

Abstract
BACKGROUND
Endomorphin-1 and endomorphin-2 are endogenous peptides that are highly selective for μ-opioid receptors. However, studies of their functional efficacy and selectivity are controversial. In this study, we systematically compared the effects of intrathecal (i.t.) administration of endomorphin-1 and -2 on nociception assays and G protein activation with those of [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO), a highly effective peptidic μ-opioid receptor agonist.
METHODS
Male Sprague-Dawley rats were used. Acute and inflammatory pain models were used to compare the duration and magnitude of antinociception. Agonist-stimulated [35S]GTPγS binding was used to observe the functional activity at the level of the receptor-G protein in both spinal cord and thalamic membranes. In addition, antagonists selective for each receptor type were used to verify the functional selectivity of endomorphins in the rat spinal cord.
RESULTS
After i.t. administration, endomorphin-1 and -2 produced less antinociceptive effects than DAMGO in the model of acute pain. Concentration–response curves for DAMGO-, endomorphin-1-, and endomorphin-2-stimulated [35S]GTPγS binding revealed that both endomorphin-1 and -2 produced less G protein activation (i.e., approximately 50%–60%) than DAMGO did in the membranes of spinal cord and thalamus. In addition, i.t. endomorphin-induced antinociception was blocked by μ-opioid receptor selective dose of naltrexone (P < 0.05), but not by δ- and κ-opioid receptor antagonists, naltrindole and nor-binaltorphimine (P > 0.05).
CONCLUSIONS
Endomorphins are partial agonists for G protein activation at spinal and thalamic μ-opioid receptors. Both in vivo and in vitro measurements together suggest that DAMGO is more effective than endomorphins. Spinal endomorphins’ antinociceptive efficacy may range between 53% and 84% depending on the intensity and modality of the nociceptive stimulus.
The μ-opioid receptors are G protein-coupled receptors that play a pivotal role in the analgesic effects of opioid receptor agonists used clinically.1,2 Given that intrathecal (i.t.) administration of opioids is one of the most frequently used methods of analgesia in humans,3-5 it is important to study the functions of spinal μ-opioid receptors. In particular, the peptidic μ-opioid receptor agonists are of interest due to their enzymatic degradation and low toxicity.6
Endomorphin-1 and endomorphin-2 are endogenous opioid peptides isolated from bovine and human brains, and both peptides have high affinity and selectivity for μ-opioid receptors.7,8 Endomorphins have been implicated in a broad range of physiological functions including antinociceptive, cardiovascular, respiratory, digestive, rewarding, and endocrine responses.9,10 One important issue is the efficacy of endomorphins in activating μ-opioid receptors and subsequent antinociceptive effects manifested in vivo. Previous studies measuring the degree of agonist-stimulated G protein activation have shown that endomorphins act as partial agonists in the mouse spinal cord membranes and cell lines expressing μ-opioid receptors.11,12 In contrast, other studies indicate that endomorphins are full agonists in the rat thalamic membranes and cell lines expressing μ-opioid receptors.13,14 Although the antinociceptive effects of spinally administered endomorphins have been studied in rodents,15-18 there are few studies that directly compare the relative degrees, potencies, and durations of antinociceptive effects of i.t. endomorphins with those of i.t. [d-Ala,2N-Me-Phe,4Gly5-ol]-enkephalin (DAMGO), a highly effective peptidic μ-opioid receptor agonist.17 In particular, there is no study using both in vivo and in vitro measurements at the same time to compare the intrinsic efficacy between endomorphins and DAMGO at the rat spinal μ-opioid receptors.
Therefore, the aim of the study was to systematically investigate and directly compare the spinal antinociceptive effects of DAMGO, endomorphin-1, and endomorphin-2 together in rats by using both in vitro and in vivo assays. Agonist-stimulated [35S] guanosine-5′-O-(3-thio)triphosphate (GTPγS) binding19-22 was used to observe the functional activity at the level of the receptor-G protein in both spinal cord and thalamic membranes. Acute and inflammatory pain models23,24 were used to determine the degree of antinociceptive effects of these peptidic agonists after i.t. administration. With additional antagonist studies, this study used a pharmacological approach to elucidate the relative efficacy and functional selectivity of endomorphins as antinociceptive agents in the spinal cord of rats.
METHODS
Animals
Male Sprague-Dawley rats (275–300 g) were obtained from Harlan (Indianapolis, IN) and were housed in groups of three rats per cage. All animals were allowed ad libitum access to food and water, and were maintained on a 12 h light/dark cycle with lights on at 06:30 AM in a room kept at a temperature of 22°C ± 1°C. Experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. The experimental protocols were approved by the University Committee on the Use and Care of Animals at the University of Michigan.
Procedure
Intrathecal Catheterization
For i.t. administration, the rat was implanted with an i.t. catheter (see details for surgery in Zhang et al.25). After surgery, rats were housed individually and allowed 6–7 days for recovery. Lidocaine (2%, 10 μL) was administered i.t. to confirm whether the catheter was in the correct position. Lidocaine produces a transient paralysis of the hindpaws when it is injected into the lumbar enlargement. Rats were not used in the behavioral study if they did not show paralysis after receiving i.t. lidocaine or they showed motor dysfunction. For the behavioral study, a 10-μL solution of the test compound was infused, followed by an additional 10 μL of saline for flushing the catheter over a 30-s period. In addition, the i.t. catheter placement was verified after each experiment by administering methylene blue and checking for the distribution within the lumbar subarachnoid space. Only data obtained from rats with good i.t. catheter placement were used for data analysis. The dose ranges of i.t. DAMGO (0.01–0.3 μg), endomorphin-1, and -2 (3–30 μg), were chosen based on previous studies, showing that these doses produced maximal behavioral effects without producing behavioral toxicity.16,26 It should be noted that 100 μg of i.t. endomorphin-1 and -2 produced motor impairment in our pilot studies.
Warm Water Tail-Withdrawal Assay
The antinociceptive effect was measured by using a warm water tail-withdrawal assay, which was similar to the study by Walker et al.23 Briefly, 1 wk before the test, rats were habituated to the restraint tubes three times. The rat was placed into a restraint tube with its tail hanging freely. The last 5–7 cm of the tail was immersed into thermocontainers with either 40 (innocuous) or 55°C water (noxious stimulus), and the latency for tail withdrawal was measured. A cutoff time of 15 s was imposed to prevent tissue damage, and each stimulus presentation was separated by a 2-min interval. Baseline/control latencies of tail withdrawal in both 40°C and 55°C water were obtained for each rat 10 min before i.t. administration of the test compound. Then, the latency to withdraw the tail was re-determined at 10, 20, 30, 45, 60, and 90 min after i.t. administration.
Formalin Nociceptive Assay
Formalin was used to elicit nociceptive responses (i.e., hindpaw flinches) for measuring the antinociceptive effect of the test compound.24 Briefly, rats were placed individually in Plexiglas observation chambers (55 cm long × 30 cm wide × 26 cm high) and allowed to habituate for at least 15 min. A mirror was placed behind the chamber, which provided a clear view of the rat’s paw movement. Each rat received an injection of 2% formalin in 50 μL subcutaneously into the dorsal surface of the right hindpaw, returned to the observation chamber, and numbers of flinches were recorded by individuals who did not know the experimental condition. The test compound was given i.t. 5 min before formalin injection; then effects of the test compound to inhibit formalin-induced flinches were measured. Flinching responses were scored in a 5-min interval for 40 min and manifested for two characteristic time periods; i.e., 0–10 min (phase 1) and 11–40 min (phase 2) after formalin administration.
