

Abstract
Recently we described a new synthesis of C,D-ring symmetric chlorins 11, involving 2+2 condensation of bis-formyl-dihydrodipyrrins 9 with symmetrically substituted dipyrromethane diacids 10 (Method I). However, while versatile in many aspects, Method I was unsuited to the broader goal of synthesizing fully non-symmetric chlorins of general structure 15, which requires regioselective control over the reacting centers in the A,B- and C,D-ring components. In this paper we describe four new 2+2 strategies that accomplish this differentiation (Methods II-V). Of these, Method V, which combines operational simplicity with moderate to high product yields, proved to be the most effective route, exploiting reactivity differences between the two formyl groups of A,B-rings 9 to impart excellent regioselectivity. Methods II-IV are also useful alternatives to Method V, although in some cases the appropriately functionalized precursors are less readily available. All four approaches generate single regioisomers of diversely substituted chlorins, and in every case the 2+2 condensation is accomplished in a simple, one-flask procedure without need for additives such as oxidizing agents or metals. Taken together, these methodologies provide expanded access to an array of chlorins for SAR studies that may advance the effectiveness of PDT and other applications.
Introduction
The chlorins are a class of 18π-electron aromatic tetrapyrroles, formally derived by saturation of the C2-C3 bond in ring A of porphyrins (see below). The most ubiquitous members of this class belong to the chlorophyll a (1) group of chromophores, the primary photoreceptors in photosynthesis in higher plants, algae, cyanobacteria, and other microorganisms.1 Although far less abundant than 1, other chlorins play crucial roles in both terrestrial and marine organisms. Among these, bonellin (2) is the hormone responsible for sexual differentiation in larvae of the echiuran worm Bonella viridis,2 and cyclopheophorbide (3) is representative of several closely related chlorins believed to inhibit oxidative damage in certain marine invertebrates.3 Other pheophorbides show promising anti-tumor activity.3c
“Non-natural” chlorins are also of considerable chemical and biological interest, in part because of their “tunable” photophysical properties. Owing to this capability they have come under increasing scrutiny as key components in various light-mediated applications, ranging from alternative energy sources to medicine. Light-energy conversion techniques have received particular attention, encompassing such topics as (1) the creation of artificial photosynthetic systems;4 (2) design of molecular wires or antenna arrays;4f,5 (3) production of hydrogen as an alternative fuel;4e,6 and (4) generation of electricity with chlorin-based solar cells.7 Synthetic chlorins are also being studied for applications in materials science8 and medical imaging.9 Finally, in the medical field, chlorins are emerging as more effective “second generation” photosensitizers in photodynamic therapy (PDT), a treatment method that employs visible light to trigger phototoxic reactions that eradicate malignant tissue or infections.10 While PDT is an established and effective means of treating certain cancers, there are many lesser known but increasingly important applications.11 Of particular note, PDT shows great promise in combating infectious disease, including antibiotic-resistant strains of bacteria.12
Most naturally occurring chlorins bear a full complement of substituents on the pyrrolic rings and have an asymmetric substitution pattern.1 The nature and position of these substituents can have a dramatic effect on the spectral properties of the chromophore and can affect the functions of biologically active chlorins.13 Furthermore, the ability to control substituent placement may be crucial to the successful use of synthetic chlorins in many applications, including multi-chlorin arrays.14 Despite this importance, the selective assembly of fully substituted, unsymmetrical chlorins remains a significant challenge.
The majority of de novo chlorin syntheses build upon the pioneering work of Battersby and Montforts, involving either photochemical or base-induced cyclization of tetrapyrroles 4 to afford chlorins of general structure 5 (Scheme 1).15 This strategy has been employed in the syntheses of numerous naturally occurring and “non-natural” chlorins, and it played a prominent role in early studies on vitamin B12 biosynthesis.16 It is not without shortcomings, however. Principally these relate to the limited choice of methodology for preparing tetrapyrroles 4 and the generally inefficient nature of the macrocyclization step (~20-72% yields).15,17 In addition, the photochemical process is only practical on very small scales. In a noteworthy variant of this approach, Lindsey et al. have shown that condensation of fragments 6 and 7, followed by oxidative cyclization employing Zn as a template, provides Zn-chlorins 8 in 7-45% yields (often this sequence can be performed in a single reaction vessel with only slightly lower yields).13a,14,18 As in the Battersby/Montforts strategies, though, a limiting factor in this approach is the availability of precursors of type 6 and 7, in particular with respect to synthesizing more highly substituted derivatives.13a,14,19 Finally, while numerous other strategies for synthesizing chlorins have been devised, a truly general approach remains elusive.20
Scheme 1.

Chlorin syntheses by Battersby, Montforts, and Lindsey
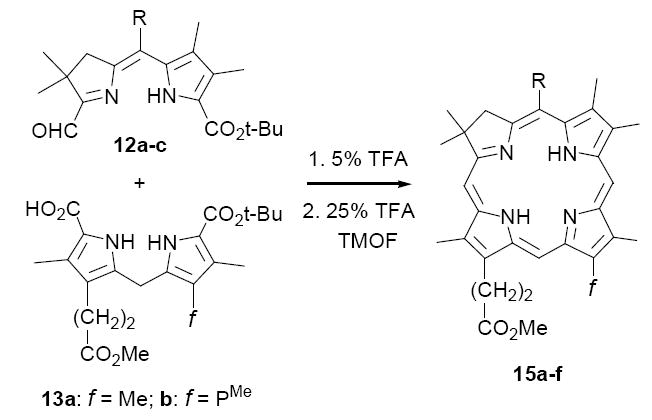
Recently we described a new synthetic approach to C,D-symmetric chlorins of general structure 11, involving acid-catalyzed condensation of A,B-ring dialdehydes 9 and C,D-ring dipyrromethanes 10 (Method I, Scheme 2).21 An attractive feature of this approach is that the cyclodehydration of 9 and 10 leads directly to the thermodynamically stable, aromatic chlorin nucleus 11, with no need for oxidation state adjustment18 or double bond isomerization(s).20a Consequently, the experimental conditions are straightforward, requiring no metal template and only routine precautions against air and light. Also, there is considerable flexibility in setting substituents about the periphery of the macrocycle. In one investigation, for example, Method I was employed in the synthesis of a diversely substituted series of twenty-five chlorins 11 designed to probe substituent effects on depth of cell membrane penetration in PDT (38-85% yields).21b However, while versatile in many aspects, in its present form Method I is unsuited to the broader goal of synthesizing unsymmetrical chlorins, which requires regioselective control over the four reacting carbon atoms in 9 and 10. To address this limitation we have been exploring variations of this “2+2” condensation strategy that incorporate the desired level of selectivity, four of which are described below (Methods II-V).
