

Abstract
Rett syndrome is a severe neurodevelopmental disease caused by mutations in the X-linked gene encoding for the methyl-CpG-binding protein MeCP2. Here, we report the identification of FOXG1-truncating mutations in two patients affected by the congenital variant of Rett syndrome. FOXG1 encodes a brain-specific transcriptional repressor that is essential for early development of the telencephalon. Molecular analysis revealed that Foxg1 might also share common molecular mechanisms with MeCP2 during neuronal development, exhibiting partially overlapping expression domain in postnatal cortex and neuronal subnuclear localization.
Main Text
In the classic form of Rett syndrome (RTT [MIM 312750]), females are heterozygous for mutations in the X-linked MECP2 gene (MIM 300005) and the few reported males have an XXY karyotype or MECP2 mutations in a mosaic state.1 A number of variants have been described including the congenital, the early-onset seizures, and the preserved speech variant.2 Soon after the discovery of MECP2 as the RTT gene, we demonstrated that the preserved speech variant is allelic to the classic form.3 More recently, we and others showed that CDKL5 (MIM 300203) is responsible for atypical RTT, namely the early-onset seizures variant.4,5 The congenital variant was initially described by Rolando in 1985.6 In this form, girls are floppy and retarded from the very first months of life. The majority of congenital variants do not bear MECP2 or CDKL5 mutations,7,8 with only four cases being reported with MECP2 mutations.9–11
Using oligo array CGH, we recently identified a de novo 3 Mb interstitial deletion of chromosome 14q12 in a 7 year-old girl.12 She showed dysmorphic features and a Rett-like clinical course, including normal perinatal period, postnatal microcephaly, seizures, and severe mental retardation. The deleted region was gene poor and contained only five genes. Among them, FOXG1 (MIM 164874) turned out to be a very interesting gene because it encodes a brain-specific transcriptional repressor. We analyzed this gene with a combination of both DHPLC and real-time quantitative PCR in a cohort of 53 MECP2/CDKL5 mutation-negative RTT patients, including seven classic, 21 preserved speech, seven early-onset seizures, one “forme fruste,” two congenital variants and 15 Rett-like cases.13 For real-time qPCR analysis, we designed primers and TaqMan probe complementary to a segment located in the middle of the single exon of the gene using Primer Express software (Applied Biosystems). Sequences of primers and probe (FAM labeled) are listed in Table S1 available online. We used an RNAase P kit as an internal reference (VIC-labeled probe, Applied Biosystems). PCR was carried out as previously described.14 The starting copy number of the unknown samples was determined with the comparative Ct method, as reported by Livak.15 By DHPLC, we identified a different de novo FOXG1 truncating mutation in the two congenital variant patients. Real-time qPCR failed to identify any microdeletion in the 53 patients.
FOXG1 encodes forkhead box protein G1, FoxG1 (formerly brain factor 1 [BF-1]), a transcriptional factor with expression restricted to fetal and adult brain and testis. FoxG1 interacts with the transcriptional repressor JARID1B and with global transcriptional corepressors of the Groucho family. The interaction with these proteins is of functional importance for early brain development.16,17 Like MeCP2, FoxG1 also indirectly associates with the histone deacetylase 1 protein.1,17 Both mutations disrupted the protein at different levels (Figure 1). In case 1, a stop-codon mutation p.W255X (c.765G→A) impaired the DNA binding because of the disruption of the forkhead domain (Figure 1D, left). Case 2 showed a 1 bp deletion c.969 delC (p.S323fsX325) causing the loss of JARID1B-interacting domain and the misfolding of the motif responsible for the Groucho binding (Figure 1D, right). Lastly, both FOXG1 mutations affected all the four brain fetal isoforms that lack the last 37 amino acids and have different C-terminal domains.18
Figure 1.