Agonist-Stimulated [35S]GTPγS Autoradiography
Agonist-stimulated [35S]GTPγS autoradiography was performed as described previously.21 Briefly, coronal sections (20 μm) were cut in the lumbar enlargement of the spinal cord of the naïve rat on a cryostat maintained at −18°C, mounted on gelatin-subbed slides, and stored at −80°C for <4 wk after collection. The sections were rinsed in the assay buffer (50 mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, and 0.1% bovine serum albumin, pH 7.4) for 10 min at 25°C, followed by preincubation in the assay buffer containing 2 mM GDP and 9.5 mU/mL adenosine deaminase for 15 min at 25°C. Then, sections were incubated in the assay buffer with GDP, adenosine deaminase, 0.04 nM [35S] GTPγS, and 10 μM of an agonist (i.e., DAMGO, endomorphin-1, or endomorphin-2) for 2 h at 25°C. Basal G protein activation was determined by incubating sections without agonists, and nonspecific binding was determined by adding 10 mM unlabeled GTPγS. After 2-h incubation, slides were rinsed twice for 2 min in ice-cold 50 mM Tris buffer (pH 7.4) and once in distilled water for 1 min. Slides were air-dried for a few hours and exposed to film for 48 h in film cassettes together with 14C Microscale standards (Amersham Biosciences Inc., Piscataway, NJ) for densitometric analysis. Then the films were developed using an autoprocessor. The slices were scanned at 600 dpi for analysis. Images were digitized and densitometric analysis was conducted by using NIH Image J software. Optical density measurements were converted to [35S]GTPγS binding values (nCi 35S per gram of tissue) by use of the 14C standards and a conversion factor.21 All three compounds were tested on eight spinal cord sections per rat in six rats.
Agonist-Stimulated [35S]GTPγS Binding in Membranes
The membranes of thalamus and spinal cords were isolated from rats. Tissues were homogenized in 20 volumes (w/vol) of ice-cold Tris–HC1 buffer (50 mM, pH 7.4) and centrifuged at 15,000g, 4°C, for 20 min. The pellet was resuspended in Tris-HCl buffer and incubated for 15 min at 37°C. Centrifugation was repeated and the final pellet resuspended in Tris–HCl buffer and stored in aliquots at −80°C. The [35S]GTPγS binding assay was modified from a previous study.22 Membranes (10 μg protein of thalamus and 25 μg protein of spinal cord in each assay tube) were incubated with 100 pM [35S] GTPγS for 60 min at 25°C, in the absence or presence of varying concentrations of agonist (DAMGO, endomorphin-1, or endomorphin-2), in GTPγS binding buffer (final concentration mM: Tris 20 mM, MgCl2 5 mM, EDTA 0.8 mM, 100 mM NaCl, GDP 80 μM, pH 7.4) in a final assay volume of 250 μL. Inhibition of agonist-stimulated [35S] GTPγS binding by naloxone (NLX, 20 nM) was evaluated by adding antagonist to the membrane 15 min before incubation with agonists. Basal binding was determined from tubes that contained the same volume of GTPγS binding buffer without agonist and antagonist. The incubation was terminated by filtrating the samples through the glass fiber filters. Filters were washed three times with ice-cold buffer (Tris 50 mM, MgCl2 5 mM, 100 mM NaCl, pH 7.4) in a Millipore filtration instrument, dried, and measured for their liquid scintillation counting. Each data point was subtracted by the basal value and converted to femtomoles per milligram of protein to determine agonist-stimulated [35S]GTPγS binding. Two thalamic and two spinal membrane preparations from a rat were tested in each binding experiment, and the experiment was repeated at least three times for each rat.
Drugs
DAMGO, endomorphin-1, endomorphin-2, naltrex-one, naltrindole, and NLX (National Institute on Drug Abuse, Bethesda, MD) and nor-binaltorphimine (Sigma-Aldrich, St. Louis, MO) were dissolved in sterile water. [35S]GTPγS (1250 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). GDP and all other biochemicals were purchased from Sigma Chemical. For the antagonist study in vivo, rats were injected with either sterile water (vehicle, 1 mL/kg), naltrexone (0.1 mg/kg) or naltrindole (3 mg/kg) subcutaneously 30 min before an i.t. administration of the agonist. Nor-binaltorphimine (10 mg/kg) was injected subcutaneously 24 h before the i.t. administration of the agonist. The dose and pretreatment time for these opioid receptor antagonists were chosen based on previous studies showing that naltrexone, naltrindole, and nor-binaltorphimine produced selective functional antagonism for μ-, δ-, and κ-opioid receptors, respectively.27-30
Data Analysis
All data were expressed as mean ± SEM from individual values for all behavioral end-points. The tail-withdrawal latencies were converted to a percentage of maximum possible effect (%MPE) by the formula: %MPE = (test latency − control latency)/(15 s − control latency) × 100, using the control baseline latency in 55°C water measured at the beginning of each experiment. Data for the time course and total flinching responses were analyzed by a two-way analysis of variance, followed by the Dunnett test for multiple post hoc comparisons (*P < 0.05 for significance). ED50 values were calculated by least-squares regression with the portion of the dose–response curves that spanned 50% of responding. For the [35S]GTPγS binding experiments, data were analyzed by a sigmoidal curve with a variable slope using GraphPad Prism (GraphPad Software Inc., San Diego, CA) to determine the EC50 value and maximum stimulation. The criterion for significance was also set at P < 0.05.
RESULTS
Figure 1 illustrates the degree and time course of antinociception produced by i.t. administered DAMGO, endomorphin-1, and endomorphin-2 in the warm water tail-withdrawal assay. There was no difference in the baseline tail-withdrawal latency among groups. All rats’ baseline withdrawal latencies in 40°C water were 15 s. The mean values of baseline withdrawal latencies in 55°C water in different groups ranged from 2.6 ± 0.4 (SEM) s to 3.2 ± 0.4 s. I.t. administration of DAMGO [F(15,120) = 8.2], endomorphin-1 [F(15,120) = 3.4], endomorphin-2 [F(15,120 = 3.7] dose-dependently produced antinociception against 55°C water (P < 0.05). The peak antinociceptive effect of all agonists occurred at the first observation period, 10 min after i.t. administration. The antinociceptive effects of i.t. DAMGO 0.3 μg lasted for 45 min after administration. In contrast, the antinociceptive effects of both endomorphins at 30 μg lasted for 20–30 min after i.t. administration. The mean values of each agonist’s potency and magnitude in producing antinociception are summarized in Table 1.
Figure 1.
