

To the editor: In a recent study of Native American mitochondrial genomes, Fagundes et al.1 claimed to have found molecular evidence that the colonization of the New World occurred well before the appearance of the Clovis cultural horizon (c. 12.6–13.2 thousand years [kyr] ago2). To support this claim, the authors performed a variety of phylogenetic analyses, including Bayesian date estimation and skyline-plot inference, using the software BEAST.3 A very similar conclusion was reached in a recent study by Achilli et al.,4 who estimated that each of the major Native American haplogroups coalesced around 19 kyr ago. A key failing of these studies, however, was an underappreciation of the importance of calibration choice. In fact, upon closer examination of the calibration techniques involved in the two studies, there appears to be little support for an American colonization event significantly antedating the earliest physical evidence of human occupation.5,6
Fagundes et al.1 employed two approaches to calibrating their date estimates. The first, which was also used by Achilli et al.4 in their study, assumed a global substitution rate of 1.26 × 10−8 subs/site/year, originally obtained by Mishmar et al.7 with the use of a human-chimpanzee calibration at 6.5 Myr. The second method was to include a chimpanzee sequence in the phylogenetic analysis, again fixing the age of the human-chimpanzee split to 6.5 Myr. The date estimates produced under the two calibration methods were very similar, which is not surprising given that they were effectively based on the same calibration. However, using only a single calibration point makes date estimates sensitive to calibration choice, particularly when fossil information has been condensed to a point estimate in spite of uncertainty over the timing for the human-chimpanzee split.
Fagundes et al.1 do consider one alternative rate, estimated exclusively from synonymous substitutions within the human phylogeny by Kivisild et al.8 They acknowledge that using this rate would have shifted their own coalescence time estimates about 5 kyr closer to the present, thereby invalidating their claim of a significantly pre-Clovis occupation of the New World. They dismiss this synonymous substitution rate, however, on the grounds that it has been questioned elsewhere9 and because it is not as widely cited as the Mishmar rate. Achilli et al.4 raised similar doubts concerning the Kivisild rate.
This uncritical dismissal of alternative rates reflects an unsettling trivialization of the effect of calibration choice. Recent observations have indicated that substitution rates estimated within species are considerably higher than those estimated on phylogenetic (interspecific) scales.10–12 The consequence of this pattern is that it is inadvisable to assume an interspecific rate in an intraspecific analysis;13 this also applies to studies of human evolution, with mounting evidence that the use of a human-chimpanzee calibration is generally inappropriate.14–17
We reanalyzed the data of Fagundes et al.1 and Achilli et al.,4 using the Bayesian phylogenetic software BEAST, in order to estimate the coalescence times of five Native American haplogroups (A2, B2, C, D1, and X2a). Rather than calibrating our analysis with either the Mishmar rate or the age of the human-chimpanzee split, we used a set of three biogeographic calibrations within the human tree to obtain a rate estimate from a data set used in our recent study of human mitochondrial substitution rates.15 Our methodology consisted of two steps, which are outlined below.
Step 1: Estimating the Coding-Region Rate
An alignment of 177 mitochondrial genomes, sampled primarily from macrohaplogroups M and N, was obtained from our previous study on human mitochondrial substitution rates.15 Details of data selection and sequence alignment were described previously.15 The aligned genomes were truncated to leave the coding region (sites 577–16023 of the Cambridge Reference Sequence18) for analysis, following both Fagundes et al.1 and Achilli et al.4
In order to estimate substitution rates and divergence times from the coding-region alignment, Bayesian phylogenetic analysis was performed with the use of BEAST 1.4.7.3 To match the settings used by Fagundes et al.,1 we used the HKY+G model of nucleotide substitution without partitioning the alignment and used a Bayesian-skyline-plot approach in order to integrate over different coalescent histories.19
Posterior distributions of parameters, including divergence times and substitution rates, were estimated by Markov chain Monte Carlo (MCMC) sampling in BEAST. In each analysis, samples were drawn every 10,000 MCMC steps from a total of 20,000,000 steps, following a discarded burn-in of 2,000,000 steps. Convergence to the stationary distribution and sufficient sampling were checked by inspection of posterior samples.
As described previously by us,15 internal calibration was conducted by specifying priors on the ages of three nodes in the tree. The time to the most-recent common ancestor (TMRCA) of haplogroup P was assumed to follow a lognormal distribution, with a minimum of 40,000 years, with a mean of 45,000 years, and with 95% of the distribution lying between 40,000 and 55,000 years. The TMRCAs of haplogroups H1 and H3 were each assumed to follow a normal distribution, with a mean of 18,000 years and an SD of 3500 years;20,21 approximately 95% of the distribution lies between 11,000 and 25,000 years. Justifications for these calibrations are described elsewhere.15
Step 2: Reanalysis of the Data of Fagundes et al. and Achilli et al
The sequences used in the studies by Fagundes et al.1 and Achilli et al.4 were collected from GenBank. These represented all 86 of the genomes analyzed by the former but only 148 of the 185 genomes analyzed by the latter. The remaining genomes, which were obtained from published studies and subsequently corrected for errors, were unavailable from Achilli et al.4 The absence of 37 genomes, all from haplogroup A2, is unlikely to have a noticeable effect on estimates of coalescence times for the total haplogroup.