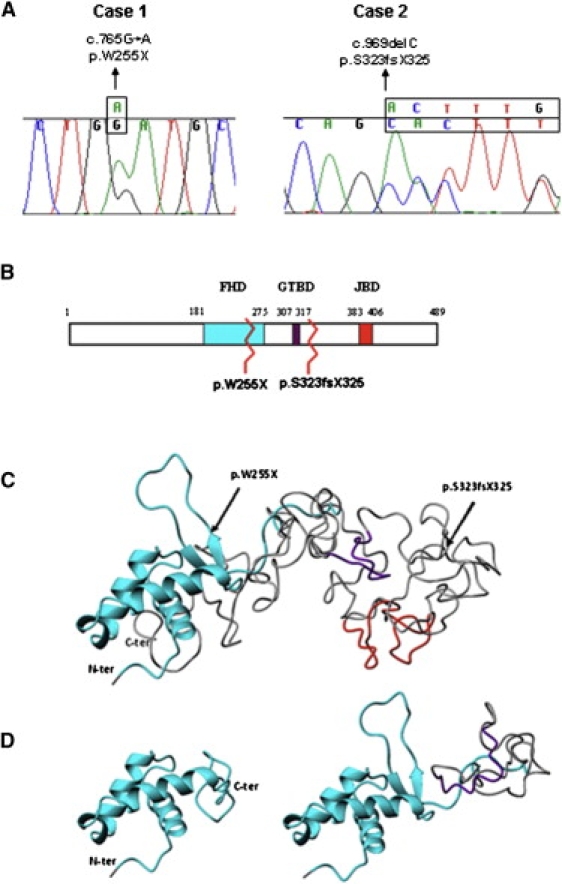
FOXG1 Mutations and Alterations of the Functional Domains
(A) Sequence tracing of FOXG1 mutations in the two patients. Mutated bases are indicated above the line.
(B) Schematic representation of FoxG1 protein. The three main functional domains are shown: the DNA binding fork-head domain in light blue (FHD), the Groucho-binding domain in violet (GTBD), and the JARID1B binding domain in red (JBD). The numbers at the top refer to the amino acid positions. Mutations are indicated by zigzag lines.
(C and D) Ribbon representation of the tertiary structure obtained with Phyre v.0.2 software. (C) shows the structure of the region containing the three functional domains of wild-type protein (amino acids 180–489). Arrows highlight the two mutations. The FHD domain (cyan) consists of three alpha helices and one beta hairpin (two beta strands and one loop), whereas the GTBD (violet) and JBD (red) domains are random coiled. (D) shows structural modification after p.W255X (left) and p.S323fsX325 (right) mutations. The p.W255X mutation determines a protein truncation just after the second beta strand leading to the loss of the beta hairpin and thus preventing DNA binding. The p.S323fsX325 mutation leaves the FHD domain intact and truncates the protein just after GTBD, inducing conformational changes that lead to its misfolding.
The two mutated individuals, aged 22 (case 1) and 7 years (case 2), fulfilled the international criteria for RTT variants.19 Pregnancy, delivery, and auxological parameters at birth were normal. Neurological and behavioral neonatal evaluations were reported as normal, but at three months, an abnormal head-circumference growing was noticed in the patients. These patients appeared to weep inconsolably, and they did not respond when called and were unable to lift their heads. Case 1 was never able to sit unaided and laid permanently in bed, whereas case 2 was barely able to sit. They were always apraxic and from 1 year of age, they showed peculiar jerky movements of the upper limbs and midline stereotypic activities, typical of RTT syndrome (Figure 2). They never acquired spoken language. Generalized convulsions appeared at 14 years in case 1 and at 2 1/2 years in case 2. Ever since cases 1 and 2 were 3 and 5 years old, respectively, an EEG showed features often found in RTT patients: a multifocal pattern with spikes and sharp waves and occasional paroxysmal activity. In both patients brain MRI showed corpus callosum hypoplasia, a finding which has already been reported in RTT.20 Currently, they show microcephaly (OFC of 49 cm in case 1, and 47 cm in case 2). They have occasional periods of deep breathing with exaggerated inspirations. Sialorrhoea, bruxism, scoliosis, and cold lower extremities as well as stypsis are present in both patients who are currently fed by mouth.
