MECP2-Tg males and four Mecp2-null males. Data are plotted as fold up- (yellow) or downregulation (blue) over WT (p<0.05, t test). Each row represents a single gene, and each column represents data for four samples from each genotype. Levels are depicted according to the color scale at the bottom. The complete list of validated genes is available in Supporting Online Material (12) (table S7). (D) Bisulfite sequencing revealed that CpG sites in the promoters of the repressed target genes Grin2a and A2bp1 are heavily methylated, while those of activated targets Sst, Gprin1, Gamt, and E2F1 are not. CpG sites are depicted as squares, with the percent methylation presented according to the scale. The data were generated from three independent animals and for each promoter ~10 clones were sequenced.
To validate the gene expression changes, we performed quantitative real-time RTPCR (reverse transcription - polymerase chain reaction) using hypothalamic RNA samples from an independent set of Mecp2-null males, MECP2-Tg males, and their respective WT littermates. We confirmed alterations in the expression of 66 genes representative of the most significantly altered gene families; of these 46 were upregulated by MeCP2 and 20 were downregulated (p < 0.05) (Fig. 1C). The complete list of validated genes is available in Supporting Online Material (12) (table S7). In order to check the CpG island methylation status of some of the validated genes, we performed bisulfite sequencing of hypothalamic DNA from WT males. We found that CpG sites in promoters of activated MeCP2 gene targets were not heavily methylated compared to those of repressed targets (Fig. 1D), consistent with the fact that in general CpG islands are unmethylated. To determine whether both activated and repressed genes are altered because they are direct MeCP2 targets, we performed chromatin immunoprecipitation (ChIP) with anti-MeCP2 antibody using hypothalamic tissue from Mecp2-null males, MECP2-Tg males, and their respective WT littermates. We used quantitative real-time PCR to assess MeCP2 binding to the promoter regions of six candidate target genes that were either activated or repressed by MeCP2. This in vivo analysis revealed that MeCP2 bound to the promoter regions of genes encoding somatostatin (Sst), opioid receptor kappa 1 (Oprk1), guanidinoacetate methyltransferase (Gamt), G protein-regulated inducer of neurite outgrowth 1 (Gprin1) (all activated by MeCP2), myocyte enhancer factor 2C (Mef2c), and ataxin 2 binding protein 1 (A2bp1) (both repressed by MeCP2). Importantly, consistent with the microarray data, MeCP2 binding was significantly enhanced in the MECP2-Tg samples compared to WT (p ≤ 0.03) (Fig. 2). To confirm that the binding of MeCP2 to its candidate primary targets (genes inversely regulated in MECP2-Tg and Mecp2-null mice) is specific, we tested MeCP2 binding to the promoter region of the gene encoding cyclin-dependent kinase-like 4 (Cdkl4), one of the genes downregulated in both mouse models (table S4). We chose this gene because the GC content of its promoter region is similar to that of the confirmed target genes. We did not detect MeCP2 at the promoter of Cdkl4 as would be predicted due to its similar regulation in both mouse models (Fig. S2).
Fig. 2.
