

Abstract
Hypomyelinating leukodystrophies (HMLs) are disorders involving aberrant myelin formation. The prototype of primary HMLs is the X-linked Pelizaeus-Merzbacher disease (PMD) caused by mutations in PLP1. Recently, homozygous mutations in GJA12 encoding connexin 47 were found in patients with autosomal-recessive Pelizaeus-Merzbacher-like disease (PMLD). However, many patients of both genders with PMLD carry neither PLP1 nor GJA12 mutations. We report a consanguineous Israeli Bedouin kindred with clinical and radiological findings compatible with PMLD, in which linkage to PLP1 and GJA12 was excluded. Using homozygosity mapping and mutation analysis, we have identified a homozygous missense mutation (D29G) not previously described in HSPD1, encoding the mitochondrial heat-shock protein 60 (Hsp60) in all affected individuals. The D29G mutation completely segregates with the disease-associated phenotype. The pathogenic effect of D29G on Hsp60-chaperonin activity was verified by an in vivo E. coli complementation assay, which demonstrated compromised ability of the D29G-Hsp60 mutant protein to support E. coli survival, especially at high temperatures. The disorder, which we have termed MitCHAP-60 disease, can be distinguished from spastic paraplegia 13 (SPG13), another Hsp60-associated autosomal-dominant neurodegenerative disorder, by its autosomal-recessive inheritance pattern, as well as by its early-onset, profound cerebral involvement and lethality. Our findings suggest that Hsp60 defects can cause neurodegenerative pathologies of varying severity, not previously suspected on the basis of the SPG13 phenotype. These findings should help to clarify the important role of Hsp60 in myelinogenesis and neurodegeneration.
Introduction
Myelin formation is one of the major processes involved in normal brain development and maturation. Intact myelination of the central nervous system (CNS) results from a highly controlled sequence of events requiring the coordinated function of numerous genes. Inborn errors affecting brain myelin are collectively recognized as leukodystrophies and can be classified into hypomyelinating disorders, in which myelin is not properly formed, and demyelinating leukodystrophies in which myelin is initially formed but is later destroyed.1 The prototype of early-onset hypomyelinating leukodystrophy is Pelizaeus-Merzbacher disease (PMD [MIM 312080]), an X-linked hypomyelinating disorder caused by mutations in the proteolipid protein 1 gene (PLP1) encoding lipophilin, the predominant myelin protein present in the CNS.2,3 Approximately 80% of patients with typical PMD carry PLP1 mutations.4 Patients with the classical form of the disease are characterized by the appearance of nystagmus and psychomotor retardation during the first months of life and then muscle hypotonia and progressive limb spasticity. Magnetic resonance imaging (MRI) of the brain in these patients typically shows diffuse hypomyelination.
Pelizaeus-Merzbacher-like disease (PMLD [MIM 608804 and MIM 260600]) is an autosomal-recessive disorder clinically indistinguishable from PMD, in which PLP1 defects are not found. Recently, mutations in GJA12 encoding the gap junction protein connexin 47 (Cx47) have been identified as causing PMLD.5,6 Nevertheless, according to recent data, GJA12 defects account for less than 10% of PMLD,7 and there are many patients of both genders who suffer from PMLD, and who carry neither PLP1 nor GJA12 mutations, suggesting the involvement of additional, as-yet-unidentified genetic defects that may also cause perturbation of normal myelinogenesis.
Molecular chaperones, also known as heat-shock proteins (HSPs), are highly conserved specialized proteins that occur in all organisms and that play a crucial role in cell maintenance and survival. Chaperones exert their biological effect by facilitating the correct folding of newly synthesized or preexisting denatured proteins, thus preventing the potential hazards of protein misfolding and intracellular aggregation.8–10 In addition to their role in protein folding, chaperones also function as regulators of protein degradation, assist in protein targeting and translocation across different cellular compartments, and intervene in signal transduction of apoptotic pathways.11,12
Chaperonins are a subgroup of molecular chaperones comprising high-molecular-weight oligomers that mediate ATP-dependent folding of proteins that are resistant to handling by simpler chaperones.13 On the basis of their sequence homology, chaperonins are divided into two distinct subgroups: Group I chaperonins (bacterial GroEL/mitochondrial Hsp60s) are found in prokaryotic cytosol and endosymbiotic organelles such as mitochondria and chloroplasts, and their function depends on cooperation with specific cochaperonin molecules (bacterial GroES/mitochondrial Hsp10s). Group II chaperonins reside in archaeal and eukaryotic cytosol and, although they are cochaperonin independent, are nevertheless assisted by other cofactors, such as prefoldin.10,11 The mitochondrial Hsp60 chaperonin is one of the most important components of the protein-folding system inside the mitochondrial matrix. Hsp60 together with its cochaperonin Hsp10 produce a large, efficient protein-editing machinery that facilitates proper folding and assembly of mitochondrial-imported proteins and corrects misfolded polypeptides generated under mitochondrial oxidative stress.14–16 Two mutations in the HSPD1 gene encoding Hsp60 have recently been found to underlie spastic paraplegia 13 (SPG13 [MIM 605280]), an autosomal-dominant spinal-cord neurodegenerative disorder of late onset, characterized by progressive weakness and spasticity of the lower limbs.17,18
SPG13 is a member of the hereditary spastic paraplegia group of disorders (SPGs). To date, 33 single-gene disorders attributed to mutations in 15 identified genes have been reported, affecting one in 10,000–100,000 persons.19,20 Traditionally, disorders within this group are classified according to both the mode of inheritance and their clinical features. They can be transmitted with autosomal-dominant, autosomal-recessive, or X-linked inheritance. The SPGs are classified as “pure” if leg spasticity is the sole clinical manifestation or as “complicated” when accompanied by additional or systemic manifestations, such as optic neuropathy, retinopathy, extrapyramidal disturbances, dementia, ataxia, mental retardation, and deafness.19 Defects in intracellular trafficking and transport in myelination processes, as well as abnormalities of mitochondrial proteins, have been implicated in the pathogenesis of SPGs.21
Mitochondria are essential organelles that house energy-producing respiratory complexes and represent the major site of cellular reactive-oxygen production. They carry out crucial anabolic reactions and important key functions during apoptotic processes. Pathogenic mutations in gene products expressed in the mitochondria, whether encoded by mitochondrial or nuclear genomic DNA, cause mitochondrial diseases.22 Given the importance of these organelles for cellular physiology, it is not surprising that mitochondrial dysfunction plays a fundamental role in neurodegenerative diseases including SPGs.23 To date, three SPGs have been reported to be associated with mutations in nuclear-encoded mitochondrial proteins, including paraplegin, Hsp60 and the REEP1 protein (SPG7 [MIM 602783], SPG13 [MIM 605280], and SPG31 [MIM 610250], respectively). The finding that these genes are widely expressed and their encoded proteins localize to mitochondria underscores the importance of mitochondrial function in neurodegenerative diseases.
In the current study, we describe an autosomal-recessive lethal hypomyelinating leukodystrophy in a large inbred Israeli Bedouin kindred. By using homozygosity mapping and mutation analysis of selected candidate genes within a linked genomic interval on 2q32.3-q33, we have identified a homozygous missense mutation, D29G, in HSPD1, encoding the mitochondrial Hsp60 chaperonin. The pathogenic effect of the D29G mutation was verified by an E. coli Hsp60-Hsp10/GroES-GroEL complementation assay. The disorder, which we have termed MitCHAP-60 disease, causes a “complicated” SPG, which is allelic to the pure autosomal-dominant SPG13. This finding provides evidence for the pivotal role of the mitochondrial Hsp60 chaperonin in the process of normal brain myelination and in the pathogenesis of hypomyelinating neurodegenerative disease.
Subjects and Methods
Patients
A highly consanguineous Israeli Bedouin kindred residing in several adjacent villages of the Galilee region of northern Israel was clinically and genetically investigated. Ten patients suffering from an autosomal-recessive fatal neurodegenerative disorder and 37 of their close relatives were included in the study. The study protocol was approved by the Institutional Ethics Review Committee and by the National Committee for Genetic Studies of the Israeli Ministry of Health, and informed consent was obtained from all participants or their legal guardians.