Figure 2.

Pictures of the Two Congenital RTT Patients
Case 1 (#156) is shown on the left; case 2 (#868) is shown on the right. They show peculiar jerky movements of the upper limbs frequently pushed in different directions accompanied by continuous flexion-extension, wringing movements of the fingers of the hands. The hands were brought together in hand-washing and hand-mouth stereotypic activities, which were intense and present all the time they were awake. Similar flexion-extension movements of the toes were noticed in the feet. The double scoliosis of case 1 is clearly evident, whereas the other girl maintained a straight vertebral column as often occurs in RTT in the first decade. Teeth grinding was present, and the tongue often protruded out from the mouth.
These two girls show neurological and neurovegetative symptoms as well as somatic features consistent with a diagnosis of congenital RTT variant. It should only be noted that a retrospective assessment concerning the possible presence of a regression was not feasible. We attempted to compare their phenotype with the four other MECP2-mutated girls described as congenital variants.9–11 However, they have been reported with very little detail, thereby hampering a posteriori clinical re-evaluation according to the revised criteria.19 According to the new criteria, in the classic form, psychomotor development may have been delayed from birth; thus, a re-evaluation of these four patients would have lead to their reclassification as classic form. Alternatively, the disruption of either MeCP2 or FoxG1 may lead to a phenotype, namely the congenital variant, indistinguishable at the level of the clinical and instrumental investigations performed.
A translocation with inversion affecting fetal isoforms of FOXG1 was recently described in a 7-year-old girl.18 She had acquired microcephaly, alalia, inability to sit and walk, and epilepsy in common with the present cases. Corpus callosum was absent, whereas in our cases, it was hypoplasic. Stereotypic hand activities were not mentioned, and tetraplegia was described.18 The clinical features of this patient have something in common with a RTT phenotype. The impairment of only fetal FOXG1 isoforms and the possible contribution of genes at the other two breakpoints of the complex rearrangement might explain the phenotypic differences.
The mouse ortholog Foxg1 has a restricted expression domain in the central nervous system coinciding with the emergence of the telencephalic structures of the brain. Its function has been extensively characterized and found to promote telencephalon development by sustaining proliferation of the progenitor pool and preventing premature cortical neural differentiation.21,22 In agreement, FoxG1 expression is found in the proliferating neuroepithelium starting from early development onward.23 This expression profile might explain the particular early onset of the neurological symptoms displayed by the patients.
Despite its early expression in telencephalon development, in this study we found that Foxg1 expression is detectable in the differentiating cortical compartment in the postnatal stages, although at lower levels with respect to the early embryonic phases (Figure 3A). This expression profile overlaps with the described MeCP2 expression domain in cortical tissues, in differentiating and mature neurons (Figures 3A and 3B). Foxg1 homozygote-mutant mice die shortly after birth with severe brain defects.24–26 Unfortunately, the severe compromised development of Foxg1 mutant telencephali has prevented the analysis of its function in more differentiated neurons. At the single-cell level, FoxG1 localizes in the nuclear compartment but is excluded from the MeCP2-positive heterochromatic foci both in nonneural and primary neurons (Figures 3C–3J). These findings suggest that, differently from MeCP2, FoxG1 is not a transcriptional repressor stably associated with heterochromatin. However, both proteins have a large colocalization domain in other nuclear compartments (Figure 3I).
Figure 3.