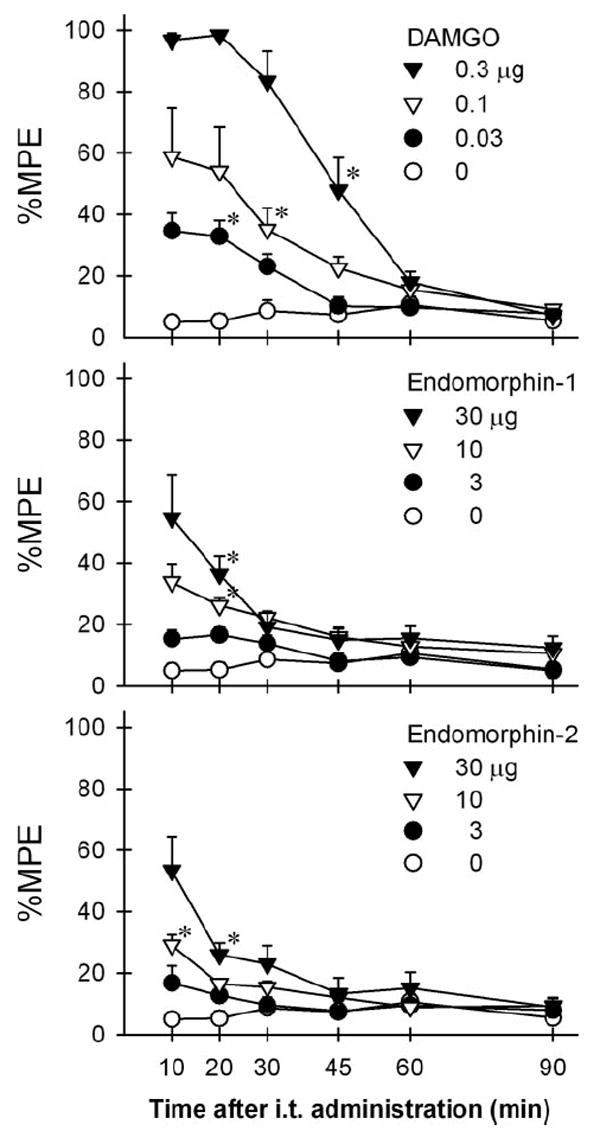
Time course of antinociception produced by intrathecally administered [d-Ala,2N-Me-Phe,4Gly5-ol]-enkephalin (DAMGO), endomorphin-1, and endomorphin-2 in rats. Antinociception was measured with a warm water tail-withdrawal assay. Abscissas: time in minutes after intrathecal administration. Ordinates: latency to withdraw the tail in 55°C water, as a percentage of the maximum possible effect (%MPE). Each value represents mean ± SEM (n = 6–8). Symbols represent different dosing conditions for each panel. The asterisks represent a significant difference from the vehicle condition from the time point, 10 min, to the corresponding time point for each dose (*P < 0.05).
Table 1.
Comparison of the Potency and Magnitude of Intrathecally Administered Endomorphin-1, -2-, and [d -Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO)-Induced Antinociception in the Rat Nociceptive Assays
| Formalin assay
|
|||||
|---|---|---|---|---|---|
| Warm water tail-withdrawal assay
|
Phase 1
|
Phase 2
|
|||
| ED50 (μg)a | %MPEb | ED50 (μg) | Max. effect
(%inhibition)c |
ED50 (μg) | Max. effect
(%inhibition) |
| DAMGO | |||||
| 0.11 (0.09–0.14) | 97 ± 2d | 0.03 (0.02–0.04) | 90 ± 5 | 0.05 (0.02–0.09) | 89 ± 7 |
| Endomorphin-1 | |||||
| 17.9 (16.1–19.9) | 55 ± 13 | 5.4 (4.8–6.0) | 84 ± 2 | 9.1 (3.6–23.0) | 81 ± 12 |
| Endomorphin-2 | |||||
| 25.9 (22.4–29.9) | 53 ± 11 | 6.0 (5.2–7.0) | 73 ± 3 | 10.2 (2.9–25.7) | 77 ± 8 |
Each value represents the mean ED50 with 95% confidence interval.
The percentage of maximum possible effect (%MPE) as evaluated at the 10-min time point in thermal antinociception (see Fig. 1 for other details).
The percentage of maximum inhibition of nociceptive responses (i.e., flinches) compared with the vehicle-treated group (Phase 1: 0–5 min and Phase 2: 10–15 min after formalin injection, see Fig. 3 for other details).
Shown are mean values and SEM of individual data of rats receiving i.t. DAMGO (0.3 μg), endomorphin-1 (30 μg), or endomorphin-2 (30 μg).
Figure 2 illustrates the effects of opioid receptor antagonists on the antinociception of i.t. administered endomorphins in the warm water tail-withdrawal assay. Post hoc comparisons indicated that only pretreatment with naltrexone 0.1 mg/kg significantly blocked antinociception of both i.t. endomorphins (P < 0.05). Neither naltrindole nor nor-binaltorphimine antagonized i.t. endomorphin-induced antinociception (P > 0.05).
Figure 2.
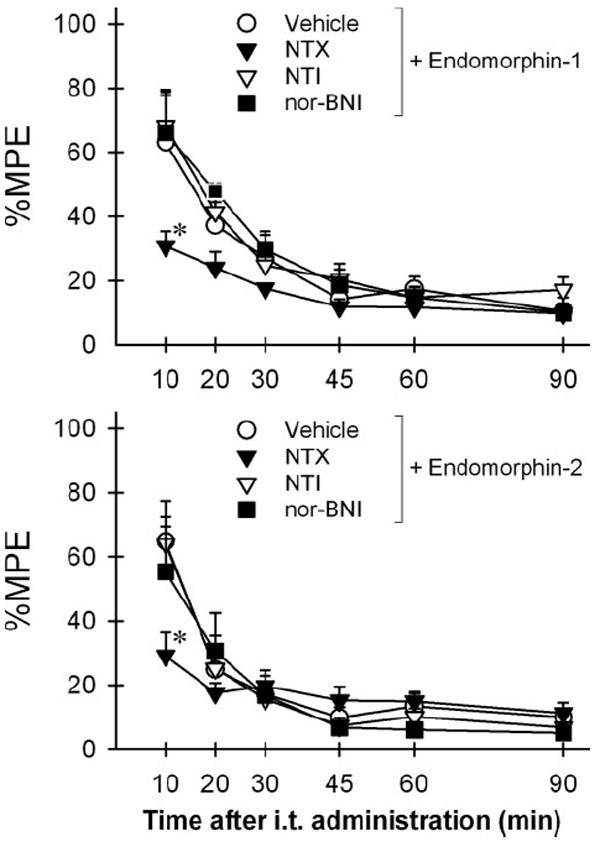
Effects of opioid receptor antagonists on the antinociception of intrathecal administration of endomorphin-1 (30 μg) and endomorphin-2 (30 μg) in the rat tail-withdrawal assay. Sterile water (vehicle 1 mL/kg), naltrexone (NTX 0.1 mg/kg), or naltrindole (NTI ± mg/kg) was administered subcutaneously 30 min before intrathecal administration of endomorphins. Nor-binaltorphimine (nor-BNI 10 mg/kg) was administered subcutaneously 24 h before endomorphins. The asterisks represent a significant difference from the vehicle-pretreated group condition (P < 0.05). See Figure 1 for other details.