Clinical and Laboratory Evaluation
All affected individuals were clinically evaluated by pediatric neurologists and clinical specialists in pediatric metabolic disease. Clinical and biochemical details of deceased family members were collected from patients' medical charts. Biochemical evaluation with standard laboratory techniques included the following in all patients: measurement of plasma pH and concentration in plasma of glucose, electrolytes, liver, and muscle enzymes, BUN, creatinine, total carbon dioxide, lactate, pyruvate, aminoacids, very-long-chain fatty acids (VLCFAs), and isotransferrin, as well as urinary aminoacids, organic acids, and cerebro-spinal fluid (CSF) lactate, pyruvate, and alanine levels. Histologic and histochemical analyses of muscle biopsy specimens from four patients were performed by standard techniques. Spectrophotometric measurements of respiratory-chain enzyme activities and citrate synthase as well as Southern-blot analyses for detection of mitochondrial DNA depletion were carried out in muscle-biopsy specimens as previously reported.24,25 Electrophysiological tests, including nerve conduction, electromyography, brain-stem auditory evoked potentials, and visual evoked potentials were performed in nine patients.
Brain Imaging
Eight patients underwent brain MRI, with 1.5 T system (GE Signa 9.1) in four patients and 0.5 T system (GE Gyrex) in the other four. All patients were evaluated by conventional T1-weighted and T2-weighted images. Fluid-attenuation inversion recovery (FLAIR), diffusion-weighted images (DWI), and single-voxel proton MR spectroscopy (MRS) were obtained in four of eight patients. MRS voxels were located in the frontal white matter (WM) and in the thalamic gray matter. Peak areas of N-acetylaspartate (NAA), choline (Cho), creatine/phosphocreatine (Cr), myoinositol (mI), and lactate (Lac) were assessed and metabolite ratios of NAA/Cr, Cho/Cr, and mI/Cr were calculated.
Homozygosity Mapping and Linkage Analysis
Genomic DNA of all participants was extracted from peripheral lymphocytes by standard methods.26 A genome-wide scan was performed with 378 fluorescently labeled microsatellite marker pairs (Version 10 Human Screening Set-ABI Dyes, Research Genetics/Invitrogen). Microsatellite markers were amplified by multiplex polymerase chain reaction (PCR) with standard protocols. Amplicons were resolved with an ABI 310 sequencer system (Perkin Elmer) and analyzed with Genscan 3.1 and Genotyper 2.1 software (Applied Biosystems).
Single-nucleotide polymorphism (SNP) genotyping of 58,495 markers was performed on Affymetrix Human Mapping 50k Xba 240 arrays (Affymetrix) according to instructions provided by the manufacturer. In brief, 250 ng of genomic DNA from each subject was processed and labeled with reagents and protocols supplied by the manufacturer. After hybridization, the arrays were scanned with the GeneChip scanner 3000 with GeneChip Operating Software (GTYPE) version 1.4. Array CEL files were processed with Gene-Chip DNA Analysis Software version 4.0. Raw data of SNP genotyping results were sorted on Microsoft Office Excel sheets, and regions of homozygosity were detected and analyzed by visual inspection.
Linkage to the GJA12 locus on 1q41-q42, verification of linkage to the locus on chromosome 2q32.3-q33.1, haplotype analysis, and fine mapping were performed by genotyping of additional microsatellite markers derived from Marshfield maps (Mammalian Genotyping Service database of the Marshfield clinic) in all affected individuals included in the study and in their close relatives.
Two-point LOD-score calculations for markers D2S2387, D2S311, D2S2327, D2S316, D2S374, and D2S115 on 2q32.3-q33.1 were performed with the SUPERLINK program.27 We assumed an autosomal-recessive mode of inheritance with penetrance of 0.99, a disease mutant gene frequency of 0.01 and a uniform distribution of allele frequencies.
Sequencing of Candidate Genes
Exploration of the entire coding sequence and intron-exon boundaries of four candidate genes (MARS2 encoding the mitochondrial methionine-tRNA synthetase 2, COQ10B encoding coenzyme Q10 homolog B, HSPD1 encoding the mitochondrial chaperonin Hsp60, and HSPE1 encoding the mitochondrial chaperonin Hsp10 [GenBank accession number NC_000002.10]) and of the intergenic region between HSPD1 and HSPE1 was initially performed in one affected child (patient VIII-6 in the pedigree, shown in Figure 1). Intronic primer pairs were designed with the Gene Runner program version 3.05 (Hastings Software), based upon DNA sequences obtained from UCSC Genome Browser. Primer sequences and annealing temperatures used in PCRs for HSPD1 are shown in Table 1. Those used for MARS2, COQ10B, HSPE1, and the intergenic region between HSPD1 and HSPE1 are available on request. Amplicons were directly sequenced with the BigDye terminator sequencing system (Perkin Elmer Applied Biosystems) on an ABI PRISM 3100 sequencer (Perkin Elmer). Sequence variations were confirmed by bidirectional sequencing.
Figure 1.
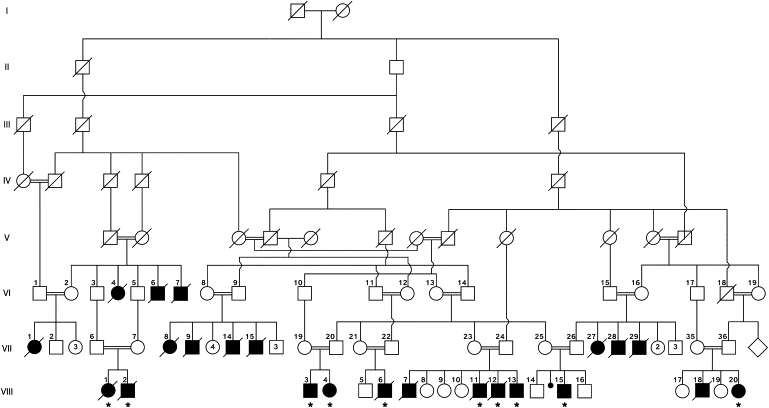
Pedigree of the Analyzed Subjects
The inheritance of the disease is compatible with an autosomal-recessive trait. Roman capitals indicate generations. Arabic numerals indicate serial numbers of study participants. Blackened symbols indicate affected individuals. Diagonal lines across symbols indicate deceased individuals. Asterisks indicate affected individuals who underwent DNA analysis.
Table 1.
Primer Sequences and Annealing Temperatures for HSPD1 Sequence Analysis
| Exon | Primer Sequence (5′→3′) |
PCR Product Size (bp) | Annealing Temp (°C) | |
|---|---|---|---|---|
| Forward | Reverse | |||
| 2 | GCTCAGCATCTCACGTTG | ATCCACCTTGAGCTGTAAG | 537 | 53 |
| 3 | TGAGACTTTGAAGTAGTTGC | ACCTACCCACCATACAGC | 535 | 51 |
| 4 | TTGACATAGAAGCCATGAG | ACCTTTGACAATGGCTAAG | 533 | 51 |
| 5 | TCAATTTGGAAGTTACTAGG | GGTAGAGTATGAAACAGCC | 488 | 54 |
| 6 | GCTGTTTCATACTCTACCC | CAGCTACTTGGGAGACTG | 388 | 55 |
| 7 | ATTAGTTGGTGTCATCCTC | ATCAAATGTGGAGGTACAC | 456 | 53 |
| 8 | CCAGATTACTTAGGGAGTCC | AGTCAGTAAGCTCATTCTTAGG | 391 | 57 |
| 9 | GGAAATGCAGTGAAAGAG | GTTAATTCCTCAAGTATTCG | 537 | 50 |
| 10 | TAGTGGAGACAGGGCTTCAC | TGTGATGCATGTTTAGCTCTG | 540 | 57 |
| 11 | ATTTCCCTACATCAATATCC | CATGAGAATTGTTGATGGG | 418 | 54 |
| 12 | GGTCTTGAACTCCTAACCTC | GTTATTGGTGAGGAACACTGC | 378 | 61 |
| Exon 2, mismatch RFLP | CTCCTCATCTCACTCGGGCTCCTGCCAAAG | GGTACCTTTGGCCCCATTGTAACGG | 124 | 64 |
Mutation Detection by Mismatch Primer Restriction Assay
A mismatch restriction-fragment-length polymorphism (RFLP) assay was constructed and utilized for mutation detection in investigated family members and in healthy controls. Because the g.1512A→G (D29G) mutation in exon 2 of HSPD1 could not be identified by any known restriction enzyme, we designed a 30-bp-long mismatching forward primer and a matching reverse primer to amplify a 124-bp-long fragment encompassing the mutation (see Table 1 for RFLP primer sequences). Table 1 shows the RFLP primer sequences, with the two consecutive mismatching C nucleotides in the forward primer located 9 and 10 bp 5′ to g.1512A highlighted in bold. Amplicons containing both the mismatching forward primer and the g.1512A→G mutation introduced a BslI (New-England Biolabs) restriction site, which digested the 124 bp product into 94 bp and 30 bp fragments. Fragments were separated by electrophoresis on 5% MetaPhor Agarose gel.
