

Summary
Opioids can modulate neuroendocrine function. Less is known about the involvement of opioid receptor subtypes in the stimulatory effects of opioids on the primate hypothalamic–pituitary–adrenal (HPA) axis. The aim of this study was to investigate the stimulatory effects of opioids selective for each receptor subtype on plasma adrenocorticotropic hormone (ACTH) and cortisol levels in both male and female monkeys. The blood collection procedure was conducted in home-caged and unanesthetized rhesus monkeys that showed low and stable basal ACTH and cortisol levels. Three opioid receptor agonists, fentanyl, U-50488H, and SNC80, were used in behaviorally active doses; they are highly selective for mu, kappa, and delta opioid receptors, respectively. Plasma samples were collected at multiple time points before and after IV administration of each compound and were quantified by radioimmunoassay. Neither fentanyl (0.0003–0.02 mg/kg) nor SNC80 (0.03–0.3 mg/kg) changed either ACTH or cortisol basal levels. In contrast, U-50488H (0.01–1 mg/kg) dose-dependently stimulated ACTH and cortisol release in both male and female monkeys. Importantly, the stimulatory effects of U-50488H on the secretion of ACTH were blocked by a selective kappa opioid receptor antagonist, nor-Binaltorphimine. The antagonist effect of nor-binaltorphimine lasted up to 20 weeks. These results indicate that only synthetic kappa, but not mu or delta opioid receptor agonists stimulate HPA axis activity after acute administration in primates.
Keywords: HPA axis, Opioid receptor agonists, Opioid receptor antagonists, ACTH, Cortisol, Primates
1. Introduction
The analgesic effect produced by opioids has long been used to treat moderate to severe pain in humans (Peng and Sandler, 1999; Martin and Eisenach, 2001). The discovery of opioid receptor subtypes, mu, kappa, and delta opioid receptors, has fostered more pharmacological studies of each receptor subtype’s physiological functions. Whereas the administration of opioid receptor agonists suppress the body’s natural response to pain, it also affects other systems in the body, such as neuroendocrine function. The hypothalamic–pituitary–adrenal (HPA) axis is an example of a neuroendocrine system that is significantly influenced by opioids. The secretion of hypothalamic releasing factors and hormones from the pituitary and adrenal glands can be stimulated or inhibited depending on which opioid receptor subtype is activated (Pechnick, 1993; Mellon and Bayer, 1998).
There are numerous contradictions in the literature regarding the effects of opioids on the HPA axis. For example, human studies using mu opioid analgesics, such as morphine, fentanyl, and buprenorphine, have indicated an inhibitory role of mu opioid receptors in the modulation of HPA activity (McDonald et al., 1959; Zis et al., 1984; Pende et al., 1986; Hoehe et al., 1988). Conversely, mu opioid receptor agonists have been shown to increase plasma adrenocorticotropic hormone (ACTH) and corticosterone levels in rodents and other non-primate species (Iyengar et al., 1987; Nikolarakis et al., 1987; Ignar and Kuhn, 1990; Gonzalvez et al., 1991; Pechnick, 1993). These differences have led to different interpretations for the effects of opioid receptor antagonists and the expected roles of endogenous opioids in the regulation of HPA axis. It is clear that differences in the species used, as well as the receptor binding selectivity, doses, and administration routes of opioid compounds could contribute to different results. Nevertheless, the use of opioid analgesics as part of anesthesia may alter the HPA response to the surgery, and the HPA responses to other stressors may be altered in individuals who take opioids for the pain medication or who abuse opioids. Therefore, it is important to study the acute effects of opioid agonists selective for each receptor subtype on regulating the HPA activity in non-human primates.
Although most opioid analgesics used for the clinical management of pain are mu opioid receptor agonists, studies using monkeys have suggested that kappa and delta opioid receptor agonists have therapeutic potential as analgesics and for other indications (Dykstra et al., 1987; Butelman et al., 1993; Negus et al., 1998; Brandt et al., 2001). Despite their clinical potential, the interaction of kappa and delta opioid receptor agonists on the HPA axis has not been extensively studied in primates. There is one human study showing that a kappa opioid receptor agonist stimulated cortisol release in men (Ur et al., 1997). The effect of a kappa opioid receptor antagonist on this effect was not determined, so it is uncertain that the stimulation was via the kappa opioid receptors. We have reported that a kappa opioid receptor-selective antagonist, nor-binaltorphimine (nor-BNI), produced a transient increase in plasma ACTH and cortisol concentrations in monkeys (Williams et al., 2003), but the duration of nor-BNI-stimulated HPA activity (i.e., 3 h) was much shorter than its typical long duration of kappa opioid receptor antagonist action (i.e., 3–5 weeks) (Butelman et al., 1993; Ko et al., 1999). Prior studies suggest non-kappa opioid receptor selective actions of nor-BNI in the period immediately after administration (Endoh et al., 1992). Given that highly selective kappa opioid receptor agonists and antagonists are available and their active doses have been documented in various monkey behavioral assays (Butelman et al., 1993, 1998; Ko et al., 1999, 2003), it was possible to determine the role of kappa opioid receptor in modulating the HPA activity in monkeys.