Scheme 2.
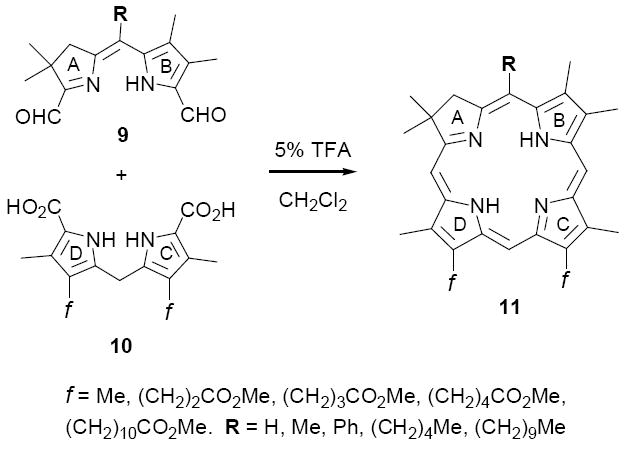
Synthesis of C,D-ring symmetric chlorins (Method I)
Results and Discussion
Syntheses of Unsymmetrical Chlorins via a “2+2” Strategy
Method II
Any “2+2” approach to unsymmetrical chlorins requires that the reactive sites of the A,B-and C,D-rings be differentiated, such that a single regioisomer is generated. The first strategy we tested for meeting this criterion is outlined in Scheme 3, in which the differentiating step involves mild acid-catalyzed condensation of C-9 protected A,B-ring precursors of type 12 with readily available22 C-11 protected C,D-ring precursors 13 (Method II). Our supposition was that seco-chlorins 14 would afford chlorins 15 upon decarboxylative formylation, with the stipulation that mono-formylation is followed by rapid cyclodehydration as opposed to oligomerization. As precedent for this approach, porphyrins have been prepared by a similar method in fair yields (30-35%),23 although in these examples post-cyclization oxidation was required to give the aromatic macrocycles. In the analogous cyclization of 14, the aromatic chlorin would be obtained directly upon cyclodehydration (cf. 16→15), with the same advantages as described above for Method I. In particular, aromatization would drive forward any equilibrium processes.
Scheme 3.
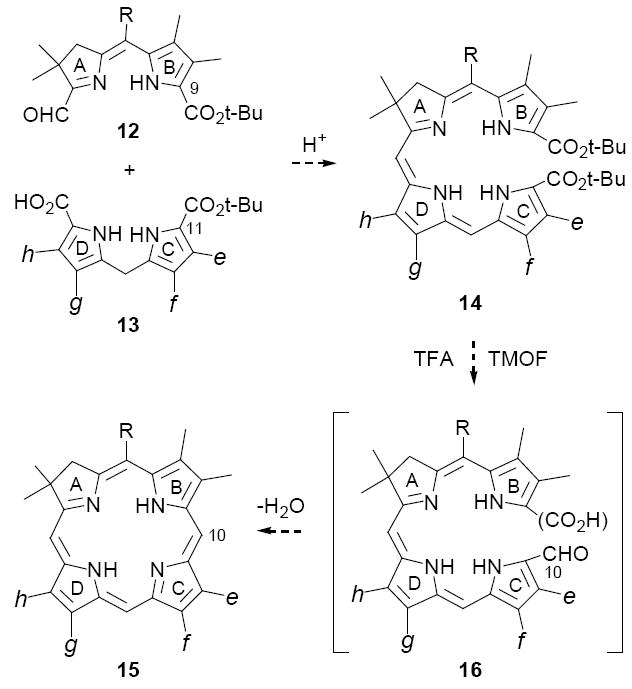
Method II for synthesizing unsymmetrical chlorins
To explore this route we prepared a group of three A,B-ring substrates 12a-c, employing methodology closely analogous to that earlier developed for synthesizing diformyl-dihydropyrrins 9 (Scheme 4). As described previously,24 dihydrodipyrrins 21 were derived in three steps, on scales ranging up to several grams, by (1) Pd(0)-mediate coupling-cyclization of alkyne acids 17 with iodopyrrole 18 to afford enelactones 19 (74-98%); (2) methylenation of the lactone carbonyl group with Petasis’s reagent (19→20; 86-92%); and (3) in situ enol-ether hydrolysis/amination (20→21; 71-82%). The desired A,B-ring aldehydes 12a-c were then obtained directly from 21 by selenium dioxide oxidation of the C-20 methyl group (65-81%).
Scheme 4.
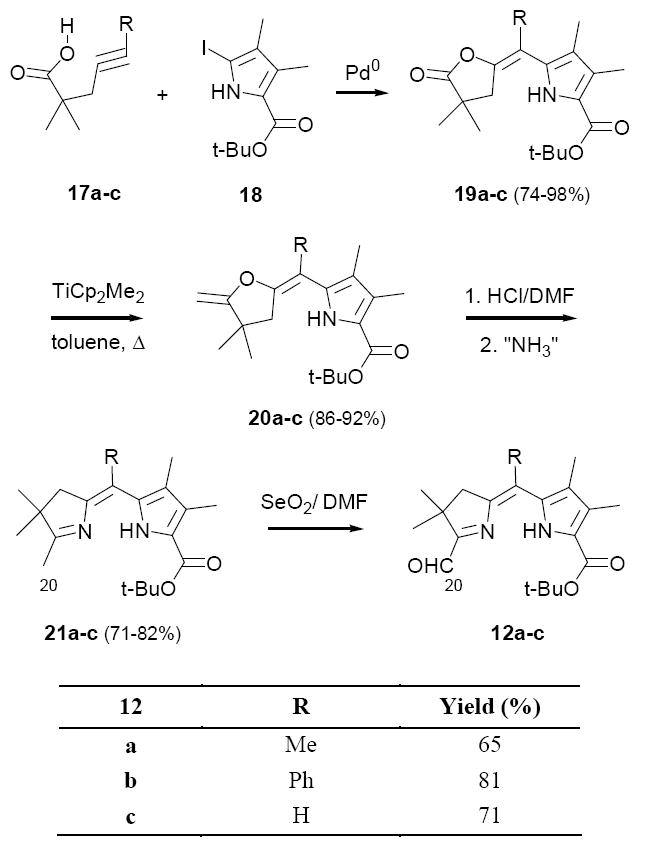
Synthesis of A,B-ring precursors 12a-c
Our initial investigations using Method II were conducted with the A,B-ring substrate 12a and unsymmetrical C,D-ring substrate 13a, and provided encouraging results. Thus, decarboxylation of 13a with p-toluenesulfonic acid (TsOH) in CH2Cl2/MeOH afforded an 84% yield of the C-19 unsubstituted dipyrromethane 22, which on coupling with 12a in the presence of TiCl4 gave a 66% yield of the moderately stable tetrapyrrole 14a (path a, Scheme 5). Next, dissolution of 14a in neat TFA, followed shortly thereafter by dilution with CH2Cl2 and excess triethyl orthoformate (TEOF), gave a 50% yield of the desired chlorin 15a as a single regioisomer. The substitution pattern of 15a was unequivocally established by NOE studies (see Supporting Information), as well as by direct comparison with its subsequently prepared regioisomer 26 (vide infra).