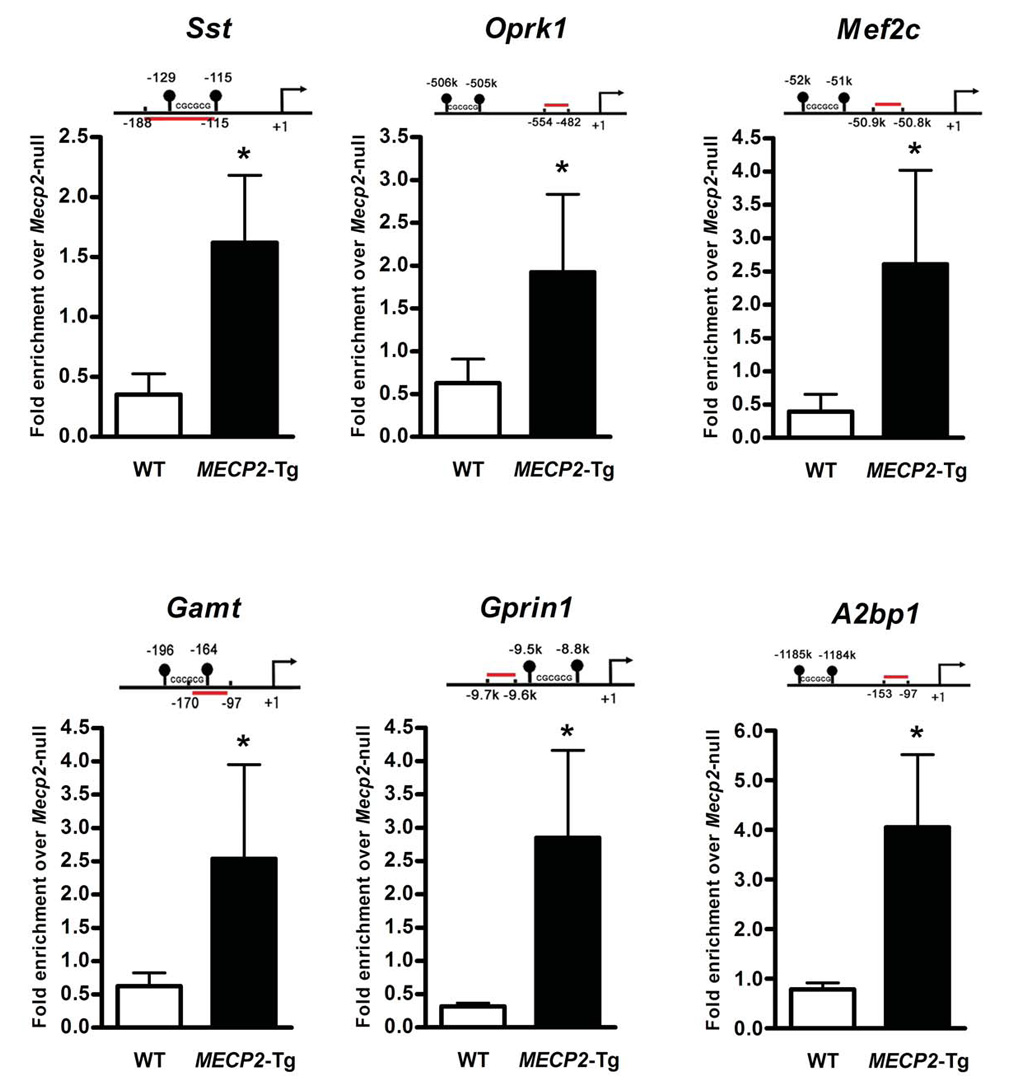
MeCP2 binds to the promoter region of six target genes. ChIP with anti-MeCP2 antibody shows that MeCP2 binds to the promoter regions of activated targets Sst, Oprk1, Gamt, and Gprin1, and repressed targets Mef2c and A2bp1. The red line indicates the location of the probe and primers (relative to the start site (+1), in bps or in kb, k) and the closest CpG islands in the promoter region are depicted. Quantitative real-time PCR values were normalized to the input and plotted as fold enrichment over Mecp2-null (N ≥ 3, * p≤0.03, t test).
Given that gain of MeCP2 causes significantly more activation rather than repression, whereas the loss of MeCP2 causes the reverse, we explored whether MeCP2 associates with coactivators in vivo. We immunopurified MeCP2 using an antibody to its C-terminus (13) from brain extracts of WT mice, and as a negative control, from Mecp2-null mice. Mass spectrometry analysis identified CREB1, a major transcriptional activator, as one of nine proteins that co-purified with MeCP2 (Fig. 3A) (table S8). We confirmed the association of MeCP2 and CREB1 by performing the reciprocal immunoprecipitation with anti-CREB1 from Neuro2a cells overexpressing MeCP2 (Fig. 3B). The data suggest that the relationship between MeCP2 and CREB1 is non-stoichiometric and that not all of CREB1 is associated with MeCP2 in complexes from brain. To determine whether MeCP2 and CREB1 co-occupy any of the promoters of activated genes in vivo, we performed sequential ChIP (seqChIP) analysis using anti-CREB1 antibody on hypothalamic chromatin that was initially immunoprecipitated with anti-MeCP2 antibody. Quantitative real-time PCR analysis showed that the promoter of an activated target gene (Sst) was enriched for CREB1 binding, consistent with MeCP2 and CREB1 being simultaneously associated with the promoter. In contrast, the promoter of a gene repressed by MeCP2 (Mef2c) was not enriched for CREB1 binding (Fig. 3C). To determine if there is functional synergy between MeCP2 and CREB1, we transfected Neuro2a cells with luciferase reporter constructs driven by the promoter of Sst (activated MeCP2 target) or the promoter of A2bp1 (repressed MeCP2 target). MeCP2 and CREB1 co-transfection resulted in significant enhancement of the Sst reporter activity, but not the A2bp1 reporter (Fig. 3D). Interestingly, Creb1 was one of the activated MeCP2 targets that we validated by quantitative real-time RT-PCR (Fig. 1C), and using ChIP analysis we found that in vivo MeCP2 binds to the promoter region of Creb1, with significantly enhanced binding in MECP2-Tg samples compared to WT (p < 0.05) (Fig. 4A). We confirmed this result and assessed MeCP2 binding to neighboring genomic regions using a custom array with probes to the Creb1 promoter (Fig. 4B). In addition, Sst and CREB1 protein levels were increased in MECP2-Tg hypothalami compared to WT, indicating that MeCP2 indeed enhances expression of Sst and Creb1 (Fig. 4C).
