

Main Text
To the Editor: Hereditary spastic paraplegia (HSP) is a hereditary condition characterized by selective retrograde degeneration of corticospinal motor axons and is therefore a model disease for studying mechanisms of axonal degeneration.1 Although HSP is genetically heterogeneous, the most common form is due to heterozygous mutations in the SPG4 gene (MIM 604277), encoding the microtubule-severing protein spastin.2 We read with great interest the report in The American Journal of Human Genetics of the identification of ZFYVE27 (also known as protrudin) as a spastin interactor in a two-hybrid screening and the finding of a missense mutation (p.G191V) in the protein in a German family with classical features of autosomal-dominant HSP3. The ZFYVE27 gene (MIM 610244) has been assigned the SPG33 gene symbol. The p.G191V mutation was found to segregate with HSP in the proband's children (one healthy and two affected individuals) and was not detected in 210 control chromosomes. The authors do not specify whether and which known HSP genes were previously excluded in this family. The p.G191V change does not fall into any functional domain but was suggested to provoke a shift in a transmembrane domain of protrudin and thus to alter proper conformation.3 Immunofluorescence experiments showed a slightly different subcellular localization of the mutant version of protrudin than of the wild-type, although these studies were based on overexpression. More interestingly, the authors showed coimmunoprecipitation experiments suggesting a reduced binding of mutant protrudin to spastin, thus implying that at the basis of the disease is a failure of protrudin to interact with spastin.3
Our interest in protrudin increased even more when a paper in Science reported a role of protrudin in Rab-11-mediated membrane trafficking.4 When overexpressed, protrudin promotes outgrowth of cellular projections in HeLa cells and neurite formation in PC12 cells and primary neurons. Conversely, protrudin downregulation inhibits neurite formation. Protrudin action occurs via preferential binding to the GDP-bound form of Rab11, suggesting that it regulates Rab11-dependent membrane recycling to induce directional membrane trafficking required for neurite formation.4 This study has important implications for our understanding of the role of protrudin in HSP and also provides an opportunity to address the functional role of the p.G191V substitution.
We assessed the functional ability of protrudinG191V to mediate outgrowth of HeLa cell projections and of neurites of NSC34 cells (an immortalized murine motoneuronal cell line). Remarkably, we found no difference in the ability of wild-type and mutant protrudin to extend neurites in NSC34 cells or to elongate HeLa cells in several independent experiments (Figures 1A–1D and not shown). Consistent with these findings, protrudinG191V maintains the ability to preferentially interact with Rab11-GDP (supplied to cells as a dominant-negative mutant, Rab11S25N) (Figure 1E). These data strongly indicate that the p.G191V change does not lead to loss of function. These results leave open the possibility that protrudinG191V malfunction is linked to its failure to interact with spastin. However, we could coimmunoprecipitate wild-type and mutated protrudin equally well with spastin, in disagreement with results of Mannan et al.3 (Figure 1F). Our data provide no clue as to the pathogenic role of the p.G191V mutation. We searched the SNP database for the ZFYVE27 gene. We were struck by the finding that the p.G191V change is listed as a polymorphism (id rs35077384) with an allele frequency of 0.038 in the APPLERA_GI:AGI_ASL population, of 0.067 in the HapMap-YRI population, of 0.011 in the HapMap-JPT population, and of 0.008 in the Hap-Map-CEU population.
Figure 1.
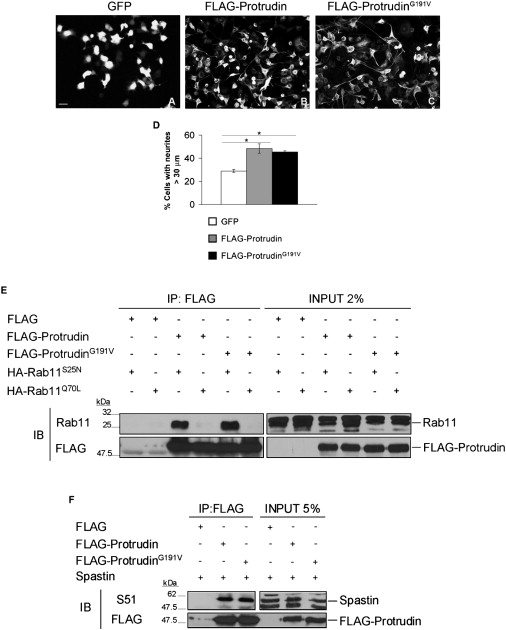
Exploring the Pathogenic Role of ProtrudinG191V
(A–C) NSC34 cells were transfected with control GFP, FLAG-protrudin, or FLAG-protrudinG191V vectors. Both wild-type protrudin and protrudinG191V stimulate neurite elongation with the same efficiency. The scale bar represents 50 μm.
(D) Quantification of the percentage of cells with neurites longer than 30 μm in the different conditions (means of at least three independent experiments ± SEM; at least 450 cells were scored per condition; ∗p < 0.05, Student's t test). The average length of neurites in these cells was 58.01 μm ± 3.56 for wild-type protrudin and 59.55 μm ± 3.66 for protrudinG191V.
(E) Coimmunoprecipitation experiments between protrudin and Rab11 show that both wild-type protrudin and protrudinG191V interact with Rab11S25N (GDP-bound form) but not with Rab11Q70L (GTP-bound form).
(F) Spastin interacts both with wild-type protrudin and protrudinG191V. To detect spastin, we used a specific antibody (S51).
All together, our results cast doubts on the implication of ZFYVE27/protrudin in HSP, unless incontrovertible pathogenic mutations are identified in other families. The classification of ZFYVE27 as an HSP disease gene is misleading to the community of geneticists interested in HSP molecular diagnosis and pathogenic studies. This stresses the importance of accurate functional validation of putative disease-causing mutations, especially when the mutation is a missense change and a single family is identified. It remains of course possible that the p.G191V change is in linkage disequilibrium with the real mutation in the original family or that it acts as a gene modifier for another HSP gene. Although we propose removing SPG33 from the list of HSP genes, we should continue to consider this gene relevant for the understanding of the pathogenesis of SPG4-related HSP.
Web Resources
URLs for databases cited in the text are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Single Nucleotide Polymorphism (SNP) database, http://www.ncbi.nlm.nih.gov/projects/SNP/
Acknowledgments
We thank K. Nakayama for sharing constructs. This work has been supported by the Italian Telethon Foundation.
References
- 1.Fink J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 2.Hazan J., Fonknechten N., Mavel D., Paternotte C., Samson D., Artiguenave F., Davoine C.S., Cruaud C., Durr A., Wincker P. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 1999;23:296–303. doi: 10.1038/15472. [DOI] [PubMed] [Google Scholar]
- 3.Mannan A.U., Krawen P., Sauter S.M., Boehm J., Chronowska A., Paulus W., Neesen J., Engel W. ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am. J. Hum. Genet. 2006;79:351–357. doi: 10.1086/504927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shirane M., Nakayama K.I. Protrudin induces neurite formation by directional membrane trafficking. Science. 2006;314:818–821. doi: 10.1126/science.1134027. [DOI] [PubMed] [Google Scholar]