Scheme 5.
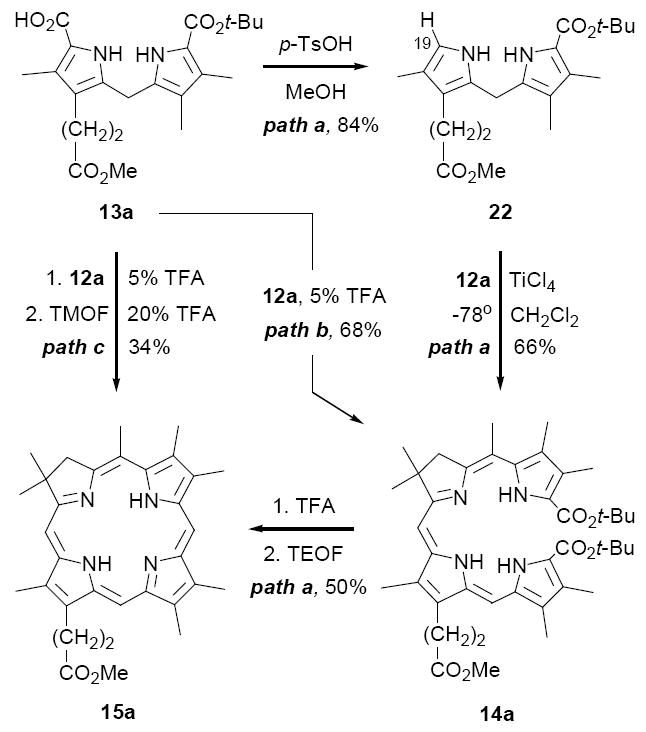
Three variations of Method II; paths a-c
Having established proof-of-concept, we were hopeful that the transformation of 13a to 15a could be streamlined to a “single pot” procedure, by-passing the isolation of the α-unsubstituted dipyrromethane 22 and seco-chlorin 14a. There was precedent for this possibility in our original studies on C,D-symmetric chlorins (cf. Scheme 2), where we found that dipyrromethane dicarboxylic acids 10 and dialdehydes 9 underwent acid-catalyzed condensation under conditions where t-butyl ester cleavage is exceedingly slow (5% TFA/CH2Cl2). The initial step of this goal, avoiding isolation of 22, was readily accomplished by treating a solution of dipyrromethane 13a and aldehyde-ester 12a in CH2Cl2 with sufficient neat TFA to bring the final concentration to 5% (path b, Scheme 5). After stirring ~1 h at rt under Ar, the anticipated seco-chlorin 14a was isolated in 68% yield. Cyclization of 14a as before then provided a route to chlorins that circumvented the separate TiCl4-catalyzed condensation of 22 and 12a. Next, a second simplification was realized by effecting the TFA-initiated condensation and cyclization steps in a single flask without isolation of intermediate 14a (path c). In this modification, a solution of 13a and 12a in 5% TFA/CH2Cl2 was stirred at rt under Ar for a period of 5 h, when it appeared that formation of 14a was complete.25 The resulting deep purple reaction was then adjusted to a concentration of 20% TFA/CH2Cl2 by addition of neat TFA and then treated with excess trimethyl orthoformate. After stirring an additional 16 h at rt, chlorin 15a was isolated in 34% yield by straightforward concentration and chromatography. This yield compares favorably with the overall yields of 28% and 34% obtained following paths a and b respectively, and in no case was evidence found for formation of regioisomeric chlorin products.26
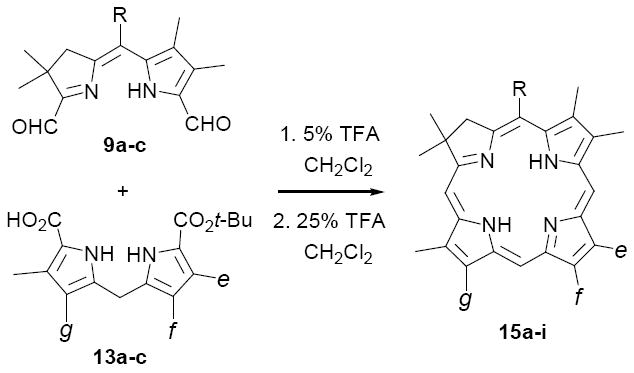
To test the generality and effectiveness of Method II, we synthesized two additional unsymmetrical chlorins 15b,c, and for comparison, three C,D-ring symmetric chlorins 15d-f previously prepared by Method I (Table 1; cf. also Scheme 2). We observed little effect of non-meso C,D-ring substituents f on the course of these syntheses. However, the nature of the A,B-ring meso-substituents R had a significant influence on reaction efficiency, with dihydrodipyrrins 12a (R=Me) and 12b (R=Ph) affording considerably higher yields of the corresponding chlorins 15a,d and 15b,e than the case with unsubstituted substrate 12c (R=H). These results are consistent with a reaction pathway involving cationic intermediates stabilized by electron donating substituents at C-5. Interestingly, Method II proved to be less efficient than Method I for synthesizing C,D-ring symmetric chlorins of type 15d-f, yields in each case being significantly lower. However, even with these limitations, Method II does provide access to unsymmetrical chlorins via the readily available precursors 12 and 13.