Figure 3 illustrates the degree and time course of antinociception produced by i.t. administered DAMGO, endomorphin-1, and endomorphin-2 in the formalin nociceptive assay. I.t. administration of DAMGO [F(44,300) = 2.2], endomorphin-1 [F(33,240) = 1.8], endomorphin-2 [F(33,240) = 1.7] dose-dependently attenuated formalin-induced flinching responses (P < 0.05). The antinociceptive effects of i.t. DAMGO 0.3 μg lasted at least for 40 min after formalin administration. In contrast, the antinociceptive effects of both endomorphins at 30 μg lasted for 30–35 min after formalin administration. The mean values of each agonist’s potency and magnitude in attenuating formalin-induced flinches are summarized in Table 1.
Figure 3.
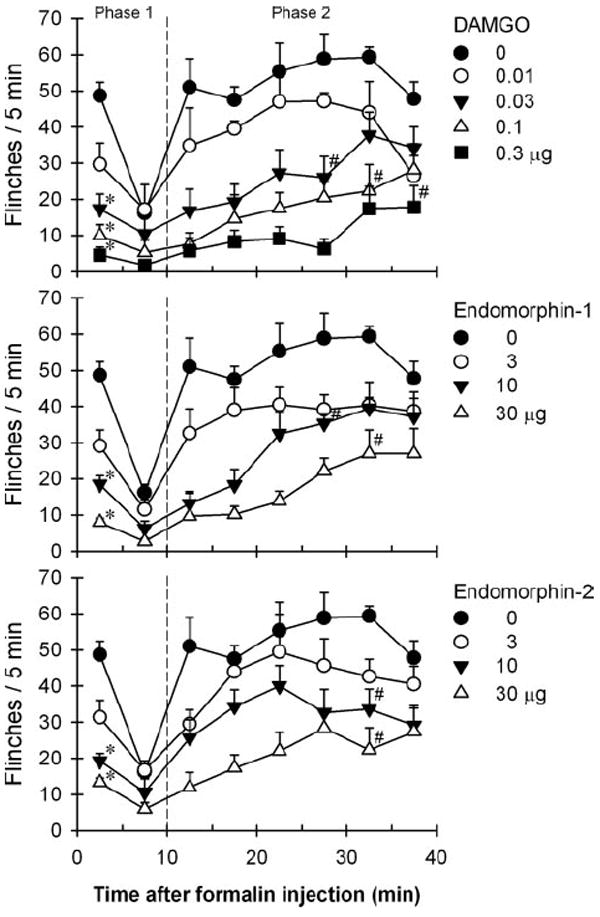
Comparison of antinociception produced by intrathecally administered [d-Ala2N-Me-Phe,4Gly5-ol]-enkephalin (DAMGO), endomorphin-1, and endomorphin-2 in the rat formalin assay. Antinociception was measured by counting formalin-induced flinching responses. Abscissas: time in minutes after formalin injection. Ordinates: number of flinches for each 5-min session. Each value represents mean ± SEM (n = 6–8). Symbols represent different dosing conditions for each panel. The asterisk represents a significant difference from the vehicle condition in the phase one of formalin-induced flinching (P < 0.05). The # represents a significant difference from the vehicle condition from the session (11–15th min) to the corresponding session in the phase two of formalin-induced flinching (P < 0.05).
Figure 4 illustrates the effects of opioid receptor antagonists on the antinociception of i.t. administered endomorphins in the formalin nociceptive assay. Post hoc comparisons indicated that only pretreatment with naltrexone 0.1 mg/kg significantly reversed antinociception of i.t. endomorphin-1 and endomorphin-2 in both phases (P < 0.05). Neither naltrindole nor nor-binaltorphimine antagonized i.t. endomorphins-induced attenuation of formalin-induced flinching responses (P < 0.05).
Figure 4.
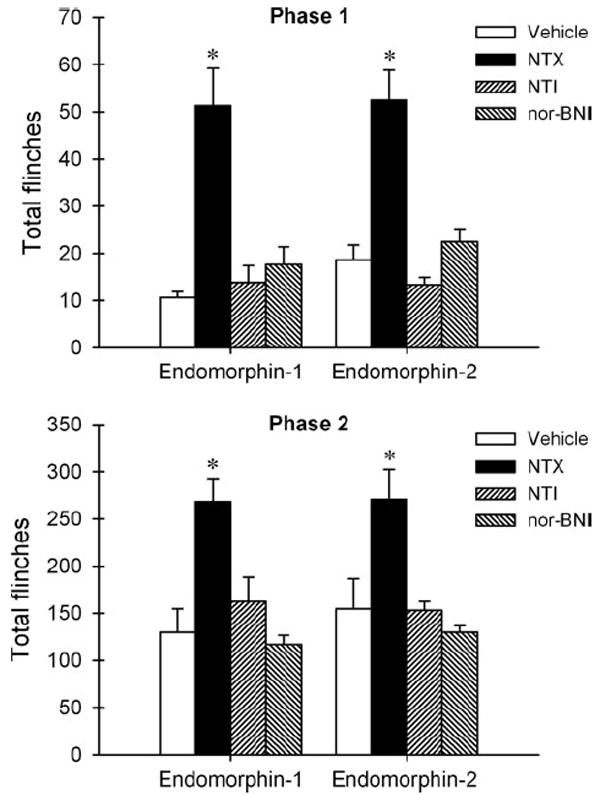
Effects of opioid receptor antagonists on the antinociception of intrathecal administration of endomorphin-1 (30 μg) and endomorphin-2 (30 μg) in the rat formalin assay. Sterile water (vehicle 1 mL/kg), naltrexone (NTX 0.1 mg/kg), or naltrindole (NTI ± mg/kg) was administered subcutaneously 30 min before intrathecal administration of endomorphins. Nor-binaltorphimine (nor-BNI 10 mg/kg) was administered subcutaneously 24 h before endomorphins. Ordinates: Total flinches during the phase 1 (0–10 min, top panel) and phase 2 (11–40 min, bottom panel) after administration of formalin. The asterisks represent a significant difference from the vehicle-pretreated group condition (P < 0.05). See Figure 3 for other details.
Figure 5 compares the degrees of agonist-stimulated [35S]GTPγS binding autoradiography in the rat spinal cord. DAMGO-, endomorphin-1-, and endomorphin-2-stimulated [35S]GTPγS bindings in the spinal cord slices were higher than the basal values (P < 0.05). There was no difference between the effects of endomorphin-1 and endomorphin-2. However, post hoc comparisons indicated that DAMGO significantly stimulated more [35S]GTPγS binding than endomorphin-1 and endomorphin-2 at the same concentration 10 μM (P < 0.05).
Figure 5.