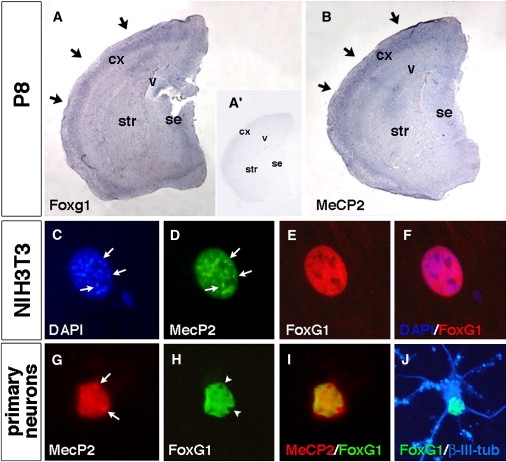
FoxG1 and MeCP2 Expression Domain in Postnatal Cortex and Neuronal Subnuclear Localization
(A and B) Expression analysis by in situ hybridization of Foxg1 and MeCP2 on P8 postnatal forebrain tissue. As shown in (A), Foxg1 expression is found in differentiating and mature cortical neurons in the definitive cortical plate (indicated by arrows in [A]) similar to the MeCP2 expression pattern (indicated by arrows in [B]). In (A′), the inset shows background staining with a sense cRNA for Foxg1 in the same in situ hybridization conditions used for (A) and (B).
(C–J) FoxG1 and MeCP2 sub-cellular localization in non-neural and primary neurons. As shown in (C)–(F), in NIH 3T3 cells, MeCP2-GFP exogenous protein has a diffuse nuclear localization with accumulation in the heterochromatic foci (indicated by arrows in [D]) as identified when compared with DAPI staining (indicated by arrows in [C]). (E) and (F) show that conversely, FoxG1-flag exogenous protein displays a widespread nuclear localization without enrichment in heterochromatic sites. (G)–(J) show FoxG1 and MeCP2 localization in 12DIV (days in vitro) primary hippocampal neurons. In (G), MeCP2 endogenous protein is accumulated in heterochromatic foci (indicated by arrows). As shown in (H), FoxG1-flag exogenous nuclear localization is excluded from heterochromatic puncta (indicated by arrowheads). As shown in (I), MeCP2 and FoxG1-flag colocalize in the nuclear compartment outside the heterochromatic foci. As shown in (J), Nuclear FoxG1-flag localization is detected in a differentiated β-III-tubulin-positive neuron. The following abbreviations are used: cx, cerebral cortex; se, septum; str, striatum; and v, ventricle.
Overall, these data suggest that FoxG1 may exert some additional functions in differentiating and mature neurons, thus sharing similarities with those described for MeCP2. These findings may provide some biological evidence for the development of similar clinical manifestations in disorders affecting the two genes. However, it is also possible that the two transcriptional regulators act on different stages of the process that leads to proper cortical development, from early cell-fate decisions to later circuit connectivity and dendritic development.
FoxG1 shares some interesting analogies with MeCP2 in its molecular functions, raising the question whether the two protein networks may interact in some circumstances and on selective common targets. Future studies will address this intriguing hypothesis. Recently, heterozygous Foxg1+/− mice were found to display subtler defects including a reduction in size of the corpus callosum together with specific patterning defects.25,27 Furthermore, heterozygous Foxg1+/− exhibit learning deficits based on fear-condition behavioral tests associated with a loss of postnatal neurogenesis in the hippocampus.27 These mice represent a very interesting animal model for further investigation about how Foxg1 haploinsufficiency may impact on brain development and neuronal maturation and function.
In conclusion, we demonstrated that FOXG1 is a previously unrecognized gene responsible for variant Rett syndrome. It is worth noting that in the revised criteria for Rett syndrome the female sex is no longer present as inclusion criteria.19 This seemed to open the door to the discovery of an autosomal gene.