The two alignments were analyzed with the use of BEAST, with the same settings as in Step 1. Instead of internal calibration, we used the posterior rate estimate in Step 1 to specify a prior distribution for the substitution rate in the present analyses. The prior rate was assumed to be normally distributed, with a mean of 2.038 × 10−8 subs/site/year and an SD of 2.064 × 10−9 subs/site/year.
The coalescence times of haplogroups A2, B2, C, D1, and X2a were estimated from the data of Fagundes et al.,1 and the coalescence times of haplogroups A2, B2, C1, and D1 were estimated from the data of Achilli et al.4
Our coalescence-time estimates are closer to the present than are those obtained by either of the original studies1,4 (Table 1) but are very similar to those estimated by Tamm et al.,22 who obtained a mean estimate of 13.9 kyr by using the Kivisild rate with a median-joining network. In contrast with the interpretations of Fagundes et al.1 and Achilli et al.,4 our date estimates are unable to exclude the hypothesis of a colonization event coincident with the archaeological dates for the Americas. There is a similar contraction in the time scale of our Bayesian skyline plot, suggesting that rapid population expansion occurred around 10–12 kyr ago (Figure 1). These results present a considerably different scenario from that visualized by Fagundes et al.1 and Achilli et al.,4 in which this population expansion commenced toward the end of the last glacial maximum in Beringia.
Table 1.
Coalescence-Time Estimates for Native American Haplogroups
| Haplogroup | Coalescence-Time Estimate (Years) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Fagundes et al. |
Achilli et al. |
|||||||
| Original Estimate |
Present Studya |
Original Estimate |
Present Studya |
|||||
| Mean | 95% HPDb | Mean | 95% HPDb | Mean | 95% CI | Mean | 95% HPDb | |
| A2 | 21,290 | (16,550 – 28,130) | 13,840 | (9,380 – 18,700) | 17,200 | (13,870 – 20,530) | 14,970 | (10,030 – 20,600) |
| B2 | 22,140 | (17,570 – 28,730) | 14,070 | (9,670 – 18,680) | 21,200 | (16,500 – 25,900) | 14,440 | (10,190 – 19,120) |
| C | 20,680 | (16,830 – 26,260) | 13,260 | (9,360 – 17,630) | 23,800 | (15,370 – 32,230) | 15,600 | (10,870 – 20,830) |
| D1 | 21,430 | (16,850 – 28,730) | 13,930 | (9,550 – 19,200) | 18,600 | (14,090 – 23,110) | 13,670 | (9,570 – 18,400) |
| X2a | 20,730 | (16,100 – 29,000) | 13,340 | (9,140 – 18,920) | – | – | ||
| Average | 20,730 | 13,690 | 20,200 | 14,670 | ||||
For calibration of these coalescence-time estimates, a normally distributed prior (mean 2.038 × 10−8 subs/site/year, SD 2.064 × 10−9) was placed on the substitution rate.
HPD: highest posterior density.
Figure 1.
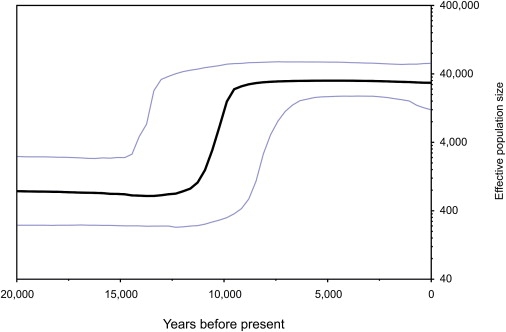
Bayesian Skyline Plot of Native American Population-History
Bayesian skyline plot, obtained with the use of BEAST, showing population history estimated from the coding regions of 86 Native American mitochondrial genomes. The vertical scale measures the effective population size, assuming a generation time of 25 years. There is evidence of population expansion commencing around 12–13 kyr before the present.