Multiple Protein Alignment
Analysis of conservation of the D29 residue in Hsp60 was performed by multiple sequence alignment of ten Hsp60 homolog proteins from various species with the ClustalW software with a Java viewer. Species-specific Hsp60 homolog sequences were derived from the MitoRes database.
E. coli Complementation Assay
The GroEL-GroES group I bacterial chaperonin complex is the E. coli homolog of mitochondrial Hsp60-Hsp10 and is essential for bacterial survival under all conditions tested.28 It was previously shown that expression of the human Hsp60-Hsp10 genes can complement deleted GroEL-GroES in E. coli strains and effectively support bacterial growth at moderate temperatures.29,30 On the basis of these findings, we performed an E. coli complementation assay, essentially as previously described,17,30 to experimentally determine the ability of the D29G-Hsp60 variant to support E. coli survival. In brief, we used a GroEL-GroES deletion allele marked with both a Chloramphenicol-resistance (CamR) cassette in the operon and a Tetracycline-resistance (TetR) cassette nearby on the chromosome to replace the wild-type (WT) GroEL-GroES operon in our test strains by T4gt7 phage-mediated transduction.30 Besides the WT GroEL-GroES operon on the chromosome, the test strains also carry plasmid pOFX (Kanamycin resistant [KanR], isopropyl-β-D-fthiogalactopyranoside [IPTG]-inducible expression) containing either the WT human Hsp60-Hsp10 set of genes or the Hsp60(D29G)-Hsp10(WT) variant. Fresh transductants were first selected for TetR by incubation on LB-agar plates containing 12.5 μg/ml of tetracycline and 500 μM IPTG at 30°C for 48 hr. These were then tested for CamR, indicating that the deletion allele had replaced the WT GroEL-GroES operon on the chromosome. Because the cotransduction frequency between the two resistance markers was normal (90%), and so that the risk of accumulating nonspecific suppressors was minimized, in subsequent experiments GroEL-GroES-deleted transductants were directly selected for CamR at 30°C in the presence of 500 μM IPTG for 48 hr. Single colonies from independent transductions (12 expressing the D29G mutant and 12 expressing WT Hsp60) were resuspended in 10 mM MgSO4, diluted serially 20-fold, and spotted on LB-agar plates containing varying amounts of IPTG inducer and 12.5 μg/ml of chloramphenicol; this was followed by incubation at 30°C, 37°C, and 38.5°C for various times.
Measurement of Hsp60 Levels
To verify equivalent levels of Hsp60 expression by E. coli containing the various plasmid constructs, we performed measurement of WT and D29G mutant Hsp60 protein levels by western blotting and quantitative immunoassay. All analyses were performed on seven independent GroEL-GroES-positive cultures (GroESL-plus) and seven independent GroEL-GroES-deleted cultures (GroESL-minus). Each group of seven cultures was transformed with either WT or D29G-Hsp60 mutant plasmid. Western blotting was performed by standard procedures,15 and quantitative immunoassay of Hsp60 was performed at room temperature with xMAP technology as previously described.31
Results
Clinical and Laboratory Evaluation
Over the past decade, 23 children of the family investigated in the current study have been diagnosed as suffering from an autosomal-recessive fatal neurodegenerative disorder of early onset, associated with hypomyelinating leukodystrophy (see pedigree in Figure 1). The clinical features of ten affected individuals are presented in Table 2. Early clinical manifestations in all patients included hypotonia, nystagmus, and psychomotor developmental delay, followed by appearance of prominent spasticity, developmental arrest, and regression. Head circumference, which was normal at birth, showed decreased growth rate. Seizures were reported in six patients. Feeding problems commonly led to malnutrition and growth failure. Death usually occurred within the first two decades of life from aspiration pneumonia or sudden death of unknown cause. One of the most extreme presentations was hydrops fetalis in one patient, who eventually died at the age of 2 years. Patients with the more severe course died before the age of 2 years, with some of them never gaining social eye contact or any other developmental milestone. These severely affected patients suffered from recurrent episodes of shallow breathing and apneic spells during acute febrile illnesses and died during such episodes. In patients who survived beyond the age of 2 years, the motor disability, which mainly included progressive limb spasticity and contractures, seemed to be more prominent than the psychointellectual impairment. Patients with greater longevity (at present, the oldest surviving patient is age 14 years of age) sometimes showed improvement of nystagmus, but all suffered from strabismus and lack of normal head control, in addition to choreoatetotic movements in some. A wide range of intrafamilial and interfamilial phenotypic heterogeneity at the level of psychomotor impairment and in the rate of neurologic and developmental deterioration was evident among affected individuals carrying the same HSPD1 mutation.
Table 2.
Clinical Characteristics of Ten MitCHAP-60 Patients
| Characteristic | Number of Patients |
|---|---|
| Affected individuals included in the study | 10 |
| Age at onset of symptoms between birth to 3 months | 10/10 |
| Deceased <2 years of age | 3/10 |
| Deceased between 5 and 12 years of age | 2/10 |
| Alive at present (11 months to 14 years of age) | 5/10 |
| No head control | 10/10 |
| Head titubation | 10/10 |
| Truncal hypotonia | 10/10 |
| Spastic paraplegia, increased tendon reflexes/positive Babinski sign | 10/10 |
| Strabismus | 10/10 |
| Nystagmus | 9/10 |
| Psychomotor retardation | 10/10 |
| Severe | 8/10 |
| Some social interaction and few words | 2/10 |
| Rate of disease progression/deterioration | |
| Rapid (died prior to 2 years of age) | 3/10 |
| Slow | 7/10 |
| Seizures | 6/10 |
| Exacerbation during febrile episodes | 10/10 |
| Episodes of abnormal breathing/apneic spells (more often in patients deceased prior to 2 years of age) | 6/10 |
| MRI showing hypomyelinating leukodystrophy | 8/8 |
| BERA results compatible with abnormal brain-stem function | 9/9 |
| Elevated urinary ethylmalonic acid levels | 5/8 |
| Elevated plasma lactate during acute illness with normal CSF lactate | 4/106/6 |
| Muscle biopsy | |
| Electron microscopy—abnormal | 1/4 |
| Cytochrome c oxidase activity—mildly reduced | 1/4 |
Routine and extensive metabolic laboratory investigation including blood glucose, electrolytes, liver and muscle enzymes, kidney-function tests, pH, bicarbonate, VLCFA, isotransferrin, serum, urinary amino acids, plasma, and CSF lactate, pyruvate, and alanine did not reveal any specific biochemical marker associated with the investigated disorder. However, in five of eight studied patients in whom we conducted urinary organic amino acid measurements, we found intermittently markedly increased urinary secretion of ethylmalonic (25–169 mmol/mol creatinine; normal ≤ 12) and to a lesser extent methylsuccinic acids. The highest level of urinary ethylmalonic acid was found in the patient who presented with hydrops fetalis. In three of five of these patients, we also found elevated levels of C4 acylcarnitine.
The plasma lactate level, which is considered as a basic screening marker for mitochondrial respiratory-chain defects, was found to be normal, except for mild elevation, i.e., 2.6–4.2 mmol/l, documented in four patients during acute encephalopathic exacerbations (normal level ≤ 2). Of note, CSF lactate pyruvate and alanine levels were normal in all six patients in whom this was measured, even during episodes of acute deterioration.
In all four muscle biopsies performed, no evidence of mitochondrial function impairment, such as ragged-red fibers or cytochrome c oxidase (COX)-deficient fibers, was found. However, in one patient examined, we found ultrastructural changes in the muscle as manifested by morphologically giant mitochondria with abnormal cristae (results not shown). Only in the most severely affected patient with hydrops fetalis did we observe mildly reduced activity of COX (40% of control level).