Acknowledgments
We would first like to thank the Rett patients and their families. This work was supported by Telethon grants GTB07001 to A.R. and GGP07181 to V.B., by the EuroRETT E-RARE network to A.R. and to V.B, and by the Emma and Ernesto Rulfo Foundation to A.R.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Italian Rett database and biobank, http://www.biobank.unisi.it/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Chahrour M., Zoghbi H.Y. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Hagberg B.A., Skjeldal O.H. Rett variants: A suggested model for inclusion criteria. Pediatr. Neurol. 1994;11:5–11. doi: 10.1016/0887-8994(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 3.De Bona C., Zappella M., Hayek G., Meloni I., Vitelli F., Bruttini M., Cusano R., Loffredo P., Longo I., Renieri A. Preserved speech variant is allelic of classic Rett syndrome. Eur. J. Hum. Genet. 2000;8:325–330. doi: 10.1038/sj.ejhg.5200473. [DOI] [PubMed] [Google Scholar]
- 4.Tao J., Van Esch H., Hagedorn-Greiwe M., Hoffmann K., Moser B., Raynaud M., Sperner J., Fryns J., Schwinger E., Gecz J. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am. J. Hum. Genet. 2004;75:1149–1154. doi: 10.1086/426460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scala E., Ariani F., Mari F., Caselli R., Pescucci C., Longo I., Meloni I., Giachino D., Bruttini M., Hayek G. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 2005;42:103–107. doi: 10.1136/jmg.2004.026237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rolando S. Rett syndrome: report of eight cases. Brain Dev. 1985;7:290–296. doi: 10.1016/s0387-7604(85)80030-9. [DOI] [PubMed] [Google Scholar]
- 7.Erlandson A., Samuelsson L., Hagberg B., Kyllerman M., Vujic M., Wahlstrom J. Multiplex ligation-dependent probe amplification (MLPA) detects large deletions in the MECP2 gene of Swedish Rett syndrome patients. Genet. Test. 2003;7:329–332. doi: 10.1089/109065703322783707. [DOI] [PubMed] [Google Scholar]
- 8.Scala E., Longo I., Ottimo F., Speciale C., Sampieri K., Katzaki E., Artuso R., Mencarelli M.A., D'Ambrogio T., Vonella G. MECP2 deletions and genotype-phenotype correlation in Rett syndrome. Am. J. Med. Genet. A. 2007;143:2775–2784. doi: 10.1002/ajmg.a.32002. [DOI] [PubMed] [Google Scholar]
- 9.Huppke P., Laccone F., Kramer N., Engel W., Hanefeld F. Rett syndrome: Analysis of MECP2 and clinical characterization of 31 patients. Hum. Mol. Genet. 2000;9:1369–1375. doi: 10.1093/hmg/9.9.1369. [DOI] [PubMed] [Google Scholar]
- 10.Monros E., Armstrong J., Aibar E., Poo P., Canos I., Pineda M. Rett syndrome in Spain: Mutation analysis and clinical correlations. Brain Dev. 2001;1(Suppl):S251–S253. doi: 10.1016/s0387-7604(01)00374-6. [DOI] [PubMed] [Google Scholar]
- 11.Smeets E., Schollen E., Moog U., Matthijs G., Herbergs J., Smeets H., Curfs L., Schrander-Stumpel C., Fryns J.P. Rett syndrome in adolescent and adult females: Clinical and molecular genetic findings. Am. J. Med. Genet. A. 2003;122:227–233. doi: 10.1002/ajmg.a.20321. [DOI] [PubMed] [Google Scholar]
- 12.Papa F.T., Mencarelli M.A., Caselli R., Katzaki E., Sahpieri K., Meloni I., Ariani F., Longo I., Maggio A., Balestri P. A 3 Mb deletion in 14912 causes severe mental retardation, mild facial dysmorphisms and Rett-like features. Am. J. Med. Genet. A. 2008 doi: 10.1002/ajmg.a.32413. in press. [DOI] [PubMed] [Google Scholar]
- 13.Sampieri K., Meloni I., Scala E., Ariani F., Caselli R., Pescucci C., Longo I., Artuso R., Bruttini M., Mencarelli M.A. Italian Rett database and biobank. Hum. Mutat. 2007;28:329–335. doi: 10.1002/humu.20453. [DOI] [PubMed] [Google Scholar]
- 14.Ariani F., Mari F., Pescucci C., Longo I., Bruttini M., Meloni I., Hayek G., Rocchi R., Zappella M., Renieri A. Real-time quantitative PCR as a routine method for screening large rearrangements in Rett syndrome: Report of one case of MECP2 deletion and one case of MECP2 duplication. Hum. Mutat. 2004;24:172–177. doi: 10.1002/humu.20065. [DOI] [PubMed] [Google Scholar]
- 15.Livak, K. (1997). ABI Prism 7700 Sequence Detection System.