Fig. 3.
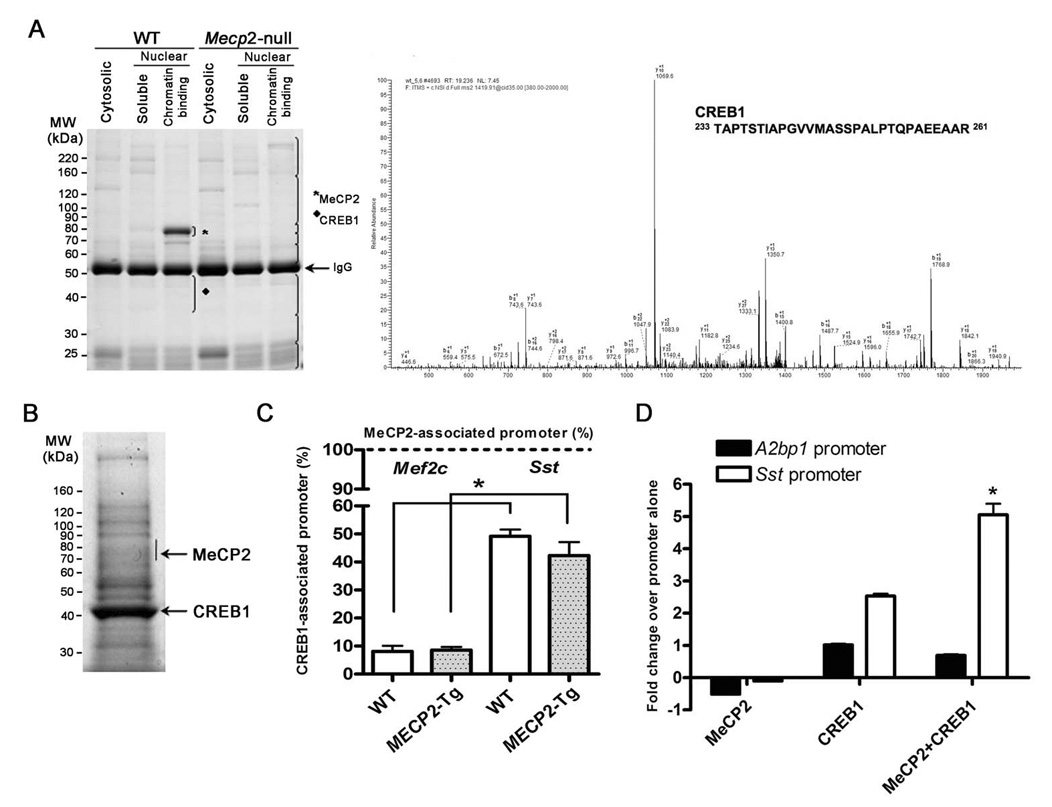
Physical and functional cooperation between MeCP2 and CREB1. (A) Specific interaction between MeCP2 and CREB1 in chromatin. Mouse brain tissue was fractionated into cytosolic, nuclear, and chromatin fractions for protein extraction from WT and Mecp2-null mice. Protein extracts were immunoprecipitated with an anti-MeCP2 antibody and resolved by SDS-PAGE. Each lane was divided into eight sections (brackets) and analyzed by mass spectrometry. Proteins identified in the WT but not in the knockout sample are considered specific MeCP2 interacting proteins. MeCP2 was identified only in the chromatin fraction from WT mice (★). A representative nano-HPLC/MS/MS spectrum is shown that identifies CREB1 as one of the specific MeCP2 binding proteins in the 35–50 kDa region in SDS-PAGE indicated by υ. The complete list of interacting proteins is available in Supporting Online Material (12) (table S8). (B) MeCP2 was detected after immunoprecipitation with anti-CREB1 from Neuro2a cells, using mass spectrometry analysis of the indicated band. (C) MeCP2 associates with CREB1 at the promoter of an activated but not a repressed target. SeqChIP analysis detects co-occupancy of MeCP2 and CREB1 at the promoter of the activated MeCP2 target Sst, but not at the promoter of the repressed target Mef2c. The primary ChIP was performed with an anti-MeCP2 antibody and the secondary ChIP was done with an anti-CREB1 antibody. Quantitative real-time PCR values were