Table 1.
Chlorins prepared by Method II
| 15 | Substituentsa | Yield (%) | Method I Yield (%)b |
|---|---|---|---|
| a | f = Me; R = Me | 38 | – |
| b | f = Me; R = Ph | 59 | – |
| c | f = Me; R = H | 11 | – |
| d | f = PMe; R = Me | 37 | 73 |
| e | f = PMe; R = Ph | 38 | 60 |
| f | f = PMe; R = H | 12 | 63 |
PMe = CH2CH2CO2Me.
see reference 21b
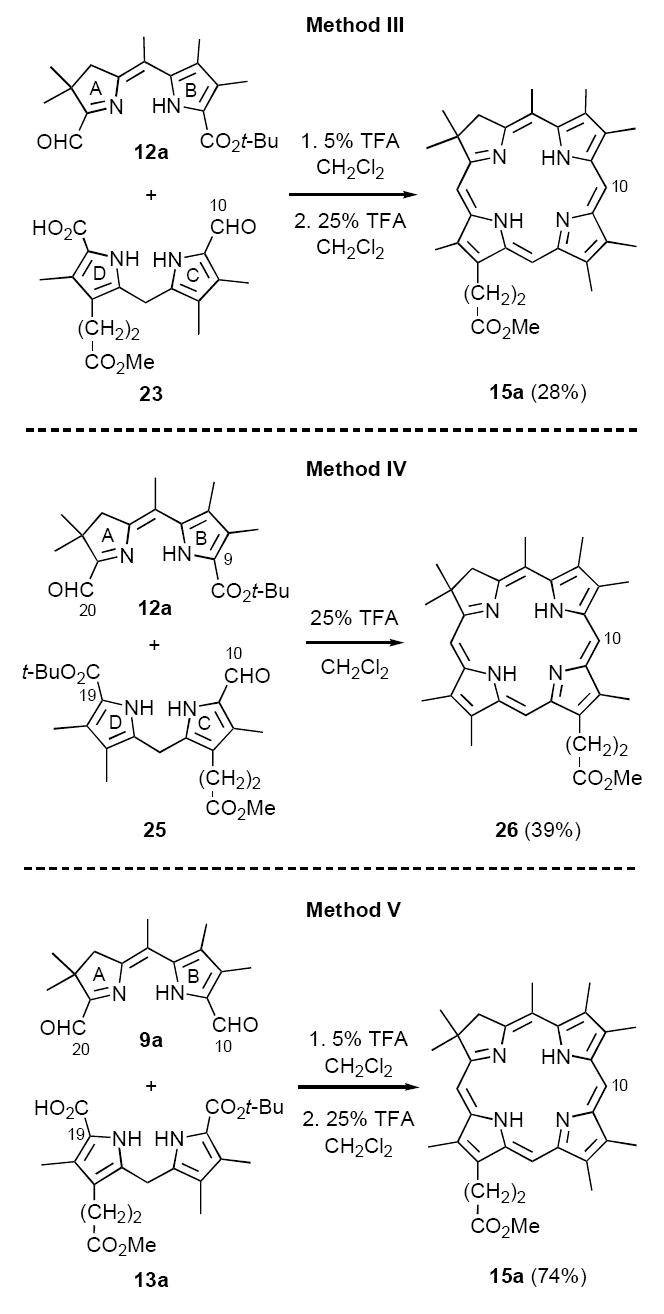
Most of our efforts at optimizing Method II centered on the decarboxylative formylation process leading from tetrapyrroles 14 to presumed chlorin intermediates 16 (cf. Scheme 3), which we had reason to believe might be problematic. In particular, the initial step in such transformations involves ipso-protonation (or formylation) on the carboxyl-bearing pyrrole ring, followed by re-aromatization with loss of CO2.27 Consequently, acid-catalyzed decarboxylation (or decarboxylative formylation) of electron deficient pyrroles is typically very slow.28 In the present case, tetrapyrrole 14 is a vinylogous amidine presumably favoring N-protonation in ring A or D, either of which lowers the likelihood of ipso-electrophilic attack on rings B or C. Indeed, in earlier mechanistic studies we observed the same phenomenon with dihydrodipyrrins having a basic pyrroline ring nitrogen.21b Since this step in Method II could not be optimized further, we examined three additional approaches to unsymmetrical chlorins that avoided decarboxylative formylation on basic ring systems (Methods III-V, Scheme 6). Methods III and IV achieve regioselectivity in similar fashion to Method II, except that in both cases a formyl group is incorporated in the C,D-ring fragment. In contrast, Method V employs the same bis-formyl A,B-ring precursor 9 previously utilized in our syntheses of C,D-ring symmetric chlorins (Method I, cf. Scheme 2), regioselectivity being dictated by the far greater reactivity of the formyl group in ring A.
Scheme 6.
Design and testing of Methods III-V
Method III
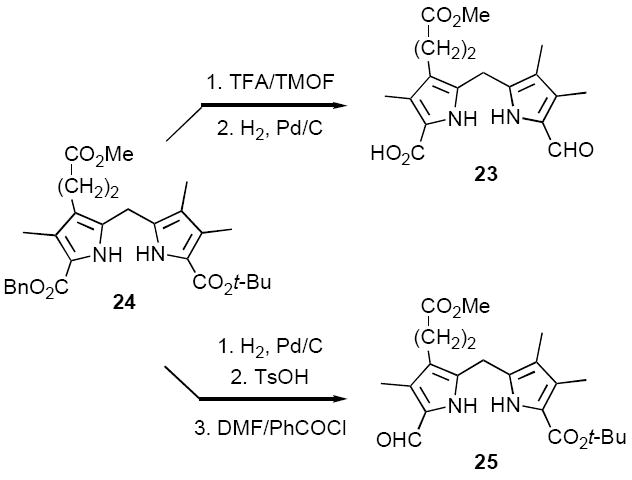
Our synthetic design for Method III is similar in concept to Method II (cf. Schemes 3 and 5), except the aldehyde destined to become C-10 is incorporated early on (Scheme 6). To test this approach, model aldehyde-acid 23 was synthesized from the known dipyrromethane 24,29 by a two step sequence consisting of decarboxylative formylation (in this case routine) followed by benzyl ester hydrogenolysis (Scheme 7). The next step called for mild acid-catalyzed condensation of 23 with the A,B-ring precursor 12a, followed by t-butyl ester hydrolysis and cyclodehydration. Each of these steps had close precedent in path c of Method II (cf. Scheme 5), although self-condensation of 23 was a potential complication. Blank experiments, however, indicated that side reactions of this nature could be controlled. In the event, condensation of 12a and 23 in 5% TFA/CH2Cl2, followed by adjustment to 25% TFA/CH2Cl2, afforded chlorin 15a in 28% yield, with no evidence for byproducts arising from self-condensation (Scheme 6). During this sequence a polar intermediate was observable by TLC, whose crude NMR spectrum was consistent with the anticipated tetrapyrrole condensation product of 12a and 23. However, several attempts at isolating and fully characterizing this compound were unsuccessful, making further optimization difficult. Thus, while clearly a viable means of synthesizing unsymmetrical chlorins, Method III had no advantages over Method II in terms of simplicity or effectiveness.