No major abnormalities were found in nerve conduction and electromyography studies. Brain-stem auditory-evoked-potential studies showed increased latency of wave I and absence or small size of later components, denoting brain-stem abnormalities. Visual evoked potentials were abnormal with slowing latency and aberrant wave shape in two of three studied patients.
MRI/MRS
Because all parents declined autopsy of their deceased children, no brain specimens were available for histopathologic analysis. MRI enables noninvasive visualization of abnormal myelination and was therefore used as a diagnostic tool for anatomic evaluation of the investigated neurodegenerative brain disorder. In all patients examined, the youngest of whom was a 5-month-old infant, all cerebral and cerebellar white-matter structures as well as the corticospinal tracts were of high signal intensity on T2-weighted images (Figures 2A–2C). There was no evidence of normal myelination, regardless of the age of the patient. All patients exhibited a thin corpus callosum and showed various degrees of ventricular enlargement. Enlargement of sulci and subarachnoid spaces, mainly in the frontal and parietal areas, were observed in six of eight patients. The degree of widening of cerebral sulci was in concordance with the severity of the clinical symptoms. The brain stem was thin in all patients (Figure 2A). In five of eight patients, the cerebellum and vermis were smaller than normal for age. DWI disclosed high signal intensity along the corticospinal tracts with decreased values on apparent diffusion coefficient (ADC) maps, indicating restricted diffusion. One patient had two MRI studies, the first at the age of 10 months and the second at 2.5 years of age. In the latter study, the cisterna magna and cerebellar and vermian sulci were wider in size as compared to those of the earlier study, suggesting progressive cerebellar atrophy. The cerebral hemispheres did not exhibit significant changes in size or intensity, and the cerebral subarachnoid spaces maintained their original size. On MRS studies performed on the frontal white matter, increased mI/Cr ratios and normal Cho/Cr ratios were found in all patients. The NAA/Cr ratio was normal or slightly reduced. Abnormal accumulation of lactate was detected in only two patients with neonatal disease presentation. MRS detected slightly reduced NAA/Cr ratio in the thalamus (gray matter) (Figure 2E).
Figure 2.
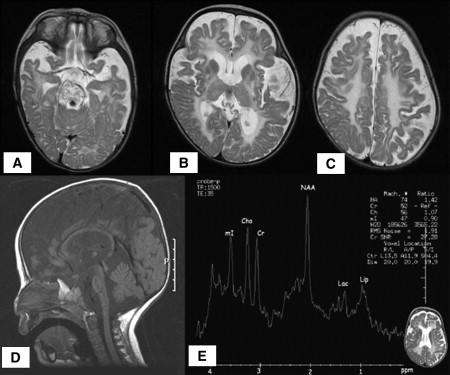
MRI/MRS Brain Imaging
(A–C) Axial T2WI in a 17-month-old patient showing high intensity of the white matter consistent with the complete absence of myelin.
(D) Sagital T1WI of a 16-month-old patient demonstrating a small brain stem and atrophy of the cerebellum and vermis.
(E) Proton MR spectroscopy MRS (TE = 35) of a 16-month-old patient disclosing a reduced NAA/Cr ratio and increased mI/Cr ratio in the white matter, a normal Cho/Creatine ratio consistent with hypomyelination rather than with myelin destruction, and peaks for lactate and lipids, consistent with defective oxidative phosphorylation.
HSPD1 Mapping on 2q32.3-q33.1
Genotyping of polymorphic DNA markers flanking GJA12 on 1q41-q42 failed to reveal linkage with the disease phenotype, thus ruling out a connexin 47 defect as the cause for the investigated disorder in this kindred. A microsatellite-based genome-wide scan with the 378 Research Genetics/Invitrogen marker set performed in six affected children (subjects VIII-3, VIII-6, VIII-11, VIII-12, VIII-15, and VIII-20 in the pedigree, shown in Figure 1) failed to reveal linkage to a specific chromosomal locus. Accordingly, we turned to a 50,000-SNP-marker-microarray genome-wide scan, which was performed in three affected children (subjects VIII-3, VIII-6, and VIII-13). The call rate was > 97%, and visual inspection of SNP genotypes revealed a 4.58 Mb long continuous segment of shared homozygosity among all three patients, spanning 103 consecutive SNPs between markers rs1478129 and rs1379020 (at physical positions 194,382,571 and 198,963,363, respectively), on chromosome 2q32.3-q33.1. In two of the three analyzed patients (VIII-3 and VIII-13), the shared regions of homozygosity were contained within very large stretches of homozygosity of 14.1 Mb and 47.7 Mb, respectively. We also found five additional significantly smaller regions of shared homozygosity on chromosomes 1, 2, 4, 17, and 20, ranging in length between 1.0–1.5 Mb. However, because none of these were contained within larger stretches of individual homozygosity, we first analyzed the region of suggestive linkage on 2q32.3-q33.1. This region includes the microsatellite marker D2S117 (at physical position 195,327,044), contained in the Research Genetics/Invitrogen marker set. Genotyping of D2S117 failed to show linkage to the disease, thus narrowing down the linked interval to a 3.6-Mb-long segment within 2q32.3-q33.1. Linkage to the locus on 2q32.3-q33.1 was confirmed by genotyping of six additional microsatellite markers contained within the homozygous interval in ten affected children and their close relatives (Figure 3). The maximum two-point LOD score was 6.4 for marker D2S374 at a recombination fraction (θ) = 0.00 (Table 3). All affected individuals share a common homozygous haplotype between markers D2S117 and D2S1384, supporting a common ancestral origin for the linked locus on 2q32.3-q33.1.
Figure 3.
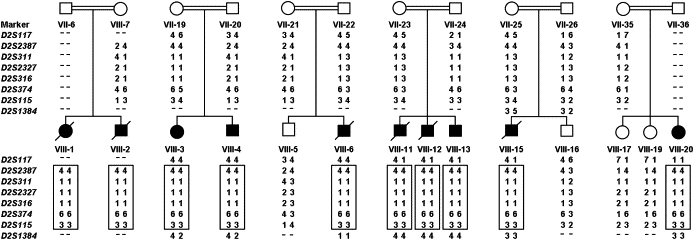
Haplotype Analysis
Confirmation of linkage was performed by genotyping of eight polymorphic microsatellite markers within the SNP-based candidate region on 2q32.3-q33.1 in ten affected children and their close relatives. Blackened symbols and diagonal lines across symbols indicate affected and deceased individuals, respectively. Numbers below symbols correspond to the position of individuals within the pedigree (see Figure 1 for details). Hyphens indicate nongenotyped markers. The shared disease-associated haplotype is shown in boxes.
Table 3.
Two-Point LOD Scores for Markers on 2q32.3-q33.1
| Locus | LOD at θ = 0.00 |
|---|---|
| D2S117 | 3.9516 |
| D2S2387 | 4.4752 |
| D2S311 | 3.23.4 |
| D2S2327 | 3.5143 |
| D2S316 | 3.5791 |
| D2S374 | 6.3972 |
| D2S115 | 4.0681 |
Mutation Analysis
Of the 23 genes contained within the linked region (NCBI Map Viewer, build 36.2), 18 are of known function and five are predicted protein coding genes of unknown role. Of the 18 known genes, eight were ruled out as disease-associated candidates on the basis of their known function or tissue expression (data derived from Online Mendelian Inheritance in Man [OMIM] and GeneCards databases). Of the ten remaining candidates, we first chose to sequence four genes implicated in normal mitochondrial function, on the basis of increasing evidence suggesting the potential role of mitochondrial dysfunction in the pathogenesis of neurodegenerative disorders.32–35
Sequencing was initially performed on genomic DNA derived from patient VIII-6. Sequence analysis of all coding exons and intron-exon boundaries of MARS2 COQ10B and HSPE1 revealed no sequence variations. Sequence analysis of HSPD1 encoding Hsp60 revealed two sequence variations: (1) a homozygous missense mutation g.1512A→G in exon 2 at position 86 of the cDNA sequence, causing an aspartic acid→glycine exchange at amino acid 29 of the Hsp60 protein sequence (D29G) (Figure 4A) and (2) a known homozygous synonymous SNP g.2981A→G (rs8539, according to NCBI dbSNP database) in exon 3 at position 273 of the cDNA sequence, encoding a lysine residue at position 91 of the Hsp60 protein sequence and resulting in no amino acid change. Sequencing of the HSPD1-HSPE1 intergenic region containing the bidirectional promoter region for these two mitochondrial chaperonins revealed a previously described homozygous sequence variation g.3175G→C36 (rs1116734 according to NCBI dbSNP database).