normalized to the input and plotted as percent of secondary over primary ChIP (N = 3, * p<0.001, two-way ANOVA). (D) Functional synergy between MeCP2 and CREB1 at the promoter of an activated target. Luciferase assay in Neuro2a cells reveals synergistic activation at the promoter of the activated MeCP2 target Sst but not the repressed target A2bp1 (N = 3, * p<0.002, two-way ANOVA).
Fig. 4.
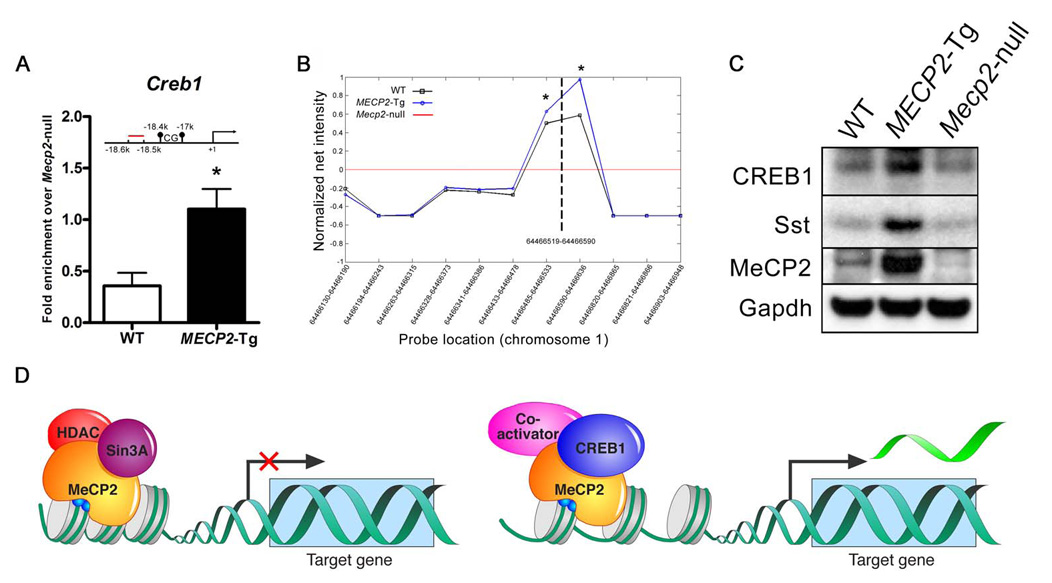
Creb1 is a direct MeCP2 target. (A) ChIP analysis revealed binding of MeCP2 to the promoter region of Creb1. Quantitative real-time PCR values were normalized to the input and plotted as fold enrichment over Mecp2-null (N = 4, * p<0.05, t test). (B) ChIP analysis using a custom array with probes to the Creb1 promoter. Genomic locations of the array probes are indicated on the X-axis and the dashed vertical line indicates location of the quantitative real-time PCR probe. The red line represents the normalized net signal from the Mecp2-null samples. For the WT and MECP2-Tg samples, normalized net intensities from each probe are plotted and are significantly different from the Mecp2-null near the predicted MeCP2 binding site (* p<0.02). (C) CREB1 and Sst protein levels are increased in MECP2-Tg hypothalami compared to WT. Western blot is representative of three animals from each genotype. (D) MeCP2 can function as a transcriptional activator and repressor in the hypothalamus (CpG sites are depicted as blue circles irrespective of their methylation status).