Scheme 7.
Synthesis of C,D-rings 24 and 25 for Methods III and IV.
Method IV
Method IV constituted a relatively minor variation on Method III, substituting a t-butyl ester in C,D-ring precursor 25 for the carboxylic acid blocking group employed in 23 (Scheme 6). We were also interested in preparing the regioisomeric chlorin 26 to compare directly with the chlorin 15a synthesized previously using both Method II and III. The synthesis of 25 was simplified by the fact that it could be prepared from the same dipyrromethane precursor 24 previously employed in the synthesis of C,D-ring precursor 23 (Scheme 7). In the case of 25, benzyl ester hydrogenolysis and decarboxylation of 24 were effected first, followed by formylation under very mild Vilsmeier-Haack conditions to ensure that the t-butyl ester remained intact.30
As in the analogous step in Method III, self-condensation of C,D-ring precursor 25 was a potential complication in Method IV, in particular under the more strongly acidic conditions required for t-butyl ester cleavage (25% TFA/CH2Cl2). However, we were again confident that the far more electrophilic C-20 formyl group in ring A of 12a would react preferentially. A second point worth noting pertains to the t-butyl ester group at C-9 in ring B. While we fully expected this group to undergo concomitant ester cleavage, we had previously found that decarboxylation in such ring systems is sluggish, due to the electron withdrawing effect of the protonated pyrroline ring A.21b This argument formed the basis for regiochemical control in the condensation of 12a and 25 and for avoiding self condensation of A,B-ring precursor 12a. Thus, we expected that relatively rapid deprotection of C-19, followed by condensation with the highly reactive C-20 aldehyde, would afford a bilin intermediate that is properly disposed for ring closure to the corresponding chlorin. This turned out to be the case, as stirring of 12a and 25 in 25% TFA/CH2Cl2 for 24 h provided chlorin 26 in 39% yield (Method IV, Scheme 6). Direct comparison of chlorins 26 and 15a then demonstrated conclusively that there had been no crossover in regiochemical control in Methods II-IV. While this result was satisfying, evidence was accumulating that an even more straightforward approach to unsymmetrical chlorins was feasible employing diformyl ring-A,B precursors 9 (Method V).
Method V
In our previous chlorin studies we demonstrated that diformyl derivatives 9 are excellent A,B-ring precursors for synthesizing C,D-symmetric chlorins 11 (cf. Scheme 2).21 We had assumed, though, that these materials lacked the unambiguous differentiation at C-10 and C-20 necessary to impart regioisomeric selectivity. The validity of this assumption was now called into question, based upon the substantial reactivity differences we had uncovered between pyrrole- and pyrroline-type formyl groups (vide supra). Thus, it now seemed clear that electron rich pyrroles would undergo selective condensation with the more reactive pyrroline aldehyde found on ring A in 9, as opposed to the vinylogous amide-like formyl group present in ring B. This approach was first tested with dialdehyde 9a and dipyrromethane 13a, which were stirred together for 5 h at rt in 5% TFA/CH2Cl2 to effect decarboxylation at C-19 and initial condensation. After stirring an additional 16 h in 25% TFA/CH2Cl2. chlorin 15a was isolated in 74% yield without any effort at optimization.31 Furthermore, this methodology proved to be quite general. Both C,D-ring symmetric and unsymmetrically substituted chlorins were accessible by this route, with yields ranging from 22-87% (Table 2). As can be seen, the C-5 meso-substituent still exerted some influence on the reaction efficiency, with the trend mirroring that previously noted for Method II. That is, meso-H derivatives 15c,f,i were consistently formed in lower yields than the corresponding meso-Me or meso-Ph chlorins, again suggesting the intermediacy of cationic species. Importantly, however, the yields of all chlorins were significantly improved over those obtained by Method II, including the meso-H series which were brought into a preparatively useful range (note that the 22% yield for 15i is atypically low, even for meso-H substituted derivatives). Finally, direct comparison of Methods I and V was possible for the series of C,D-ring symmetric chlorins 15d-f. In these examples the yields for meso-Me and meso-phenyl chlorins 15d,e were somewhat higher employing Method V (78% and 73%, respectively; vs 73% and 60% for Method I), presumably due to better control over competing oligomerization. Interestingly, though, meso-H chlorin 15f did not follow this trend, and was better prepared using Method I (38% vs 63%).
Table 2.
Chlorins prepared by Method V
| 15 | Substituentsa | Yield (%) | Method I Yield (%)b |
|---|---|---|---|
| a | e,f = Me; g = PMe; R = Me | 74 | – |
| b | e,f = Me; g = PMe; R = Ph | 87 | – |
| c | e,f = Me; g = PMe; R = H | 39 | – |
| d | e = Me; f,g = PMe; R = Me | 78 | 73 |
| e | e = Me; f,g = PMe; R = Ph | 73 | 60 |
| f | e = Me; f,g = PMe; R = H | 38 | 63 |
| g | e = PMe; f,g = Me; R = Me | 38 | – |
| h | e = PMe; f,g = Me; R = Ph | 77 | – |
| i | e = PMe; f,g = Me; R = H | 22 | – |
PMe = CH2CH2CO2Me.
see reference 21b.