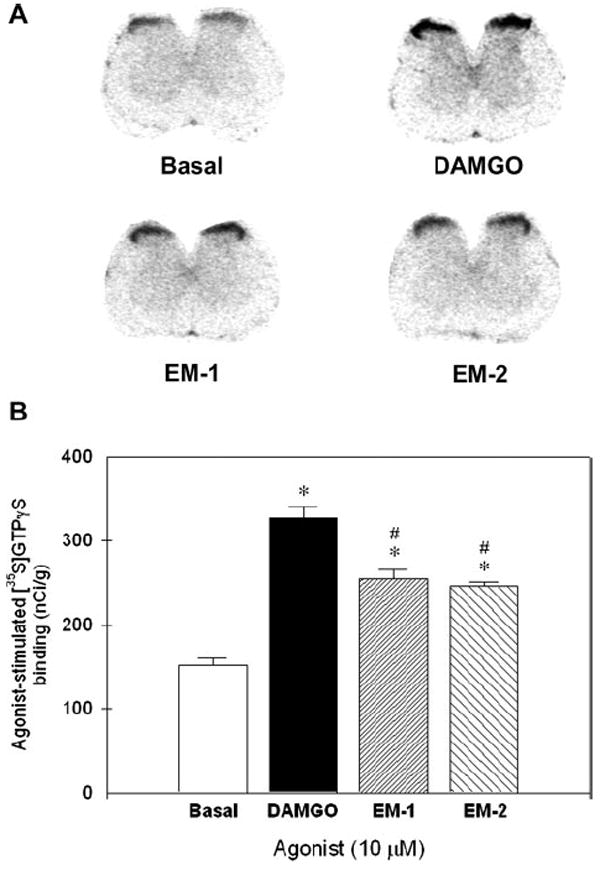
Stimulation of [35S] guanosine-5′-O-(3-thio) triphosphate (GTPγS) binding autoradiography by [d-Ala,2N-Me-Phe,4Gly5-ol]-enkephalin (DAMGO), endomorphin-1 (EM-1), and endomorphin-2 (EM-2) in the rat spinal cord. Top panel (A): Representative autoradiograms illustrating the maximal basal and agonist-stimulated [35S]GTPγS binding in the rat spinal cord sections. Bottom panel (B): Quantification of agonist-stimulated [35S]GTPγS binding in the rat spinal cord sections. The asterisk represents a significant difference from the basal value (P < 0.05). The # represents a significant difference from the value of DAMGO-treated group.
Figure 6 illustrates the degrees of [35S]GTPγS binding stimulation by DAMGO, endomorphin-1, and endomorphin-2 in the rat spinal cord membranes. DAMGO, endomorphin-1, and endomorphin-2 concentration-dependently increased the binding of [35S]GTPγS bindings in the spinal cord membranes. Although all agonists showed similar potencies in stimulating [35S]GTPγS binding, the degree of DAMGO-stimulated [35S]GTPγS binding was significantly higher than those by endomorphins in the rat spinal cord (Table 2). Addition of NLX (20 nM) produced approximately 8–10 fold rightward shifts in the concentration-response curves of DAMGO, endomorphin-1 and endomorphin-2 (Table 2). Similarly, the degree of DAMGO-stimulated [35S]GTPγS binding was significantly higher than those by endomorphins in the rat thalamic membranes. Addition of NLX (20 nM) also produced approximately 8–10 fold rightward shifts in the concentration–response curves of DAMGO, endomorphin-1, and endomorphin-2 in the thalamic membranes (Table 2).
Figure 6.
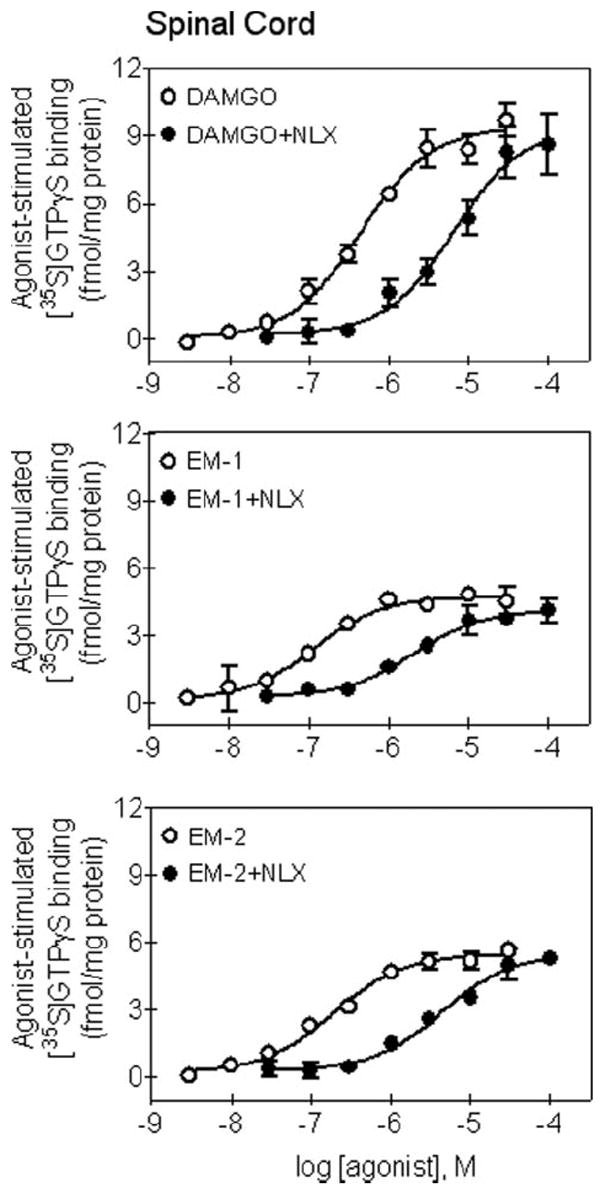
Stimulation of [35S] guanosine-5’-O-(3-thio) triphosphate (GTPγS) binding by [d-Ala,2N-Me-Phe,4Gly5-ol]-enkephalin (DAMGO), endomorphin-1 (EM-1), and endomorphin-2 (EM-2) in the rat spinal cord membranes. Concentration-response curves of agonist were determined in the absence or presence of an antagonist, naloxone (NLX). Shown are mean ± SEM from three independent experiments and each were conducted in duplicate. See Table 2 for other details.
Table 2.
Effects of Endomorphins on [35S] Guanosine-5′-O-(3-thio)triphosphate (GTPγS) Binding in the Rat Spinal Cord and Thalamus Membranes Compared with [d -Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO)
| Spinal cord
|
Thalamus
|
|||||||
|---|---|---|---|---|---|---|---|---|
| EC50 (μM)
|
Max. stimulation (fmol/mg)a |
EC50 (μM)
|
Max. stimulation (fmol/mg)
|
|||||
| Agonist | Agonist + naloxone | %DAMGOb | Agonist | Agonist + naloxone | %DAMGO | |||
| DAMGO | 0.30 ± 0.8c | 3.3 ± 1.1 | 9.0 ± 0.7 | 100 | 0.31 ± 0.7 | 2.9 ± 0.6 | 31.6 ± 2.1 | 100 |
| Endomorphin-1 | 0.31 ± 0.2 | 2.5 ± 1.0 | 4.5 ± 0.3 | 50 ± 3 | 0.35 ± 0.2 | 2.3 ± 0.6 | 18.9 ± 2.2 | 60 ± 7 |
| Endomorphin-2 | 0.37 ± 0.6 | 3.9 ± 1.1 | 4.7 ± 0.5 | 52 ± 6 | 0.37 ± 0.6 | 3.0 ± 0.7 | 16.3 ± 0.4 | 52 ± 2 |
Each value represents [35S]GTPγS (femtomoles per mg of protein) over basal binding in each membrane.