In contrast to prior studies that failed to detect many transcriptional changes due to MeCP2 dysfunction, we found that interrogating a discrete brain region, the hypothalamus, is very effective for uncovering MeCP2 target genes. Consistent with the role of MeCP2 as an activity-dependent regulator of gene expression, it would be predicted that MeCP2 plays a critical role in neurons that must constantly respond to new physiologic states. It is of interest that the list of putative targets includes genes that cause one or more RTT phenotypes when disrupted. One MeCP2 repression target is the gene encoding ataxin 2 binding protein 1 (A2bp1), which regulates splicing of neuronal genes. Disruption of A2BP1 has been identified in patients with mental retardation and epilepsy (14) and a recent study associated A2BP1 disruption with autism susceptibility (15). Gamt, a target activated by MeCP2, encodes the enzyme guanidinoacetate methyltransferase involved in creatine biosynthesis. Interestingly, patients with GAMT deficiency suffer severe mental retardation, absent or limited speech development, seizures, and hypotonia (16–18).
Beyond demonstrating that altered transcriptional regulation occurs in mouse models of MECP2 disorders, a key finding from our study is that MeCP2 acts as both an activator and a repressor (Fig. 4D), and that at least in the hypothalamus it is more of an activator. Several lines of evidence support our conclusions. First, the strongest evidence comes from the genetic data, whereby all the common transcriptional changes occur in opposite directions in Mecp2-null versus MECP2-Tg mice (except for twenty one genes; tables S3 and S4). Second, we detect MeCP2 binding at the promoters of genes that are altered in opposite directions in the two models (Sst, Oprk1, Gamt, Gprin1, Mef2c, and A2bp1), but not at the promoter of a gene downregulated in both mouse models, leading us to propose that these are direct MeCP2 targets. Third, the fact that MeCP2 binding to these targets is increased in the MECP2-Tg mice compared to WT, argues against the possibility that excess MeCP2 is titrating corepressors away from promoters, and supports a model where MeCP2 directly recruits activating factors to promoters. Finally, the binding of MeCP2 to promoters of activated genes and the association of MeCP2 with the transcription factor CREB1 at an activated promoter (but not at a repressed promoter) supports a role for MeCP2 as an activator in certain contexts. While it is possible that the gene expression changes shared by both mouse models are secondary to the physiological properties of hypothalamic neurons, we believe this is unlikely to explain expression changes in all 2,582 genes, especially because many are not activity-regulated. Although we cannot exclude the possibility that some of the inversely altered genes we found in the two mouse models are due to MeCP2 repressing a transcriptional repressor (resulting in activation of the targets of such a repressor), this also seems unlikely, since we did not find a repressor in the list of repressed MeCP2 targets. On the other hand, we found that MeCP2 activates the major transcriptional activator CREB1 (Fig. 1C, table S7, Fig. 4, A, B and C). A recent study showed that the CREB-induced miRNA, miR132, represses MeCP2 translation (19). This finding along with our data that MeCP2 activates Creb1, proposes a negative regulatory loop: MeCP2 activating Creb1 results in an increase in CREB1 levels, which in turn induces miR132 leading to decreased MeCP2 levels. In addition, ChIP-chip analysis using SH-SY5Y cells showed that MeCP2 associates more frequently with promoters that are also associated with RNA polymerase II (20), further supporting that the function of MeCP2 extends beyond transcriptional repression.
Our findings resolve inconsistencies in the literature regarding MeCP2 and Bdnf. Chen et al. and Martinowich et al. demonstrated that MeCP2 binds the Bdnf promoter in neuronal cultures (21, 22). However, the in vivo data showing that Bdnf is downregulated both at the RNA and protein levels in Mecp2-null mice (23) were confusing in light of the proposed role of MeCP2 as a repressor. Our finding that Bdnf is upregulated in MECP2-Tg and downregulated in Mecp2-null animals (Fig. 1C, table S7), and the ample evidence that Bdnf is a direct target of MeCP2 (21, 22), is consistent with the role of MeCP2 as an activator on the Bdnf promoter, and reconciles the existing data on MeCP2 and Bdnf.