Conclusion
In summary, we have built upon our 2+2 synthesis of C,D-ring symmetric chlorins to develop four new strategies for the preparation of chlorins that are fully asymmetric in their substitution pattern. Method V, which combines operational simplicity with moderate to high product yields, proved to be the most effective route, with reactivity differences between the two formyl groups of A,B-rings 9 imparting excellent regioselectivity. Methods II-IV are also useful alternatives to Method V if the appropriately functionalized precursors are readily available. All four approaches generate single regioisomers of diversely substituted chlorins, and in every case the 2+2 condensation is accomplished in a simple, one-flask procedure without need for additives such as oxidizing agents or metals. Taken together, these methodologies provide expanded access to an array of chlorins for SAR studies that may advance the effectiveness of PDT and other applications.
Experimental Section
Representative procedures for the syntheses of chlorins by Methods II-V
2,2,5,7,8,12, 13,18-octamethylchlorin-17-propionic acid methyl ester (15a)
Method II
A solution of 12a (10.1 mg, 29 μmol) and 13a (12.2 mg, 29 μmol) in CH2Cl2 (2.6 mL) was treated with TFA (130μL) and stirred at rt in the dark for 5 h. TFA (520 μL) and TMOF (63 μL, 578 μmol) were then added, and the solution was stirred for an additional 16 h. The solvent was removed by rotary evaporation. The residue was redissolved in CH2Cl2 and washed with cold, saturated aq KHCO3, dried over Na2SO4, filtered, and concentrated. The oil was purified by flash chromatography (silica gel, EtOAc:hexanes = 1:4, 1% NEt3) to give 15a (5.1 mg, 34%) as a green film; Rf (1:4 EtOAc/hexanes) 0.30; IR(thin film) 3325, 1730 cm-1; 1H NMR (500 MHz, CDCl3) δ -2.52 (br s, 1H), -2.05 (br s, 1H), 2.07 (s, 6H), 3.22 (t, J = 8.1, 2H), 3.41 (s, 3H), 3.42 (s, 3H), 3.43 (s, 3H), 3.48 (s, 3H), 3.53 (s, 3H), 3.74 (s, 3H), 3.80 (s, 3H), 4.33 (t, J = 7.9, 2H), 4.44 (s, 2H), 8.85 (s, 1H), 9.62 (s, 1H), 9.72 (s, 1H); 13C NMR (500 MHz, CDCl3) δ 11.6, 12.00, 12.02, 16.6, 21.4, 21.9, 32.1, 37.1, 46.0, 52.1, 52.4, 91.1, 97.5, 98.8, 105.3, 128.6, 129.5, 131.5, 133.4, 135.1, 135.3, 136.4, 137.2, 137.4, 138.3, 149.8, 151.8, 162.0, 171.8, 173.9; UV-vis (CHCl3): λmax(ε, mol-1cm-1) = 399(151,000), 501(12,000), 651(42,000) nm; HRMS (EI) Calcd for C32H38N4O2: 510.2995; found: 510.3002.
Method III
A solution of 12a (20.3 mg, 58 μmol) and 23 (20.3 mg, 58 μmol) in CH2Cl2 (5.5 mL) was cooled to 0°C and treated with TFA (280 μL, 5 v/v%). After stirring for 1h, an additional 1.1 mL (20 v/v%) of TFA was added, the ice bath was removed, and the reaction was stirred at rt for 1.5 h. The solution was cooled to 0°C and quenched with NEt3 (2.6 mL). The mixture was washed sequentially with water (3 × 5 mL) and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, EtOAc:hexanes = 1:4, 1% NEt3) to give 15a (8.3 mg, 28%) as a green solid that was identical to the material prepared by Method II.
Method V
A solution of 9a (16.9 mg, 62 μmol) and 13a (26.0 mg, 62 μmol) in CH2Cl2 (5.7 mL) was treated with TFA (280μL, 5 v/v %) and stirred at rt in the dark for 5 h. More TFA (1.1 mL, 20 v/v %) was then added, and the solution was stirred for an additional 16 h. The solvent was removed by rotary evaporation. The residue was redissolved in CH2Cl2 and washed with cold, saturated aq KHCO3, dried over Na2SO4, filtered, and concentrated. The product was purified by flash chromatography (silica gel, EtOAc:hexanes = 1:4, 1% NEt3) to give 15a (23.5 mg, 74%) as a green film that was identical to the material prepared by Method II.
2,2,5,7,8,12, 17,18-octamethylchlorin-13-propionic acid methyl ester (26)
Method IV
A solution of 12a (12.4 mg, 36 μmol) and 25 (15 mg, 36 μmol) in CH2Cl2 (3.3 mL) was cooled to 0°C and treated with TFA (820 μL, 15 v/v%). The mixture was stirred at rt in the dark for 24 h, then cooled to 0°C and quenched with NEt3 (1.5 mL). The solution was washed sequentially with water (3 × 5 mL) and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, EtOAc:hexanes = 1:4, 1% NEt3) to give 26 (7.2 mg, 39%) as a green solid, decomp. > 240°C. Rf (3:7 EtOAc/hexanes) 0.49; IR(thin film) 3311, 1731, 1150, 917 cm-1; UV-vis (12% CH2Cl2 in MeOH): λmax (ε L mol-1 cm-1) = 642 (13000), 591, 501, 358 nm; 1H NMR (500 MHz, CDCl3) δ -2.43 (br s, 1H), -2.05 (br s, 1H), 2.05 (s, 6H), 3.18 (t, J = 8.09, 2H), 3.44 (s, 3H), 3.46 (s, 3H), 2.49 (s, 3H), 3.50 (s, 3H), 3.53 (s, 3H), 3.74 (s, 3H), 3.86 (s, 3H), 4.19 (t, J = 8.09, 2H), 4.46 (s, 2H), 8.78 (s, 1H), 9.54 (s, 1H), 9.75 (s, 1H); 13C NMR (500 MHz, CDCl3) δ 11.6, 11.7, 11.9, 12.0, 16.6, 21.5, 22.2, 32.0, 37.5, 46.1, 51.9, 52.3, 90.7, 97.3, 99.3, 105.1, 128.3, 129.6, 131.1, 133.3, 134.9, 135.3, 137.8, 138.29, 138.34, 138.5, 148.9, 150.4, 161.8, 172.3, 174.3. HRMS (EI) Calcd for C32H38N4O2: 510.2995; found: 510.2993.