Each value represents the degree of agonist-stimulated [35S]GTPγS binding compared with the maximum value of DAMGO-stimulated [35S]GTPγS binding.
Shown are mean values and SEM of individual data of the rat membranes.
DISCUSSION
The behavioral study showed that i.t. endomorphin-1 and endomorphin-2 were less potent and produced less antinociceptive effects than i.t. DAMGO in the rat model of acute pain (Fig. 1 and Table 1). The doses of i.t. endomorphin-1 and -2 used in this study were similar to those reported by other researchers using different types of pain models.16,17 However, these early studies did not thoroughly compare the degrees of antinociception produced by i.t. endomorphins with that of DAMGO, a highly effective peptidic μ-opioid receptor agonist. The degree of antinociceptive effects measured herein may depend on the intensity and modality of noxious stimuli.31,32 Nevertheless, by using the same noxious stimulus (i.e., 55°C water) in rats, the present study demonstrates that i.t. administration of endomorphins act as partial agonists for antinociception in vivo as compared with i.t. DAMGO.
The functional study of G protein activation in the rat spinal cord further supported the different degrees of intrinsic efficacy between endomorphins and DAMGO. The [35S]GTPγS binding assay and autoradiography both revealed that endomorphin-1 and -2 produced less G protein activation in the rat spinal cord and thalamus (Figs. 5 and 6). Such a finding is consistent with those reported in previous [35S]GTPγS binding studies using different brain membranes or cell lines.11,12,21 However, this finding is different from a study showing that endomorphins and DAMGO had similar efficacy in stimulating [35S]GTPγS binding in the rat thalamic membranes.14 The magnitudes of opioid agonist-induced [35S]GTPγS binding may depend on the receptor density in a specific region of the central nervous system.21,22,33 Therefore, the large amount of membranes (e.g., 100 vs 10 μg) used in one study14 may saturate the measurement of GTPγS binding and reduce the potential differences of efficacy measurement among tested ligands. Nevertheless, the present findings are consistent with most in vitro studies, indicating that endomorphin-1 and -2 are partial agonists for G protein activation at the μ-opioid receptors.11,12,21
The degree of antinociception produced by spinal opioids depends on the agonist’s intrinsic efficacy and the intensity of the nociceptive stimulus.34,35 For example, high efficacy μ-opioid agonists produce maximal effects in antinociceptive assays using both low- and high-intensity nociceptive stimuli, whereas the maximal effects of low efficacy μ-opioid agonists decrease when the intensity of the nociceptive stimulus increases.31,32,34,35 The present study could not test a higher dose of endomorphins due to observed side effects16; thus, it may be difficult in vivo to demonstrate partial antinociceptive effects of endomorphins. Nevertheless, the same dose of DAMGO (0.3 μg) produced 89%–97% of maximal effects in both nociceptive assays. In contrast, the highest tested dose of endomorphins (i.e., 30 μg) produced 73%–84% of maximal effects in the model of inflammatory pain and the same dose of endomorphins produced only 53%–55% of maximal effects in the model of acute pain (Table 1). Such findings may suggest that DAMGO is more effective in vivo than endomorphins, and that endomorphins’ relative efficacy may range between 53% and 84%, depending on the intensity and modality of the nociceptive stimulus. This study is the first using both in vivo and in vitro measurements together to demonstrate the potential partial agonist actions of endomorphins for antinociception and its findings are consistent with a growing body of evidence indicating that the degree of antinociception produced by an opioid agonist depends on its intrinsic efficacy and the nociceptive stimulus intensity.31,32,34,35
Antagonist studies verified that i.t. endomorphin-induced antinociception was mainly mediated by spinal μ-opioid receptors in rats (Figs. 2 and 4). The doses for opioid receptor antagonists, naltrexone, naltrindole, and nor-binaltorphimine, used herein were able to produce selective functional antagonism for μ-, δ-, and κ-opioid receptors, respectively.27-30 Lack of effects of naltrindole and nor-binaltorphimine against i.t. endomorphin-induced antinociception suggested that the antinociceptive effects of i.t. endomorphins were independent from spinal δ- and κ-opioid receptors. In addition, NLX treatment produced similar degrees of parallel rightward shifts of the concentration-response curve for DAMGO- and endomorphin-stimulated [35S]GTPγS binding in the spinal cord and thalamic membranes (Table 2). Such a finding also supports the notion that the μ-opioid receptor is the key mediator for i.t. endomorphin-induced antinociception at the level of the receptor-G protein in the rat spinal cord. Interestingly, mouse studies indicated that i.t. or intracerebro-ventricular administration of endomorphin-2 increased the release of dynorphin A1-17 which subsequently acted on κ-opioid receptors to produce antinociception.18,36 Although a κ-opioid receptor antagonist, nor-binaltorphimine, did not antagonize the antinociception produced by i.t. endomorphins in the present study, more studies will be needed to elucidate the species differences in terms of the release of endogenous opioid peptides under different pain conditions.
The duration of antinociception produced by i.t. endomorphins was shorter than that of i.t. DAMGO (i.e., 20 vs 45 min) against acute nociceptive stimulus. Studies have shown that endomorphins are vulnerable to enzymatic cleavage and several enzymes participate in the enzymatic degradation of endomorphins.37-39 Interestingly, endomorphin-1 seems more resistant to enzymatic degradation than endomorphin-2 in vivo.40 However, most studies and the present study did not find a significant difference in the duration between i.t. endomorphin-1- and endomorphin-2-induced antinociception.15,16,36,41 It is worth noting that the duration of i.t. DAMGO-induced itch/scratching (i.e., 3–4 h) in monkeys is much longer than i.t. DAMGO-induced itch/scratching and antinociception (<1 h) in rodents.42-44 It is possible that the enzymatic degradation pathway of endomorphins may be different or/and slower in the primate cerebrospinal fluid, and it will be valuable to further investigate the duration and magnitude of antinociception and itch/scratching by i.t. endomorphins in primates.
This study demonstrated that i.t. administration of endomorphins produced antinociceptive effects against acute and inflammatory nociceptive stimuli in rats. More importantly, both in vivo and in vitro measurements together validated that endomorphin-1 and -2 are partial agonists selectively acting at the spinal μ-opioid receptors. Studies determining DAMGO- and endomorphin-stimulated [35S]GTPγS binding in spinal cord and thalamic membranes are valuable because they provide a pharmacological explanation for the spinal actions of i.t. DAMGO and endomorphins. In particular, it supports the use of the GTPγS binding assay for exploring and predicting the efficacy of opioid compounds on antinociception against the high intensity of nociceptive stimuli. The pharmacological properties of endomorphins in terms of the receptor binding selectivity and efficacy are unique because other discovered endogenous opioid peptides are not selective for any specific opioid receptor type.45,46 This study provides a sound pharmacological basis for studying newly developed endomorphin analogs that may have longer duration of action or/and higher intrinsic efficacy in the future.
Acknowledgments
The authors thank Wayne Yang and Tristan D. Edwards for excellent technical assistance.
Supported by US Public Health Service Grants DA-000254 & DA-013685, and Taiwan National Science Council Grant NSC-96-2413-H-004-019.