In the previous study, we utilized indwelling IV catheters in monkeys to show reliable and low basal ACTH and cortisol levels before drug administration (Williams et al., 2003). This procedure provides the opportunity to detect stimulatory effects of test compounds in awake, behaving monkeys. The aim of this study was, therefore, to determine whether selective mu, kappa, and delta opioid receptor agonists increased plasma ACTH and cortisol levels in monkeys. Selective receptor antagonists were used to determine if the predicted stimulatory effects were selective for mu, kappa, or delta opioid receptors.
2. Materials and methods
2.1. Subjects
Eight adult male and female rhesus monkeys (Macaca mulatta), weighing 6.2–12.9 kg, were used in this experiment. These monkeys had intact gonads and adrenal glands. The monkeys were housed individually with free access to water and were fed approximately 25–30 biscuits (Purina Monkey Chow; Ralston Purina, St. Louis, MO) and fresh fruit daily. These monkeys had only received opioid receptor antagonists in our previous study (Williams et al., 2003). They were not involved in any drug self-administration studies and they had never received any opioid receptor agonist before the present study. The female monkeys had normal menstrual cycles, ranging between 28 and 32 days. All monkeys were housed in facilities accredited by the American Association for the Accreditation of Laboratory Animal Care. The studies were conducted in accordance with the University Committee on the Use and Care of Animals in the University of Michigan and the Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington DC, revised 1996).
2.2. Apparatus
The monkeys were housed individually in 83 × 76 × 91 cm3 deep stainless steel cages (Bryan Research Equipment Corporation, Bryan, TX). Initially, the monkeys were acclimated to wearing Teflon mesh jackets (Lomir, Malone, NY) that were attached to the backs of the cages by flexible, stainless steel tether arms. Once habituated to the cage and the restraint device (i.e., steel tether), the monkeys had indwelling silastic catheters inserted into their femoral vein during aseptic surgery using 10 mg/kg ketamine and 2 mg/kg xylazine anesthesia. Upon placement in the vein, the catheter was maneuvered subcutaneously to the midscapular region where it exited the monkey. The catheter was then threaded through and protected by the stainless steel tether and exited via the back of the cage. Such an apparatus enabled the monkey to remain conscious and to move with minimal restraint during blood collection. The setup and surgical procedures are described in detail elsewhere (Winger et al., 1975).
2.3. Drugs
Fentanyl (National Institute on Drug Abuse, Bethesda, MD), U-50488H (Upjohn Company, Kalamazoo, MI), and nor-BNI (Sigma-Aldrich, St. Louis, MO) were all dissolved in sterile water. SNC80 (Dr. K.C. Rice, National Institute on Drug Abuse, Bethesda, MD) was dissolved in 3% lactic acid in sterile water. All compounds were administered intravenously through the indwelling IV catheter at a volume of 0.1 mL/kg.
2.4. Experimental protocols
The first part of the study was to characterize the effects of opioid receptor agonists on the HPA axis. Four male and four female monkeys (n = 8) were used in these experiments. A selective mu opioid receptor agonist, fentanyl (0.0003–0.02 mg/kg), a kappa opioid receptor agonist, U-50488H (0.01–1 mg/kg), and a delta opioid receptor agonist, SNC80 (0.03–0.3 mg/kg) at different doses were given in a random order. Each single dosing procedure was conducted once per week. These doses of opioid receptor agonists were chosen based on previous studies showing that they were active doses in various behavioral assays in monkeys (Butelman et al., 1993, 1998; Negus et al., 1998; Ko et al., 1998, 2002). The second part of the study was to determine the receptor mechanism of U-50488H-evoked ACTH and cortisol release. In this antagonist experiment, one group of four monkeys (two males and two females) received IV the selective kappa opioid receptor antagonist nor-BNI 3.2 mg/kg and the other group of four monkeys (two males and two females) received IV vehicle (sterile water, 0.1 mL/kg). The stimulatory effect of U-50488H (0.3 mg/kg) on ACTH and cortisol level was determined 7 days before, and re-determined at 1 and 3 days, and 1, 3, 6, 9, 12, 16, 20, 24, and 30 weeks after the pretreatment. The dose of U-50488H was chosen because it was the smallest dose that produced maximal effects in the first part of the study. The dose of nor-BNI was chosen based on a previous study showing that nor-BNI produced a selective, long-lasting kappa opioid receptor antagonist effect at this dose (Butelman et al., 1993).