In two more recent studies using Bayesian-skyline-plot analysis of the mitochondrial coding region, Kitchen et al.23 and Atkinson et al.14 estimated that population expansion in the Americas began 15 kyr and 18 kyr ago, respectively. However, the time scale of Kitchen et al.23 was based on a substitution rate obtained by Ingman et al.24 with a human-chimpanzee calibration. The Ingman rate is slightly faster than the Mishmar rate because the former assumed a date of 5 Myr for the divergence whereas the latter used a value of 6.5 Myr; nevertheless, both rate estimates are interspecific in nature, meaning that the estimate of Kitchen et al.23 can be grouped with those of Fagundes et al.1 and Achilli et al.4 The contrast between our estimated chronology and that of Atkinson et al.,14 who used a similar methodology involving biogeographic calibration, is most likely due to the effect of rate variation among lineages. The internal diversity of mtDNA haplogroup Q, which was employed as the sole calibration in the study by Atkinson et al.,14 is substantially less than that of the similarly aged haplogroup P,25 which we used as one of three biogeographic calibrations. Therefore, a rate estimate calibrated with the use of haplogroup Q is likely to yield a comparatively slower substitution rate than one calibrated with the use of haplogroup P. In turn, this appears to have led to an overestimate of the antiquity of the population expansion in the Americas in the study by Atkinson et al.14
The differences among all of the various analyses predominantly derive from different approaches to calibration. In our recent study of human mitochondrial substitution rates,15 we found evidence to support the validity of the calibration technique used by Kivisild et al.8 to infer a synonymous substitution rate. This, in turn, lends support to the date estimates obtained by Tamm et al.,22 which are very similar to the results of the present study. We also note that our estimated time for population expansion overlaps with both the end of the Younger Dryas, ∼11.3 kyr ago, and the chronology for the rapid spread of the Clovis culture, ∼13 kyr ago.2 If we accept the alternative hypothesis that the genetic signal for demographic expansion is coincident with the last glacial maximum, then it is difficult to explain why there is not another population increase associated with the Clovis archaeological horizon. The older dates also require additional explanation for the absence of archaeological evidence in the Americas during this phase and for why populations should be showing significant signals of expansion under such unfavorable climatic conditions.
The outcomes of our reanalysis illustrate the crucial role of calibrations in obtaining robust date estimates and highlight the wide range of rate estimates currently used for calibration despite evidence to suggest that some of these might be misleading. Although our own estimates are unable to exclude the hypotheses presented by Fagundes et al.1 and Achilli et al.,4 they also demonstrate that it is not possible to rule out a scenario in which the timing of the colonization of the Americas closely matches that suggested by the current archaeological evidence. Improvements in the precision of the coalescence-time estimates with the use of our approach will be possible with increased availability of sequence data, especially from ancient DNA, which is able to offer precise calibrations within the human tree.16 Methods that are currently in development will be able to utilize multi-locus data in order to recover complex population histories.14 Finally, we hope that the identification of well-supported calibrations within the human tree will encourage a movement away from uncritical usage of the human-chimpanzee calibration.
Web Resources
The URL for data presented herein are as follows:
GenBank, www.ncbi.nlm.nih.gov/Genbank/
References
- 1.Fagundes N.J., Kanitz R., Eckert R., Valls A.C., Bogo M.R., Salzano F.M., Smith D.G., Silva W.A., Zago M.A., Ribeiro-dos-Santos A.K. Mitochondrial population genomics supports a single pre-Clovis origin with a coastal route for the peopling of the Americas. Am. J. Hum. Genet. 2008;82:583–592. doi: 10.1016/j.ajhg.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goebel T., Waters M.R., O'Rourke D.H. The late Pleistocene dispersal of modern humans in the Americas. Science. 2008;319:1497–1502. doi: 10.1126/science.1153569. [DOI] [PubMed] [Google Scholar]
- 3.Drummond A.J., Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Achilli A., Perego U.A., Bravi C.M., Coble M.D., Kong Q.P., Woodward S.R., Salas A., Torroni A., Bandelt H.J. The phylogeny of the four pan-American MtDNA haplogroups: Implications for evolutionary and disease studies. PLoS ONE. 2008;3:e1764. doi: 10.1371/journal.pone.0001764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dillehay T.D. Smithsonian Institute Press; Washington, DC: 1997. Monte Verde: A Late Pleistocene Settlement in Chile: Archeological Context. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert M.T., Jenkins D.L., Gotherstrom A., Naveran N., Sanchez J.J., Hofreiter M., Thomsen P.F., Binladen J., Higham T.F., Yohe R.M. DNA from pre-Clovis human coprolites in Oregon, North America. Science. 2008;320:786–789. doi: 10.1126/science.1154116. [DOI] [PubMed] [Google Scholar]