References
- 1.Inturrisi CE. Clinical pharmacology of opioids for pain. Clin J Pain. 2002;18:S3–13. doi: 10.1097/00002508-200207001-00002. [DOI] [PubMed] [Google Scholar]
- 2.Zollner C, Stein C. Opioids. Handb Exp Pharmacol. 2007;177:31–63. doi: 10.1007/978-3-540-33823-9_2. [DOI] [PubMed] [Google Scholar]
- 3.Bennett G, Serafini M, Burchiel K, Buchser E, Classen A, Deer T, Du Pen S, Ferrante FM, Hassenbusch SJ, Lou L, Maeyaert J, Penn R, Portenoy RK, Rauck R, Willis KD, Yaksh T. Evidence-based review of the literature on intrathecal delivery of pain medication. J Pain Symptom Manage. 2000;20:S12–36. doi: 10.1016/s0885-3924(00)00204-9. [DOI] [PubMed] [Google Scholar]
- 4.Dougherty PM, Staats PS. Intrathecal drug therapy for chronic pain: from basic science to clinical practice. Anesthesiology. 1999;91:1891–918. doi: 10.1097/00000542-199912000-00044. [DOI] [PubMed] [Google Scholar]
- 5.Rathmell JP, Lair TR, Nauman B. The role of intrathecal drugs in the treatment of acute pain. Anesth Analg. 2005;101:S30–43. doi: 10.1213/01.ANE.0000177101.99398.22. [DOI] [PubMed] [Google Scholar]
- 6.Negri L, Melchiorri P, Lattanzi R. Pharmacology of amphibian opiate peptides. Peptides. 2000;21:1639–47. doi: 10.1016/s0196-9781(00)00295-3. [DOI] [PubMed] [Google Scholar]
- 7.Zadina JE, Hackler L, Ge LJ, Kastin AJ. A potent and selective endogenous agonist for the μ-opiate receptor. Nature. 1997;386:499–502. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]
- 8.Hackler L, Zadina JE, Ge LJ, Kastin AJ. Isolation of relatively large amounts of endomorphin-1 and endomorphin-2 from human brain cortex. Peptides. 1997;18:1635–39. doi: 10.1016/s0196-9781(97)00259-3. [DOI] [PubMed] [Google Scholar]
- 9.Horvath G. Endomorphin-1 and endomorphin-2: pharmacology of the selective endogenous μ-opioid receptor agonists. Pharmacol Ther. 2000;88:437–63. doi: 10.1016/s0163-7258(00)00100-5. [DOI] [PubMed] [Google Scholar]
- 10.Fichna J, Janecka A, Costentin J, Do Rego JC. The endomorphin system and its evolving neurophysiological role. Pharmacol Rev. 2007;59:88–123. doi: 10.1124/pr.59.1.3. [DOI] [PubMed] [Google Scholar]
- 11.Alt A, Mansour A, Akil H, Medzihradsky F, Traynor JR, Woods JH. Stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding by endogenous opioids acting at a cloned mu receptor. J Pharmacol Exp Ther. 1998;286:282–88. [PubMed] [Google Scholar]
- 12.Narita M, Mizoguchi H, Oji GS, Tseng EL, Suganuma C, Nagase H, Tseng LF. Characterization of endomorphin-1 and -2 on [35S]GTPγS binding in the mouse spinal cord. Eur J Pharmacol. 1998;351:383–387. doi: 10.1016/s0014-2999(98)00395-1. [DOI] [PubMed] [Google Scholar]
- 13.Burford NT, Tolbert LM, Sadee W. Specific G protein activation and mu-opioid receptor internalization caused by morphine, DAMGO and endomorphin-1. Eur J Pharmacol. 1998;342:123–26. doi: 10.1016/s0014-2999(97)01556-2. [DOI] [PubMed] [Google Scholar]
- 14.Fichna J, do-Rego JC, Kosson P, Schiller PW, Costentin J, Janecka A. [35S]GTPγS binding stimulated by endomorphin-2 and morphiceptin analogs. Biochem Biophys Res Commun. 2006;345:162–68. doi: 10.1016/j.bbrc.2006.04.079. [DOI] [PubMed] [Google Scholar]
- 15.Stone LS, Fairbanks CA, Laughlin TM, Nguyen HO, Bushy TM, Wessendorf MW, Wilcox GL. Spinal analgesic actions of the new endogenous opioid peptides endomorphin-1 and endomorphin-2. Neuroreport. 1997;8:3131–35. doi: 10.1097/00001756-199709290-00025. [DOI] [PubMed] [Google Scholar]
- 16.Horvath G, Szikszay M, Tomboly C, Benedek G. Antinociceptive effects of intrathecal endomorphin-1 and -2 in rats. Life Sci. 1999;65:2635–41. doi: 10.1016/s0024-3205(99)00532-9. [DOI] [PubMed] [Google Scholar]
- 17.Przewlocka B, Mika J, Labuz D, Toth G, Przewlocki R. Spinal analgesic action of endomorphins in acute, inflammatory and neuropathic pain in rats. Eur J Pharmacol. 1999;367:189–196. doi: 10.1016/s0014-2999(98)00956-x. [DOI] [PubMed] [Google Scholar]
- 18.Tseng LF, Narita M, Suganuma C, Mizoguchi H, Ohsawa M, Nagase H, Kampine JP. Differential antinociceptive effects of endomorphin-1 and endomorphin-2 in the mouse. J Pharmacol Exp Ther. 2000;292:576–83. [PubMed] [Google Scholar]
- 19.Emmerson PJ, Clark MJ, Mansour A, Akil H, Woods JH, Medzihradsky F. Characterization of opioid agonist efficacy in a C6 glioma cell line expressing the μ opioid receptor. J Pharmacol Exp Ther. 1996;278:1121–27. [PubMed] [Google Scholar]
- 20.Selley DE, Sim LJ, Xiao R, Liu Q, Childers SR. μ-opioid receptor-stimulated guanosine-5′-O-(gamma-thio)-triphosphate binding in rat thalamus and cultured cell lines: signal transduction mechanisms underlying agonist efficacy. Mol Pharmacol. 1997;51:87–96. doi: 10.1124/mol.51.1.87. [DOI] [PubMed] [Google Scholar]
- 21.Sim LJ, Liu Q, Childers SR, Selley DE. Endomorphin-stimulated [35S]GTPγS binding in rat brain: Evidence for partial agonist activity at μ-opioid receptors. J Neurochem. 1998;70:1567–76. doi: 10.1046/j.1471-4159.1998.70041567.x. [DOI] [PubMed] [Google Scholar]
- 22.Ko MCH, Lee H, Harrison C, Clark MJ, Song HF, Naughton NN, Woods JH, Traynor JR. Studies of μ-, κ-, and δ-opioid receptor density and G protein activation in the cortex and thalamus of monkeys. J Pharmacol Exp Ther. 2003;306:179–86. doi: 10.1124/jpet.103.050625. [DOI] [PubMed] [Google Scholar]
- 23.Walker EA, Zernig G, Young EA. In vivo apparent affinity and efficacy estimates for mu opiates in a rat tail-withdrawal assay. Psychopharmacology. 1998;136:15–23. doi: 10.1007/s002130050534. [DOI] [PubMed] [Google Scholar]
- 24.Kovelowski CJ, Ossipov MH, Hruby VJ, Porreca F. Lesions of the dorsolateral funiculus block supraspinal opioid delta receptor mediated antinociception in the rat. Pain. 1999;83:115–22. doi: 10.1016/s0304-3959(99)00083-4. [DOI] [PubMed] [Google Scholar]