Lastly, our data provide insight into the molecular mechanism underlying MECP2 disorders. The transcriptional changes suggest that the duplication phenotype is due to MeCP2 gain of function (hypermorph), rather than loss of function, and that at the molecular level, RTT is due mostly to loss of transcriptional activation, rather than derepression. In terms of clinical relevance, our results suggest that patients with RTT will have to be treated differently than patients with MECP2 duplications. The finding that MeCP2 regulates a large number of genes, at least in the hypothalamus, suggests a need for therapeutic strategies that focus on restoring neuronal function rather than restoring the activity of individual gene products affected by MeCP2 dysfunction. It might prove challenging to restore the level of each of these genes at the same time, thus an alternative approach will be to identify proteins or pathways that suppress MeCP2 dysfunction phenotypes, or bypass MeCP2, and restore neuronal homeostasis.
Supplementary Material
Footnotes
Summary sentence: MeCP2 acts as a transcriptional activator and repressor in the hypothalamus.
References
- 1.Amir RE, et al. Nat Genet. 1999;23:185. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 2.Moretti P, Zoghbi HY. Curr Opin Genet Dev. 2006;16:276. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. Nat Genet. 2001;27:322. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 4.Collins AL, et al. Hum Mol Genet. 2004;13:2679. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 5.Nan X, et al. Nature. 1998;393:386. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 6.Jones PL, et al. Nat Genet. 1998;19:187. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 7.Chao H, Zoghbi HY, Rosenmund C. Neuron. 2007;56:1. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tudor M, Akbarian S, Chen RZ, Jaenisch R. Proc Natl Acad Sci U S A. 2002;99:15536. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Axelrod FB, Chelimsky GG, Weese-Mayer DE. Pediatrics. 2006;118:309. doi: 10.1542/peds.2005-3032. [DOI] [PubMed] [Google Scholar]
- 10.Gardina PJ, et al. BMC Genomics. 2006;7:325. doi: 10.1186/1471-2164-7-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srinivasan K, et al. Methods. 2005;37:345. [Google Scholar]
- 12.Materials and methods are available as supporting material on Science Online.
- 13.Zhou Z, et al. Neuron. 2006;52:255. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhalla K, et al. J Hum Genet. 2004;49:308. doi: 10.1007/s10038-004-0145-4. [DOI] [PubMed] [Google Scholar]
- 15.Martin CL, et al. Am J Med Genet B Neuropsychiatr Genet. 2007;144:869. doi: 10.1002/ajmg.b.30530. [DOI] [PubMed] [Google Scholar]
- 16.Caldeira Araujo H, et al. Am J Med Genet A. 2005;133:122. doi: 10.1002/ajmg.a.30226. [DOI] [PubMed] [Google Scholar]
- 17.Verbruggen KT, et al. Eur J Pediatr. 2007;166:921. doi: 10.1007/s00431-006-0340-8. [DOI] [PubMed] [Google Scholar]
- 18.Sykut-Cegielska J, Gradowska W, Mercimek-Mahmutoglu S, Stockler-Ipsiroglu S. Acta Biochim Pol. 2004;51:875. [PubMed] [Google Scholar]
- 19.Klein ME, et al. Nat Neurosci. 2007;10:1513. doi: 10.1038/nn2010. [DOI] [PubMed] [Google Scholar]
- 20.Yasui DH, et al. Proc Natl Acad Sci U S A. 2007;104:19416. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen WG, et al. Science. 2003;302:885. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 22.Martinowich K, et al. Science. 2003;302:890. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 23.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. Neuron. 2006;49:341. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 24.We thank members of the Zoghbi lab for their thoughtful comments, Z. Yin for technical assistance, M. Shinawi and A. Tawil for technical advice, Y. Klisch for the art work, and J. Lim, J. Neul and M. Greenberg for reagents. The Baylor College of Medicine Microarray Core Facility performed the microarray experiments. This work was funded by National Institutes of Health/National Institute of Neurological Disorders and Stroke grant NS057819 (HZ), National Institute of Child Health and Human Development Mental Retardation and Developmental Disabilities Research Center HD024064 (HZ), the International Rett Syndrome Foundation, and the Simons Foundation. H. Zoghbi is a Howard Hughes Medical Institute investigator. The microarray data have been deposited in the NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE11150.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.