Figure 4.
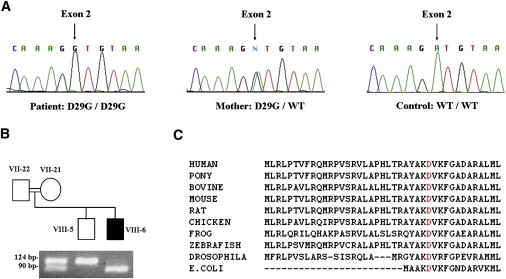
HSPD1 Mutation Analysis
(A) Genomic DNA sequence analysis of HSPD1 reveals a homozygous A→G transition at position g.1512 (indicated by an arrow) in exon 2, resulting in an aspartic-acid-to-glycine substitution at position 29 of the Hsp60 protein (D29G). The mutation is carried in a homozygous state by an affected child (left panel) and in a heterozygous state by his mother (middle panel). A healthy control shows the wild-type (WT) sequence (right panel).
(B) Confirmation of the D29G mutation in an affected individual and his close relatives by mismatch PCR-RFLP. Numbers adjacent to symbols correspond to the position of individuals within the pedigree (see Figure 1 for details). PCR amplification was performed as described in the text. Amplicons containing both the mismatching forward primer and the D29G mutation in exon 2 of HSPD1 introduce a recognition site for BslI, which digests the 124 bp PCR product into 94 bp and 30 bp fragments. Homozygous carriers of the mutation display a 94 bp fragment (the additional 30 bp fragment is not shown), healthy individuals carrying the wild-type sequence show a 124-bp fragment, and heterozygous carriers of the mutation display two fragments of 124 bp and 94 bp.
(C) ClustalW analysis of the Hsp60 N-terminal region encompassing the mutation site demonstrates that the D29 residue (highlighted in red) is highly conserved across Hsp60 homologs from various species. The D29 residue in human Hsp60 corresponds to D5 in the E. coli homolog protein because of the lack of a mitochondrial-targeting sequence in bacteria.
Of the three identified sequence variations, only the D29G mutation in HSPD1 segregated completely with the disease; all ten affected children were homozygous for the mutation and all parents were heterozygous, whereas healthy siblings were either heterozygous or wild-type in a manner consistent with haplotype analysis. Most unaffected individuals were homozygous for both of the known SNPs, g.2981A→G in exon 3 of HSPD1 and g.3175G→C in the HSPD1-HSPE1 intergenic region, thus ruling out segregation of these sequence variations with the disease.
The D29G variant of HSPD1 was verified by the mismatch restriction assay (Figure 4B), and the RFLP pattern was compatible with exon 2 sequencing results in all investigated family members. The D29G mutation was not present in 678 chromosomes of healthy ethnically matched control samples.
The D29 residue was found to be highly conserved across Hsp60 homologs from nine different nonhuman species (Figure 4C) and is contained within a conserved domain adjacent to the first 26 N-terminal residues composing the mitochondrial matrix targeting sequence (according to Hsp60 predicted domains derived from Swiss-Prot database, accession number P10809). In an alignment of human Hsp60 with the E. coli Hsp60 homolog GroEL, the human D29 residue corresponds to D5 in E. coli GroEL. Inspection of the crystal structure of E. coli GroEL37 shows that D5 is located within the first β strand of the GroEL equatorial domain, facing the interior of the chaperonin central cavity.
E. coli Complementation Assay and Measurement of Hsp60 Levels
E. coli B178 carrying a plasmid with either WT Hsp60-Hsp10 or Hsp60(D29G)-Hsp10(WT) was the recipient in transduction experiments replacing the WT GroEL-GroES operon on the chromosome with a deleted version marked with a CamR gene. The cotransduction frequency with a nearby TetR marker was 90% for both WT (90/100) and D29G-Hsp60-expressing cells (88/100). This indicates that the plasmid-encoded WT Hsp60 and Hsp60(D29G) mutant proteins both support E. coli survival under these conditions.
More extensive complementation assays showed that all E. coli transductants that lost the GroEL-GroES locus and were kept alive with the pOFX mutant Hsp60(D29G)-Hsp10(WT) plasmid grew significantly more slowly and were more temperature sensitive than the corresponding Hsp60(WT)-Hsp10(WT) cells under all conditions tested, except at 30°C on 500 μM IPTG plates, in which growth was similar. At lower IPTG concentrations, however, the D29G mutants grew much more slowly than the wild-type, even at 30°C (Figure 5).
Figure 5.
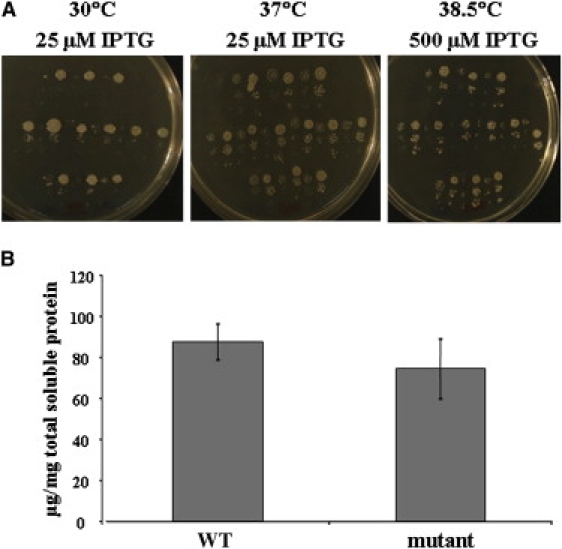
E. coli Complementation Assay
(A) Impaired growth of GroEL-GroES-deleted E. coli cells complemented with the D29G-Hsp60 variant under various conditions. Single, fresh transductant colonies (12 expressing the wild-type Hsp60 and 12 expressing the mutant D29G-Hsp60) were resuspended in 10 mM MgSO4; 20-fold serial dilutions were made, alternately spotted on LB agar plates containing varying amounts of IPTG inducer and 12.5 μg/ml of chloramphenicol, and incubated at various temperatures. Growth impairment of serial dilutions of mutant as compared to wild-type transductans alternating from left to right at 30°C in 25 μM IPTG (left panel), at 37°C in 25 μM IPTG (middle panel) and at 38.5°C in 500 μM IPTG (right panel) is shown.
(B) Measurement of Hsp60 expression in GroEL-GroES-deleted E. coli transductants by immunoassay. The mean expression levels of Hsp60 in eight individual bacterial transductants, four expressing the wild-type Hsp60 (WT) and four expressing the mutant D29G-Hsp60 (mutant), were found to be comparable (p value for two-sample Student's t test = 0.47). Error bars indicate the standard error of the mean.
Measurements of intracellular Hsp60 levels by quantitative immunoassay (Figure 5) and by western-blot analysis (data not shown) revealed comparable levels of WT and D29G-Hsp60 mutant protein expression, thus ruling out the possibility of increased temperature sensitivity of mutant E. coli transductants due to reduced D29G-Hsp60 expression.
All these experiments taken together clearly show that bacteria expressing the D29G variant grow considerably more slowly than WT cells under most conditions tested, and especially at elevated temperatures (37°C and 38.5°C), indicating that the D29G variant is significantly compromised in its chaperone function.
Discussion
In the current study, we describe an autosomal-recessive hypomyelinating leukodystrophy caused by a missense mutation in the mitochondrial Hsp60 chaperonin, which we have accordingly termed MitCHAP-60 disease. The ten patients studied exhibited the following shared cardinal features: (1) rotatory nystagmus, which was the presenting symptom in most patients; (2) progressive spastic paraplegia within the first months of life, followed by a variable rate of neurological deterioration and regression; (3) severe motor impairment, with none of the affected children ever gaining normal head control; (4) profound mental retardation; and (5) hypomyelinating leukodystrophy as evidenced by brain MRI.