Supplementary Material
Supporting Information Available: Complete Ref. 11c, experimental details, characterization data, and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Chart 1.
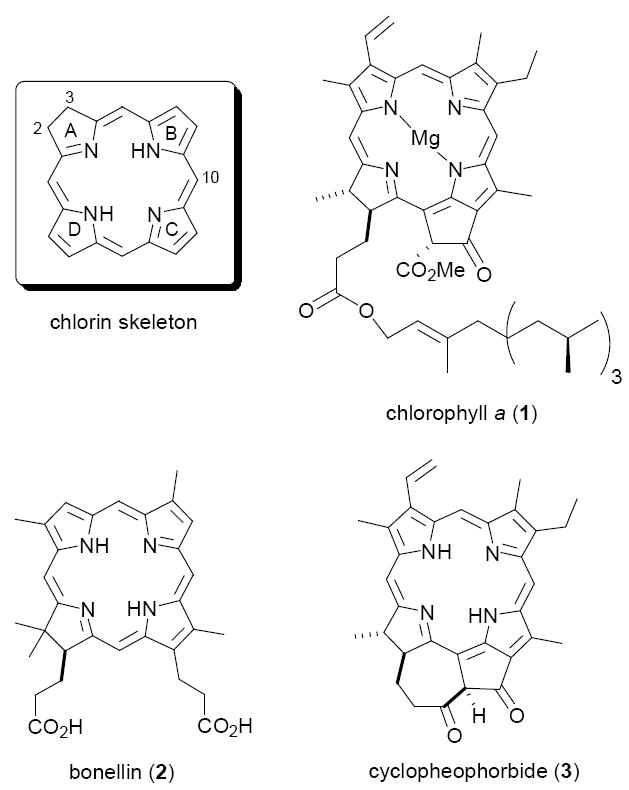
The chlorin skeleton and some natural chlorins
Acknowledgments
Financial support of this work by the National Institutes of Health, NIGMS Grant No. GM38913 is gratefully acknowledged. We thank Dr. William P. Roberts for his work during the initial development of Method II.
References
- 1.Montforts F-P, Glasenapp-Breiling M. In: Progress in the Chemistry of Organic Natural Products. Herz W, Falk H, Kirby GW, editors. Vol. 84. Springer; Wein, New York: 2002. pp. 1–51. [DOI] [PubMed] [Google Scholar]
- 2.Agius L, Ballantine JA, Ferito V, Jaccarini V, Murray-Rust P, Pelter A, Psaila AF, Schembri PJ. Pure Appl Chem. 1979;51:1847. [Google Scholar]
- 3.(a) Karuso P, Berguquist PR, Buckleton JS, Cambie RC, Clark GR, Rickard CEF. Tetrahedron Lett. 1986;27:2177. [Google Scholar]; (b) Watanabe N, Yamamoto K, Ihshikawa H, Yagi A. J Nat Prod. 1993;56:305. [Google Scholar]; (c) Wongsinkongman P, Brossi A, Wang H-K, Bastow KF, Lee K-H. Bioorg Med Chem. 2002;10:583. doi: 10.1016/s0968-0896(01)00305-4. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wasielewski MR. Chem Rev. 1992;92:435. [Google Scholar]; (b) Kay A, Grätzel M. J Phys Chem. 1993;97:6272. [Google Scholar]; (c) Kay A, Humphry-Baker R, Grätzel M. J Phys Chem. 1994;98:952. [Google Scholar]; (d) Wasielewski MR, Wiederrecht GP, Svec WA, Niemczyk MP. Sol Energy Mater Sol Cells. 1995;38:127. [Google Scholar]; (e) Itoh T, Asada H, Tobioka K, Kodera Y, Matsushima A, Hiroto M, Nishimura H, Kamachi T, Okura I, Inada Y. Bioconjugate Chem. 2000;11:8. doi: 10.1021/bc990045t. [DOI] [PubMed] [Google Scholar]; (f) Röger C, Müller MG, Lysetska Y, Holzwarth AR, Würthner F. J Am Chem Soc. 2006;128:6542. doi: 10.1021/ja0584469. [DOI] [PubMed] [Google Scholar]
- 5.(a) Taniguchi M, Ra D, Kirmaier C, Hindin E, Schwartz JK, Diers JR, Knox RS, Bocian DF, Lindsey JS, Holten D. J Am Chem Soc. 2003;125:13461. doi: 10.1021/ja035987u. [DOI] [PubMed] [Google Scholar]; (b) Tamiaki H, Miyatake T, Tanikaga R, Holzwarth AR, Schaffner K. Angew Chem Int Ed Engl. 1996;35:772. [Google Scholar]; (c) Balaban TS. Acc Chem Res. 2005;38:612. doi: 10.1021/ar040211z. [DOI] [PubMed] [Google Scholar]
- 6.(a) Saiki Y, Amao Y. Bioconjugate Chem. 2002;13:898. doi: 10.1021/bc025506g. [DOI] [PubMed] [Google Scholar]; (b) Takeuchi Y, Amao Y. Bioconjugate Chem. 2003;14:268. doi: 10.1021/bc0255844. [DOI] [PubMed] [Google Scholar]; (c) Himeshima N, Amao Y. Green Chem. 2005;7:742. [Google Scholar]
- 7.(a) Amao Y, Yamada Y, Aoki K. J Photochem Photobiol A. 2004:47. [Google Scholar]; (b) Takeuchi Y, Amao Y. J Jpn Pet Inst. 2004;47:355. [Google Scholar]; (c) Amao Y, Yamada Y. J Jpn Pet Inst. 2004;47:406. [Google Scholar]; (d) Amao Y, Yamada Y. Biosens Bioelectron. 2007;22:1561. doi: 10.1016/j.bios.2006.07.006. [DOI] [PubMed] [Google Scholar]; (e) Stromberg JR, Marton A, Kee HL, Kirmaier C, Diers JR, Muthiah C, Taniguchi M, Lindsey JS, Bocian DF, Meyer GJ, Holten D. J Phys Chem C. 2007;111:15464. [Google Scholar]
- 8.Milgrom LR. The Colours of Life. Chapter 7 Oxford University; Oxford, UK: 1997. [Google Scholar]
- 9.Licha K. Top Curr Chem. 2002;222:1. [Google Scholar]
- 10.Bonnett R. Chemical Aspects of Photodynamic Therapy. Gordon and Breach; Amsterdam, The Netherlands: 2000. pp. 177–289. [Google Scholar]
- 11.(a) Wickens J, Blinder KJ. Drug Safety. 2006;29:189. doi: 10.2165/00002018-200629030-00003. [DOI] [PubMed] [Google Scholar]; (b) Gold MH. Dermatol Clin. 2007;25:1. doi: 10.1016/j.det.2006.09.004. [DOI] [PubMed] [Google Scholar]; (c) Kereiakes DJ, et al. Circulation. 2003;108:1310. doi: 10.1161/01.CIR.0000087602.91755.19. [DOI] [PubMed] [Google Scholar]