- 25.Zhang C, Tall JM, Meyer RA, Raja SN. Antiallodynic effects of systemic and intrathecal morphine in the spared nerve injury model of neuropathic pain in rats. Anesthesiology. 2004;100:905–11. doi: 10.1097/00000542-200404000-00021. [DOI] [PubMed] [Google Scholar]
- 26.Miaskowski C, Sutters KA, Taiwo YO, Levine JD. Antinociceptive and motor effects of delta/mu and kappa/mu combinations of intrathecal opioid agonists. Pain. 1992;49:137–44. doi: 10.1016/0304-3959(92)90200-U. [DOI] [PubMed] [Google Scholar]
- 27.Takemori AE, Ho BY, Naeseth JS, Portoghese PS. Nor-binaltorphimine, a highly selective kappa-opioid antagonist in analgesic and receptor binding assays. J Pharmacol Exp Ther. 1988;246:255–8. [PubMed] [Google Scholar]
- 28.Chang KJ, Rigdon GC, Howard JL, McNutt RW. A novel, potent and selective nonpeptidic delta opioid receptor agonist BW373U86. J Pharmacol Exp Ther. 1993;267:852–7. [PubMed] [Google Scholar]
- 29.Lee H, Naughton NN, Woods JH, Ko MCH. Characterization of scratching responses in rats following centrally administered morphine or bombesin. Behav Pharamcol. 2003;14:501–8. doi: 10.1097/01.fbp.0000095082.80017.0f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang HN, Torregrossa MM, Jutkiewicz EM, Shi YG, Rice KC, Woods JH, Watson SJ, Ko MCH. Endogenous opioids upregulate brain-derived neurotrophic factor mRNA through δ- and μ-opioid receptors independent of antidepressant-like effect. Eur J Neurosci. 2006;23:984–94. doi: 10.1111/j.1460-9568.2006.04621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parsons CG, Headley PM. Spinal antinociceptive actions of mu-and kappa-opioids: the importance of stimulus intensity in determining “selectivity” between reflexes to different modalities of noxious stimulus. Br J Pharmacol. 1998;98:523–32. doi: 10.1111/j.1476-5381.1989.tb12626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan D, Cook CD, Picker MJ. Sensitivity to the discriminative stimulus and antinociceptive effects of mu opioids: role of strain of rat, stimulus intensity, and intrinsic efficacy at the mu opioid receptor. J Pharmacol Exp Ther. 1999;289:965–75. [PubMed] [Google Scholar]
- 33.Maher CE, Selley DE, Childers SR. Relationship of mu opioid receptor binding to activation of G-proteins in specific rat brain regions. Biochem Pharmacol. 2000;59:1395–401. doi: 10.1016/s0006-2952(00)00272-0. [DOI] [PubMed] [Google Scholar]
- 34.Saeki S, Yaksh TL. Suppression of nociceptive responses by spinal mu opioid agonists: effects of stimulus intensity and agonist efficacy. Anesth Analg. 1993;77:265–74. doi: 10.1213/00000539-199308000-00010. [DOI] [PubMed] [Google Scholar]
- 35.Dirig DM, Yaksh TL. Differential right shifts in the dose-response curve for intrathecal morphine and sufentanil as a function of stimulus intensity. Pain. 1995;62:321–8. doi: 10.1016/0304-3959(95)00006-E. [DOI] [PubMed] [Google Scholar]
- 36.Sakurada S, Hayashi T, Yuhki M, Orito T, Zadina JE, Kastin AJ, Fujimura T, Murayama K, Sakurada C, Sakurada T, Narita M, Suzuki T, Tan-No K, Tseng LF. Differential antinociceptive effects induced by intrathecally administered endomorphin-1 and endomorphin-2 in the mouse. Eur J Pharmacol. 2001;427:203–10. doi: 10.1016/s0014-2999(01)01238-9. [DOI] [PubMed] [Google Scholar]
- 37.Tomboly C, Peter A, Toth G. In vitro quantitative study of the degredation of endomorphins. Peptides. 2002;23:1573–80. doi: 10.1016/s0196-9781(02)00100-6. [DOI] [PubMed] [Google Scholar]
- 38.Sakurada C, Sakurada S, Hayashi T, Katsuyama S, Tan-No K, Sakurada T. Degradation of endomorphin-2 at the supraspinal level in mice is initiated by dipeptidyl peptidase IV: an in vitro and in vivo assay. Biochem Pharmacol. 2003;66:653–61. doi: 10.1016/s0006-2952(03)00391-5. [DOI] [PubMed] [Google Scholar]
- 39.Janecka A, Kruszynski R, Fichna J, Kosson P, Janecki T. Enzymatic degradation studies of endomorphin-2 and its analogs containing N-methylated amino acids. Peptides. 2006;27:131–35. doi: 10.1016/j.peptides.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 40.Fujita T, Kumamoto E. Inhibition by endomorphin-1 and endomorphin-2 of excitatory transmission in adult rat substantia gelatinosa neurons. Neuroscience. 2006;139:1095–105. doi: 10.1016/j.neuroscience.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 41.Soignier RD, Vaccarino AL, Brennan AM, Kastin AJ, Zadina JE. Analgesic effects of endomorphin-1 and endomorphin-2 in the formalin test in mice. Life Sci. 2000;67:907–12. doi: 10.1016/s0024-3205(00)00689-5. [DOI] [PubMed] [Google Scholar]
- 42.Hao JX, Yu W, Wiesenfeld-Hallin Z, Xu XJ. Treatment of chronic allodynia in spinally injured rats: effects of intrathecal selective opioid receptor agonists. Pain. 1998;75:209–17. doi: 10.1016/s0304-3959(97)00221-2. [DOI] [PubMed] [Google Scholar]
- 43.Ko MCH, Song MS, Edwards T, Lee H, Naughton NN. The role of central mu opioid receptors in opioid-induced itch in primates. J Pharmacol Exp Ther. 2004;310:169–76. doi: 10.1124/jpet.103.061101. [DOI] [PubMed] [Google Scholar]
- 44.Yamaguchi T, Kitagawa K, Kuraishi Y. Itch-associated response and antinociception induced by intracisternal endomorphins in mice. Jpn J Pharmacol. 1998;78:337–43. doi: 10.1254/jjp.78.337. [DOI] [PubMed] [Google Scholar]
- 45.Simon EJ. Opioid receptors and endogenous opioid peptides. Med Res Rev. 1991;11:357–74. doi: 10.1002/med.2610110402. [DOI] [PubMed] [Google Scholar]
- 46.Zhang S, Tong Y, Tian M, Dehaven RN, Cortesburgos L, Mansson E, Simonin F, Kieffer B, Yu L. Dynorphin A as a potential endogenous ligand for four members of the opioid receptor gene family. J Pharmacol Exp Ther. 1998;286:136–41. [PubMed] [Google Scholar]