The use of homozygosity mapping and then mutation analysis of candidate genes within the linked locus on 2q32.3-q33.1 enabled us to pinpoint a homozygous missense mutation (D29G) in HSPD1, encoding the mitochondrial Hsp60, as responsible for MitCHAP-60 disease. The finding of the D29G missense mutation was surprising, given the fact that disease-causing mutations in human Hsp60 have so far been exclusively identified in individuals suffering from SPG13, a relatively mild and late-onset form of hereditary SPG transmitted in an autosomal-dominant pattern.17,18 SPG13 represents an uncomplicated form of hereditary SPG. To date, two pathogenic heterozygous missense mutations in HSPD1 have been identified in families with SPG13: V98I in seven affected adults from a French family and Q461E in a 58-year-old woman from a Danish family. In both families, the disease was transmitted as a late-onset autosomal-dominant trait, with variable penetrance. Whereas the V98I-Hsp60 mutation was associated with an age-dependent, highly penetrant disorder, the Q461E mutant exhibited much lower penetrance, as suggested by the finding of asymptomatic heterozygous carriers in their sixth or seventh decade.17,18 These differences in penetrance correlated with the results of E. coli complementation assays with the two identified HSPD1 mutations; these results showed that the V98I variant was completely unable to support the survival of GroEL-GroES deleted bacteria, whereas the Q461E variant only mildly compromised bacterial growth. The results of these functional tests suggest the existence of a genotype-phenotype correlation between the functional pathogenicity of HSPD1 mutations in the complementation assay and clinical disease severity or penetrance.
The current HSPD1 missense mutation identified in MitCHAP-60 results in a substitution of aspartic acid for glycine at amino acid 29 of the Hsp60 protein. Several findings support the causative role of the D29G mutation in the pathogenesis of MitCHAP-60 disease. These include (1) complete segregation of the mutation with the disease-associated phenotype in all investigated family members; (2) the absence of the mutation in a large number of ethnically matched controls; (3) the high degree of conservation of the D29 residue across Hsp60 homologs of various nonhuman species, from vertebrates to bacteria, suggesting its functional importance; and (4) the E. coli complementation-assay results, which clearly demonstrate the markedly reduced ability of the D29G-Hsp60 mutant protein to support E. coli survival at all temperatures, but especially at the higher ones. Taken together, all these findings strongly suggest that the homozygous state of the g.1512A→G mutation of HSPD1 resulting in the D29G mutation compromises Hsp60 protein function and is responsible for the severe phenotype of MitCHAP-60 disease.
The fact that E. coli survival was not completely abolished by the D29G-Hsp60 variant suggests that the D29G substitution may exert a less severe effect on Hsp60 function, as compared to the previously described Hsp60-V98I variant causing SPG13. This result is not surprising, given the fact that all heterozygous carriers of the D29G mutation in our study are completely asymptomatic, with some of the carriers reaching their ninth decade of life, suggesting that a single D29G mutant allele does not significantly impair overall mitochondrial Hsp60 activity.
The mechanisms by which HSPD1 mutations in general and the D29G-Hsp60 variant in particular may compromise normal Hsp60-chaperonin function is not well understood. According to the inferred structures of GroEL, the E. coli homolog of Hsp60, all bacterial equivalents of the disease causing Hsp60 substituted residues that have been identified so far (i.e., V98, Q461, and D29) are located within the equatorial domain of the chaperonin molecule.37,17,18 In vitro studies have shown that the V98I variant Hsp60 protein forms heptameric chaperonin complexes but displays strongly reduced chaperone activity with malate dehydrogenase as a substrate.31 The specific effect of the D29G mutation on Hsp60 conformational structure and chaperonin activity remains to be delineated. Moreover, it is unclear whether the two additional and apparently nonpathogenic SNPs identified in HSPD1 of MitCHAP-60 patients (i.e., g.2981A→G in exon 3 and g.3175G→C in the HSPD1-HSPE1 intergenic region) also exert a modifier effect on Hsp60 conformation and function, especially in combination with the D29G mutation.
Of special interest are the potential mechanisms by which different mutations in HSPD1 may lead to such dramatic differences in both the pattern of inheritance and the disease phenotype between SPG13 and MitCHAP-60. It has been previously postulated that the autosomal-dominant inheritance of SPG13 results from a dominant-negative effect exerted by defective Hsp60 monomers on proper assembly of the whole oligomeric chaperonin molecule.17 However, recent in vivo experiments in a bacterial model system indicate that heterologous complexes consisting of both wild-type and V98I mutant Hsp60 subunits are formed and that incorporation of mutant subunits does not result in a strong dominant-negative effect.31 One would also not expect the D29G subunits to exert any significant dominant-negative effect in mixed oligomers with the Hsp60 WT because there is no observable phenotype in the heterozygous state. Consistent with our functional analyses, different patterns of inheritance of MitCHAP-60 and SPG13 may thus be explained in the following way: The D29G mutation, which exerts a relatively milder effect on Hsp60 function, only manifests in the homozygous state, whereas the V98I mutation, having a more severe effect on Hsp60 activity, produces a phenotype in the heterozygous state. Nevertheless, homozygosity for the mild mutation causes a more severe phenotype than heterozygosity for the severe one.
The link between mitochondrial derangements and neurodegenerative diseases is well known32–35 and served as the initial impetus to consider the HSPD1 gene as a candidate for MitCHAP-60. Because Hsp60 is a mitochondrial chaperonin, it is reasonable to assume that neuronal damage is initiated by some form of mitochondrial dysfunction. Experiments in yeast have shown that null mutations of Hsp60 render the cells nonviable, secondary to severe mitochondrial-protein-folding defects, whereas those with conditional mutations accumulate misfolded proteins that are incapable of forming active oxidative-phosphorylation enzyme complexes.38 In another recent in vitro experiment using neuronal cells, Hsp60 was shown to exert a protective effect on vital mitochondrial respiratory-chain enzymes against poisoning by β-amyloid, indicating the preventive effect of Hsp60 against mitochondrial damage.39 Of interest is the finding that defects in paraplegin, another nuclear-encoded mitochondrial protein with chaperone-like function, are the cause of SPG7, a complicated form of autosomal-recessive SPG.40 Taken together, all these findings suggest that mitochondrial chaperonin defects exert a profound impact on overall mitochondrial biogenesis, integrity, and function. The absence of some of the classical clinical and laboratory manifestations of impaired electron-transport-chain activity in MitCHAP-60 disease, such as evidence of elevated lactic-acid production or reduced activity of respiratory-chain complexes, is suggestive of diverse cellular effects of mitochondrial dysfunction besides impaired oxidative phosphorylation for production of ATP. Among others, these include exaggerated production of reactive-oxygen species or apoptosis, which may underlie the neuronal-disease phenotype exhibited by the MitCHAP-60 patients and is reminiscent of the pathogenesis of Leber's hereditary optic atrophy.35
It is possible that one of the most important means of understanding the precise mechanisms leading to neurodegeneration secondary to Hsp60 defects depends on identifying the biochemical interactions of the Hsp60-Hsp10 mitochondrial machinery with its various protein substrates, both under physiologic conditions as well as during biophysical stress or toxic cellular-protein overload. Unfortunately, the full range of Hsp60 substrates has not yet been delineated in human mitochondria. It is reasonable to assume that various Hsp60 disease-causing mutations differ in the type and extent of substrate-processing defects they exert. Identifying those substrates that are critically dependent on Hsp60 function may shed light on disease mechanisms related to their native biological function. In this regard, it is interesting that our MitCHAP-60 patients were found to have intermittently increased excretion of ethylmalonic acid (EMA), mainly during acute febrile episodes. EMA is considered a biochemical marker for short-chain acyl-CoA dehydrogenase deficiency (SCAD deficiency [MIM 201470]). In vitro studies have shown that interaction of Hsp60 with SCAD is crucial for correct folding of the latter.41,42 Thus, the increased urinary EMA in MitCHAP-60 patients may indicate either a causative or sentinel role of abnormal biogenesis of SCAD proteins associated with the Hsp60 function defect.
Although most cellular Hsp60 is localized to the mitochondria, ∼15%–20% of the protein is located at extramitochondrial sites.43 Growing evidence connects cytosolic accumulation of Hsp60 with apoptosis under both physiologic and disease states.44–46 The rostral ventrolateral medulla (RVLM) of the brain stem governs central cardiovascular regulatory integrity, and its disruption leads to brain-stem death. With an experimental model of endotoxemia, it has been shown that redistribution of Hsp60 from the mitochondria to the cytosol in the RVLM exerts a neuroprotective effect against fatal cardiovascular depression by inhibiting apoptosis.44,45 During acute febrile illnesses, several MitCHAP-60 patients suffered from episodes ranging from disordered breathing with apneic spells to cardiac dysrrhythmias with unexplained sudden death. These phenotypic features combined with brain stem and medullary atrophy depicted in brain MRI images could reflect in vivo abnormal regulation of RVLM caused by compromised cytosolic Hsp60 function.