- 12.(a) Wainwright M. J Antimicrob Chemother. 1998;42:13. doi: 10.1093/jac/42.1.13. [DOI] [PubMed] [Google Scholar]; (b) Soukos NS, Ximeniz-Fyvie LA, Hamblin MR, Socransky SS, Hasan T. Antimicrob Agents Chemother. 1998;42:2595. doi: 10.1128/aac.42.10.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jori G, Brown SB. Photochem Photobiol Sci. 2004;3:403. doi: 10.1039/b311904c. [DOI] [PubMed] [Google Scholar]; (d) Ferro S, Ricchelli F, Mancini G, Tognon G, Jori G. J Photochem Photobiol B. 2006;83:98. doi: 10.1016/j.jphotobiol.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Laha JK, Muthiah C, Taniguchi M, McDowell BE, Ptaszek M, Lindsey JS. J Org Chem. 2006;71:4092. doi: 10.1021/jo060208o. and references cited therein. Taniguchi M, Ptaszek M, McDowell BE, Boyle PD, Lindsey JS. Tetrahedron. 2007;63:3850. doi: 10.1016/j.tet.2007.02.040.Montforts F-P, Gerlach B, Höper F. Chem Rev. 1994;94:327.Milgrom LR. The Colours of Life. Chapter 1 Oxford University; Oxford: 1997.
- 14.Balasubramanian T, Strachan J-P, Boyle PD, Lindsey JS. J Org Chem. 2000;65:7919. doi: 10.1021/jo000913b. [DOI] [PubMed] [Google Scholar]
- 15.(a) Battersby AR, Dutton CJ, Fookes CJR, Turner SPD. J Chem Soc Perkin Trans. 1988;1:1557. [Google Scholar]; (b) Montforts F-P. Angew Chem Int Ed Engl. 1981;20:778. [Google Scholar]
- 16.For an overview see: Battersby AR. Acc Chem Res. 1986;19:147.
- 17.(a) Battersby AR, Dutton CJ, Fookes CJR. J Chem Soc Perkin Trans. 1988;1:1569. [Google Scholar]; (b) Battersby AR, Turner SPD, Block MH, Sheng Z-C, Zimmerman SC. J Chem Soc Perkin Trans. 1988;1:1577. [Google Scholar]
- 18.(a) Strachan J-P, O’Shea DF, Balasubramanian T, Lindsey JS. J Org Chem. 2000;65:3160. doi: 10.1021/jo991942t. [DOI] [PubMed] [Google Scholar]; (b) Taniguchi M, Ra D, Mo G, Balasubramanian T, Lindsey JS. J Org Chem. 2001;66:7342. doi: 10.1021/jo0104835. [DOI] [PubMed] [Google Scholar]; (c) Ptaszek M, McDowell BE, Taniguchi M, Kim H-J, Lindsey JS. Tetrahedron. 2007;63:3826. doi: 10.1016/j.tet.2007.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taniguchi M, Kim MN, Ra D, Lindsey JS. J Org Chem. 2005;70:275. doi: 10.1021/jo048440m. [DOI] [PubMed] [Google Scholar]
- 20.For other representative synthetic approaches to chlorins, see: Burns DH, Li YH, Shi DC, Caldwell TM. J Org Chem. 2002;67:4536. doi: 10.1021/jo020105f.Galezowski M, Gryko DT. J Org Chem. 2006;71:5942. doi: 10.1021/jo060545x.
- 21.(a) Jacobi PA, Lanz S, Ghosh I, Leung SH, Lower F, Pippin D. Org Lett. 2001;3:831. doi: 10.1021/ol006983m. [DOI] [PubMed] [Google Scholar]; (b) O’Neal WG, Roberts WP, Ghosh I, Wang H, Jacobi PA. J Org Chem. 2006;71:3472. doi: 10.1021/jo060041z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.For example, see: Jackson AH, Pandey RK, Rao KRN, Roberts E. Tetrahedron Lett. 1985;26:793. and ref. 23b
- 23.(a) Li W, Lash TD. Tetrahedron Lett. 1998;39:8571. [Google Scholar]; (b) Zhang B, Lash TD. Tetrahedron Lett. 2003;44:7253. [Google Scholar]
- 24.O’Neal WG, Roberts WP, Ghosh I, Jacobi PA. J Org Chem. 2005;70:7243. doi: 10.1021/jo050907l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shorter reaction periods were found to be less effective since the final reaction mixtures were invariably contaminated with unidentified pigments that complicated isolation of chlorin. The 5 h reaction time for this step eliminated these pigments.
- 26.These yields are considered optimized with respect to temperature effects, reaction time, concentration, reagent quantity, and the presence of various metal templates, which had no beneficial effect.
- 27.Jackson AH. In: Pyrroles. The Chemistry of Heterocyclic Compounds. Part 1. Jones RA, editor. Vol. 48. John Wiley & Sons; New York: 1990. p. 315. [Google Scholar]
- 28.Jackson AH. In: Pyrroles. The Chemistry of Heterocyclic Compounds. Part 1. Jones RA, editor. Vol. 48. John Wiley & Sons; New York: 1990. pp. 297–301. [Google Scholar]
- 29.Pandey RK, Rezzano IN, Smith KM. J Chem Res Miniprint. 1987;8:2171. [Google Scholar]
- 30.Chong R, Clezy PS, Liepa AJ, Nichol AW. Aust J Chem. 1969;22:229. [Google Scholar]
- 31.Trace amounts of an isomeric chlorin could be detected by UV analysis, but the structure of this compound was not conclusively identified.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Complete Ref. 11c, experimental details, characterization data, and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.