- 7.Mishmar D., Ruiz-Pesini E., Golik P., Macaulay V., Clark A.G., Hosseini S., Brandon M., Easley K., Chen E., Brown M.D. Natural selection shaped regional mtDNA variation in humans. Proc. Natl. Acad. Sci. USA. 2003;100:171–176. doi: 10.1073/pnas.0136972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kivisild T., Shen P., Wall D.P., Do B., Sung R., Davis K., Passarino G., Underhill P.A., Scharfe C., Torroni A. The role of selection in the evolution of human mitochondrial genomes. Genetics. 2006;172:373–387. doi: 10.1534/genetics.105.043901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bandelt H.-J., Kong Q.P., Richards M., Macaulay V. Estimation of mutation rates and coalescence times: Some caveats. In: Bandelt H.-J., Macaulay V., Richards M., editors. Human Mitochondrial DNA and the Evolution of Homo sapiens. Springer; Berlin: 2006. pp. 47–90. [Google Scholar]
- 10.Burridge C.P., Craw D., Fletcher D., Waters J.M. Geological dates and molecular rates: Fish DNA sheds light on time dependency. Mol. Biol. Evol. 2008;25:624–633. doi: 10.1093/molbev/msm271. [DOI] [PubMed] [Google Scholar]
- 11.Ho S.Y.W., Phillips M.J., Cooper A., Drummond A.J. Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol. Biol. Evol. 2005;22:1561–1568. doi: 10.1093/molbev/msi145. [DOI] [PubMed] [Google Scholar]
- 12.Ho S.Y.W., Shapiro B., Phillips M., Cooper A., Drummond A.J. Evidence for time dependency of molecular rate estimates. Syst. Biol. 2007;56:515–522. doi: 10.1080/10635150701435401. [DOI] [PubMed] [Google Scholar]
- 13.Ho S.Y.W., Larson G. Molecular clocks: When times are a-changin'. Trends Genet. 2006;22:79–83. doi: 10.1016/j.tig.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Atkinson Q.D., Gray R.D., Drummond A.J. mtDNA variation predicts population size in humans and reveals a major Southern Asian chapter in human prehistory. Mol. Biol. Evol. 2008;25:468–474. doi: 10.1093/molbev/msm277. [DOI] [PubMed] [Google Scholar]
- 15.Endicott P., Ho S.Y.W. A Bayesian evaluation of human mitochondrial substitution rates. Am. J. Hum. Genet. 2008;82:895–902. doi: 10.1016/j.ajhg.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kemp B.M., Malhi R.S., McDonough J., Bolnick D.A., Eshleman J.A., Rickards O., Martinez-Labarga C., Johnson J.R., Lorenz J.G., Dixon E.J. Genetic analysis of early holocene skeletal remains from Alaska and its implications for the settlement of the Americas. Am. J. Phys. Anthropol. 2007;132:605–621. doi: 10.1002/ajpa.20543. [DOI] [PubMed] [Google Scholar]
- 17.Stoneking M., Sherry S.T., Redd A.J., Vigilant L. New approaches to dating suggest a recent age for human mtDNA ancestor. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1992;337:167–175. doi: 10.1098/rstb.1992.0094. [DOI] [PubMed] [Google Scholar]
- 18.Anderson S., Bankier A.T., Barrell B.G., de Bruijn M.H., Coulson A.R., Drouin J., Eperon I.C., Nierlich D.P., Roe B.A., Sanger F. Sequence and organisation of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 19.Drummond A.J., Rambaut A., Shapiro B., Pybus O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 20.Achilli A., Rengo C., Magri C., Battaglia V., Olivieri A., Scozzari R., Cruciani F., Zeviani M., Briem E., Carelli V. The molecular dissection of mtDNA haplogroup H confirms that the Franco-Cantabrian glacial refuge was a major source for the European gene pool. Am. J. Hum. Genet. 2004;75:910–918. doi: 10.1086/425590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gamble C., Davies W., Pettitt P., Richards M. Climate change and evolving human diversity in Europe during the last glacial. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004;359:243–253. doi: 10.1098/rstb.2003.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamm E., Kivisild T., Reidla M., Metspalu M., Smith D.G., Mulligan C.J., Bravi C.M., Rickards O., Martinez-Labarga C., Khusnutdinova E.K. Beringian standstill and spread of Native American founders. PLoS ONE. 2007;2:e829. doi: 10.1371/journal.pone.0000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitchen A., Miyamoto M.M., Mulligan C.J. A three-stage colonization model for the peopling of the Americas. PLoS ONE. 2008;3:e1596. doi: 10.1371/journal.pone.0001596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ingman M., Kaessmann H., Paabo S., Gyllensten U. Mitochondrial genome variation and the origin of modern humans. Nature. 2000;408:708–713. doi: 10.1038/35047064. [DOI] [PubMed] [Google Scholar]
- 25.Hudjashov G., Kivisild T., Underhill P.A., Endicott P., Sanchez J.J., Lin A.A., Shen P., Oefner P., Renfrew C., Villems R. Revealing the prehistoric settlement of Australia by Y chromosome and mtDNA analysis. Proc. Natl. Acad. Sci. USA. 2007;104:8726–8730. doi: 10.1073/pnas.0702928104. [DOI] [PMC free article] [PubMed] [Google Scholar]