Hypomyelination has been reported in association with several established disorders, including the prototype PMD, PMLD, Cockayne syndrome type B (CSB [MIM 133540]), hypomyelination, atrophy of the basal ganglia and cerebellum, and hypomyelination and congenital cataract syndrome (HCC [MIM 610532]), among others.47 Analysis of mutations in PLP1 encoding the major protein of CNS myelin has revealed the previously unappreciated role of glial myelination in the maintenance of CNS integrity. There is a large overlap in the clinical and MRI features of MitCHAP-60, PMD, and PMLD,48 suggesting that the HSPD1, PLP1, and GJA12 gene products, among others, may comprise components of a common pathway, whose disruption may lead to hypomyelination and SPG. Notwithstanding the similarity of these MRI findings, there are in fact significant differences among the various hypomyelinating disorders. Of note is the fact that, as opposed to PMLD patients affected by GJA12 mutations, in whom some degree of myelination restricted to the corticospinal tract can be identified by MRI,6 our MitCHAP-60 patients exhibit complete absence of brain myelination. This difference in the extent of brain myelin deficiency could explain the relatively slow progression of spasticity in patient with GJA12 defects, in contrast with the early and severe spasticity in MitCHAP-60 patients. Interestingly, PMD and its milder variant spastic paraplegia 2 (SPG2 [MIM 312920]) can serve as an example of allelic dysmyelinating disorders sharing great similarity with our newly identified allelic couple—MitCHAP-60 disease/SPG13.49 This phenotypic resemblance between MitCHAP-60/SPG13 and PMD/SPG2 allelic couples leads us to consider that common pathogenic mechanisms may contribute to these disorders.
In conclusion, we have identified a homozygous missense mutation (D29G), not previously described, in the HSPD1 gene encoding the mitochondrial chaperonin Hsp60 as the cause of a severe neurodegenerative disorder herein termed MitCHAP-60 disease. This allows genetic counseling and early prenatal diagnosis in a large consanguineous population in northern Israel. MitCHAP-60 is allelic to SPG13, constituting a continuum of neurological hypomyelinating syndromes with a wide spectrum of clinical severity caused by a common genetic defect. Our work has led to the inclusion of Hsp60 to the list of proteins involved in myelinogenesis and further emphasizes the role of chaperones in neurodegenerative disorders. It also contributes to the evidence that mitochondrial integrity plays a major part in a well-orchestrated myelinogenic program and in the pathogenesis of neurodegenerative disorders.
Supplemental Data
A table containing genome-wide scan data using 50K-SNP-marker microarray is available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ClustalW software, http://www.hongyu.org/software/clustal.html
GeneCards, http://www.genecards.org/
Mammalian Genotyping Service database of the Marshfield Clinic,
http://research.marshfieldclinic.org/genetics/GeneticResearch/compMaps.asp
MitoRes database, http://www2.ba.itb.cnr.it/MitoNuc/index.php
NCBI dbSNP database, http://www.ncbi.nlm.nih.gov/SNP/
NCBI Map Viewer (build 36.2), http://www.ncbi.nlm.nih.gov/mapview/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Superlink online, http://bioinfo.cs.technion.ac.il/superlink-online
The ExPASy proteomics server of the Swiss Institute of Bioinformatics (Swiss-Prot), http://us.expasy.org/cgi-bin/niceprot.pl?P10809
UCSG Genome Browser, http://genome.ucsc.edu/
Acknowledgments
We are very grateful to the families with MitCHAP-60 disease for participating in this study. We thank Eli Sprecher and Sarah Selig for discussions and useful suggestions, Guennady Yudkovsky for technical assistance, and the National Laboratory for the Genetics of Israeli Populations for providing control DNA samples for RFLP analysis. This research was supported by grants from the Barry Rose Fund and the Veronique Elek Fund of the Canadian Technion Society, and the Rosalinde and Arthur Gilbert Foundation of the American Technion Society.
References
- 1.Van der Knaap M.S., Breiter S.N., Naidu S., Hart A.A., Valk J. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology. 1999;213:121–133. doi: 10.1148/radiology.213.1.r99se01121. [DOI] [PubMed] [Google Scholar]
- 2.Shiffmann R., Boesflug-Tanguy O. An update on the leukodystrophies. Curr. Opin. Neurol. 2001;14:789–794. doi: 10.1097/00019052-200112000-00018. [DOI] [PubMed] [Google Scholar]
- 3.Garbern J., Cambi F., Shy M., Kamholz J. The molecular pathogenesis of Pelizaeus-Merzbacher disease. Arch. Neurol. 1999;56:1210–1214. doi: 10.1001/archneur.56.10.1210. [DOI] [PubMed] [Google Scholar]
- 4.Cailloux F., Gauthier-Barichard F., Mimault C., Isabelle V., Courtois V., Giraud G., Dastugue B., Boespflug-Tanguy O. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur. J. Hum. Genet. 2000;8:837–845. doi: 10.1038/sj.ejhg.5200537. [DOI] [PubMed] [Google Scholar]
- 5.Uhlenberg B., Schuelke M., Ruschendorf F., Ruf N., Kaindl A.M., Henneke M., Thiele H., Stoltenburg-Didinger G., Aksu F., Topaloglu H. Mutations in the gene encoding gap junction protein α-12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am. J. Hum. Genet. 2004;75:251–260. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bugiani M., Shahwan A.L., Lamantea E., Bizzi A., Bakhsh E., Moroni I., Balestrinin M.R., Uziel G., Zeviani M. GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy. Neurology. 2006;67:273–279. doi: 10.1212/01.wnl.0000223832.66286.e4. [DOI] [PubMed] [Google Scholar]
- 7.Henneke M., Combes P., Diekmann S., Bertini E., Brockmann K., Burlina A.P., Kaiser J., Ohlenbusch A., Plecko B., Rodriguez D. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology. 2008;70:748–754. doi: 10.1212/01.wnl.0000284828.84464.35. [DOI] [PubMed] [Google Scholar]
- 8.Gething M.J., Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 9.Frydman J. Folding of newly translated proteins in vivo: The role of molecular chaperones. Annu. Rev. Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 10.Horwich A.L., Fenton W.A., Chapman E., Farr G.W. Two families of chaperonin: Physiology and mechanism. Annu. Rev. Cell Dev. Biol. 2007;23:115–145. doi: 10.1146/annurev.cellbio.23.090506.123555. [DOI] [PubMed] [Google Scholar]
- 11.Hartl F.U., Hayer-Hartl M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 12.Arya R., Mallik M., Lakhotia S.C. Heat shock genes – integrating cell survival and death. J. Biosci. 2006;32:595–610. doi: 10.1007/s12038-007-0059-3. [DOI] [PubMed] [Google Scholar]
- 13.Bukau B., Horwich A.L. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 14.Ostermann J., Horwich A.L., Neupert W., Hartl F.U. Protein folding in mitochondria requires complex formation with hsp60 and ATP hydrolysis. Nature. 1989;341:125–130. doi: 10.1038/341125a0. [DOI] [PubMed] [Google Scholar]
- 15.Heyrovska N., Frydman J., Hohfeld J., Hartl F.U. Directionality of polypeptide transfer in the mitochondrial pathway of chaperone mediated protein folding. Biol. Chem. 1998;379:301–309. doi: 10.1515/bchm.1998.379.3.301. [DOI] [PubMed] [Google Scholar]
- 16.Martin J. Molecular chaperones and mitochondrial protein folding. J. Bioenerg. Biomembr. 1997;29:35–43. doi: 10.1023/a:1022407705182. [DOI] [PubMed] [Google Scholar]
- 17.Hansen J.J., Durr A., Cournu-Rebeix I., Georgopoulos C., Ang D., Nielsen M.N., Davoine C.S., Brice A., Fontaine B., Gregersen N., Bross P. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 2002;70:1328–1332. doi: 10.1086/339935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen J., Svenstrup K., Ang D., Nielsen M.N., Christensen J.H., Gregersen N., Nielsen J.E., Georgopoulos C., Bross P. A novel mutation in the HSPD1 gene in a patient with hereditary spastic paraplegia. J. Neurol. 2007;254:897–900. doi: 10.1007/s00415-006-0470-y. [DOI] [PubMed] [Google Scholar]