- 16.Tan K., Shaw A.L., Madsen B., Jensen K., Taylor-Papadimitriou J., Freemont P.S. Human PLU-1 Has transcriptional repression properties and interacts with the developmental transcription factors BF-1 and PAX9. J. Biol. Chem. 2003;278:20507–20513. doi: 10.1074/jbc.M301994200. [DOI] [PubMed] [Google Scholar]
- 17.Yao J., Lai E., Stifani S. The winged-helix protein brain factor 1 interacts with groucho and hes proteins to repress transcription. Mol. Cell. Biol. 2001;21:1962–1972. doi: 10.1128/MCB.21.6.1962-1972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shoichet S.A., Kunde S.A., Viertel P., Schell-Apacik C., von Voss H., Tommerup N., Ropers H.H., Kalscheuer V.M. Haploinsufficiency of novel FOXG1B variants in a patient with severe mental retardation, brain malformations and microcephaly. Hum. Genet. 2005;117:536–544. doi: 10.1007/s00439-005-1310-3. [DOI] [PubMed] [Google Scholar]
- 19.Hagberg B., Hanefeld F., Percy A., Skjeldal O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur. J. Paediatr. Neurol. 2002;6:293–297. doi: 10.1053/ejpn.2002.0612. [DOI] [PubMed] [Google Scholar]
- 20.Murakami J.W., Courchesne E., Haas R.H., Press G.A., Yeung-Courchesne R. Cerebellar and cerebral abnormalities in Rett syndrome: a quantitative MR analysis. AJR Am. J. Roentgenol. 1992;159:177–183. doi: 10.2214/ajr.159.1.1609693. [DOI] [PubMed] [Google Scholar]
- 21.Hanashima C., Shen L., Li S.C., Lai E. Brain factor-1 controls the proliferation and differentiation of neocortical progenitor cells through independent mechanisms. J. Neurosci. 2002;22:6526–6536. doi: 10.1523/JNEUROSCI.22-15-06526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seoane J., Le H.V., Shen L., Anderson S.A., Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 23.Tao W., Lai E. Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron. 1992;8:957–966. doi: 10.1016/0896-6273(92)90210-5. [DOI] [PubMed] [Google Scholar]
- 24.Martynoga B., Morrison H., Price D.J., Mason J.O. Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev. Biol. 2005;283:113–127. doi: 10.1016/j.ydbio.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Xuan S., Baptista C.A., Balas G., Tao W., Soares V.C., Lai E. Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron. 1995;14:1141–1152. doi: 10.1016/0896-6273(95)90262-7. [DOI] [PubMed] [Google Scholar]
- 26.Hanashima C., Fernandes M., Hebert J.M., Fishell G. The role of Foxg1 and dorsal midline signaling in the generation of Cajal-Retzius subtypes. J. Neurosci. 2007;27:11103–11111. doi: 10.1523/JNEUROSCI.1066-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen Q., Wang Y., Dimos J.T., Fasano C.A., Phoenix T.N., Lemischka I.R., Ivanova N.B., Stifani S., Morrisey E.E., Temple S. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat. Neurosci. 2006;9:743–751. doi: 10.1038/nn1694. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.