- 19.Fink J.K. The hereditary spastic paraplegias. Nine genes and counting. Arch. Neurol. 2003;60:1045–1049. doi: 10.1001/archneur.60.8.1045. [DOI] [PubMed] [Google Scholar]
- 20.Fink J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 21.Reid E. Science in motion: Common molecular pathological themes emerge in the hereditary spastic paraplegias. J. Med. Genet. 2003;40:81–86. doi: 10.1136/jmg.40.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schapira A.H.V. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- 23.Zuchner S., Vance J.M. Emerging pathways for hereditary axonopathies. J. Mol. Med. 2005;83:935–943. doi: 10.1007/s00109-005-0694-9. [DOI] [PubMed] [Google Scholar]
- 24.Chretien D., Rustin P., Bourgeron T., Rotig A., Saudubray J.M., Munnich A. Reference charts for respiratory chain activities in human tissues. Clin. Chim. Acta. 1994;228:53–70. doi: 10.1016/0009-8981(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 25.Vu T.H., Sciacco M., Tanji K., Nichter C., Bonilla E., Chatkupt S., Maertens P., Shanske S., Mendell J., Koenigsberger M.R. Clinical manifestations of mitochondrial DNA depletion. Neurology. 1998;50:1783–1790. doi: 10.1212/wnl.50.6.1783. [DOI] [PubMed] [Google Scholar]
- 26.Sambrook J., Fritsh E., Maniatis T. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. Molecular Cloning, A Laboratory Manual. pp. 9.16–9.19. [Google Scholar]
- 27.Silberstein M., Tzemach A., Dovgolevsky N., Fishelson M., Schuster S., Geiger D. On-line systems for faster linkage analysis via parallel execution on thousands of personal computers. Am. J. Hum. Genet. 2006;78:922–935. doi: 10.1086/504158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fayet O., Ziegelhoffer T., Georgopoulos C. The groES and groEL heat shock gene products of Escherichia coli are essential for bacterial growth at all temperatures. J. Bacteriol. 1989;171:1379–1385. doi: 10.1128/jb.171.3.1379-1385.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nielsen K.L., McLennan N., Masters M., Cowan N.J. A single-ring mitochondrial chaperonin (Hsp60-Hsp10) can substitute for GroEL-GroES in vivo. J. Bacteriol. 1999;181:5871–5875. doi: 10.1128/jb.181.18.5871-5875.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richardson A., Schwager F., Landry S.J., Georgopoulos C. The importance of a mobile loop in regulating chaperonin/co-chaperonin interaction. J. Biol. Chem. 2001;276:4981–4987. doi: 10.1074/jbc.M008628200. [DOI] [PubMed] [Google Scholar]
- 31.Bross P., Naundrup S., Hansen J., Nielsen M.N., Christensen J.H., Kruhøffer M., Palmfeldt J., Corydon T.J., Gregersen N., Ang D. The HSP60-(P.Val98Ile) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. J. Biol. Chem. 2008;283:15694–15700. doi: 10.1074/jbc.M800548200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mandemakers W., Morais V.A., De Strooper B. A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J. Cell Sci. 2007;120:1707–1716. doi: 10.1242/jcs.03443. [DOI] [PubMed] [Google Scholar]
- 33.Petrozzi L., Ricci G., Giglioli N.J., Siciliano G., Mancuso M. Mitochondria and neurodegeneration. Biosci. Rep. 2007;27:87–104. doi: 10.1007/s10540-007-9038-z. [DOI] [PubMed] [Google Scholar]
- 34.Haas R.H. The evidence basis for coenzyme Q therapy in oxidative phosphorylation disease. Mitochondrion. 2007;7(Suppl 1):S136–S145. doi: 10.1016/j.mito.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 35.Kwong J.Q., Beal M.F., Manfredi G. The role of mitochondria in inherited neurodegenerative diseases. J. Neurochem. 2006;97:1659–1675. doi: 10.1111/j.1471-4159.2006.03990.x. [DOI] [PubMed] [Google Scholar]
- 36.Bross P., Li Z., Hansen J., Hansen J.J., Nielsen N.M., Corydon T.J., Georgopoulos C., Ang D., Lundemose J.B., Niezen-Koning K. Single-nucleotide variations in the genes encoding the mitochondrial Hsp60/Hsp10 chaperone system and their disease-causing potential. J. Hum. Genet. 2007;52:56–65. doi: 10.1007/s10038-006-0080-7. [DOI] [PubMed] [Google Scholar]
- 37.Xu Z., Horwich A.L., Sigler P.B. The crystal structure of the asymmetric GroEL–GroES–(ADP)7 chaperonin complex. Nature. 1997;388:741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- 38.Cheng M.Y., Hartl F.U., Martin J., Pollock R.A., Kalousek F., Neupert W., Hallberg E.M., Hallberg R.L., Horwich A.L. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature. 1989;337:620–625. doi: 10.1038/337620a0. [DOI] [PubMed] [Google Scholar]
- 39.Veereshwarayya V., Kumar P., Rosen K.M., Mestril R., Querfurth H.W. Differential effects of mitochondrial heat shock protein 60 and related molecular chaperones to prevent intracellular β-amyloid-induced inhibition of complex IV and limit apoptosis. J. Biol. Chem. 2006;281:29468–29478. doi: 10.1074/jbc.M602533200. [DOI] [PubMed] [Google Scholar]
- 40.Cassari G., De Fusco M., Ciarmatori S., Zeviani M., Mora M., Fernandez P., De Michele G., Filla A., Cocozza S., Marconi R. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 41.Pedersen C.B., Bross P., Winter V.S., Corydon T.J., Bolund L., Bartlett K., Vockley J., Gregersen N. Misfolding, degradation, and aggregation of variant proteins. The molecular pathogenesis of short chain acyl-CoA dehydrogenase (SCAD) deficiency. J. Biol. Chem. 2003;278:47449–47458. doi: 10.1074/jbc.M309514200. [DOI] [PubMed] [Google Scholar]
- 42.Corydon T.J., Hansen J., Bross P., Jensen T.G. Down-regulation of Hsp60 expression by RNAi impairs folding of medium-chain acyl-CoA dehydrogenase wild-type and disease-associated proteins. Mol. Genet. Metab. 2005;85:260–270. doi: 10.1016/j.ymgme.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 43.Soltys B.J., Gupta R.S. Immunoelectron microscopic localization of the 60-kDa heat shock chaperonin protein (Hsp60) in mammalian cells. Exp. Cell Res. 1996;222:16–27. doi: 10.1006/excr.1996.0003. [DOI] [PubMed] [Google Scholar]
- 44.Chang A.Y., Chan J.Y., Chou J.L., Li F.C., Dai K.Y., Chan S.H. Heat shock protein 60 in rostral ventrolateral medulla reduces cardiovascular fatality during endotoxaemia in the rat. J. Physiol. 2006;574:547–564. doi: 10.1113/jphysiol.2006.110890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan J.Y., Cheng H.L., Chou J.L., Li F.C., Dai K.Y., Chan S.H., Chang A.Y. Heat shock protein 60 or 70 activates nitric-oxide synthase (NOS) I and inhibits NOS II-associated signaling and depresses the mitochondrial apoptotic cascade during brain stem death. J. Biol. Chem. 2007;282:4585–4600. doi: 10.1074/jbc.M603394200. [DOI] [PubMed] [Google Scholar]
- 46.Chandra D., Choy G., Tang D.G. Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: Evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. J. Biol. Chem. 2007;282:31289–31301. doi: 10.1074/jbc.M702777200. [DOI] [PubMed] [Google Scholar]
- 47.Rossi A., Biancheri R., Zara F., Bruno C., Uziel G., van der Knap M.S., Minetti C., Tortori-Donati P. Hypomyelination and congenital cataract: Neuroimaging features of a novel inherited white matter disorder. AJNR Am. J. Neuroradiol. 2008;29:301–305. doi: 10.3174/ajnr.A0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garbern J.Y. Pelizaeus-Merzbacher disease: Genetic and cellular pathogenesis. Cell. Mol. Life Sci. 2007;64:50–65. doi: 10.1007/s00018-006-6182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inoue K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics. 2005;6:1–16. doi: 10.1007/s10048-004-0207-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.