2.5. Blood sample collection and measurement
Every blood-sampling session began at 09:30 AM (i.e., the time point −30 in the figures) and the agonist or vehicle injections were given at 10:00 AM (time 0). The sessions ended at 13:00 PM for a total session time of 3.5 h. Every 15 min for the first 1.5 h and every 30 min thereafter, approximately 2 ml of blood were drawn. This regimen totaled 11 blood samples per session. Each blood sample was initially placed in a 2-ml vacutainer (Beckton Dickinson Co., Franklin Lakes, NJ) containing 0.04 ml of 7.5% EDTA and immediately placed on ice. The catheter was flushed with approximately 3 ml heparinized saline (30 U/ml) after collecting each sample. The same catheter was used for IV drug delivery and was also flushed with 3 ml heparinized saline after drug administration.
Blood samples were centrifuged at 4500 rpm for 5 min at 4 °C. The plasma was pipetted into 2-ml Cryovials (Corning Costar Co., Cambridge, MA) and stored at −80 °C until assayed for ACTH and cortisol. There was no analysis for drug blood levels. The ACTH and cortisol concentrations were measured using commercially available radioimmunoassay kits (ACTH: Nichols Institute Diagnostics, San Juan Capistrano, CA; cortisol: Diagnostic Products Co., Los Angeles, CA). The range of detection for the ACTH kit was 1–1500 pg/mL. The intra-assay and inter-assay coefficients of variation were 3–3.2% and 6–8%, respectively. The range of detection for the cortisol kit was 0.2–50 μg/dL. The intra-assay and inter-assay coefficients of variation were 3–5% and 4–6.5%, respectively.
2.6. Data analysis
The data are displayed as the time course and the area under curve (AUC). The time course data consist of the mean and SEM of actual hormone concentrations at various time points throughout the session. All data were analyzed by two-way repeated measures of ANOVA followed by the Dunnett test for multiple (post hoc) comparisons. As noted, we did not find a significant difference in the effects of agonists between male and female monkeys, so mean values for all monkeys in the same condition were used for data analysis. In addition, the trapezoidal rule was used to calculate the AUC. The reference for the calculation of the AUC for ACTH (pg min/mL) and cortisol (μg min/dL) was the second sample (−15 min time point). The AUC was then calculated for each subject and averaged across all subjects. The AUC data were analyzed by using one-way repeated measures of ANOVA followed by the Dunnett test for multiple comparisons. The significance criterion for all data was set at *p<0.05, and **p<0.01.
3. Results
Figure 1 shows the time course of ACTH release in the plasma of monkeys receiving opioid receptor agonists. Neither the mu opioid receptor agonist fentanyl [F(5,35) = 1.1, p = 0.4] nor the delta opioid receptor agonist SNC80 [F(3,21) = 1.6, p = 0.2] stimulated ACTH release. As noted, a higher dose of fentanyl was not evaluated due to the risk of respiratory arrest. A higher dose of SNC80 (i.e., 1 mg/kg) produced convulsions in the first two subjects; therefore, the effect of SNC80 was only tested up to 0.3 mg/kg. The duration of SNC80-induced convulsions was not assessed because diazepam was used immediately to treat convulsing subjects. In contrast, U-50488H dose-dependently stimulated ACTH release [F(5,35) = 31.4, p<0.01]. Post hoc comparisons indicated that both 0.3 and 1 mg/kg of U-50488H significantly increased the release of ACTH between 15 and 180 min following IV administration (p<0.01). In addition, Figure 2 illustrates the ACTH release converted to AUC across different agonists and doses in both male and female monkeys. Neither fentanyl [F(5,42) = 1.4, p = 0.3] nor SNC80 [F(3,28) = 0.8, p = 0.5] increased ACTH-AUC at any dose tested. On the other hand, U-50488H dose-dependently increased the values of ACTH-AUC [F(5,42) = 22.7, p<0.01]. Multiple comparisons indicated that U-50488H at doses between 0.3 and 1 mg/kg significantly elevated the ACTH-AUC compared with the vehicle condition (p<0.01). There was no gender difference for the effect of each drug tested herein (fentanyl: F(1,3) = 0.02, p = 0.9; U-50488-H: F(1,3) = 0.14, p = 0.7; SNC80: F(1,3) = 6.0, p = 0.1).
Figure 1.
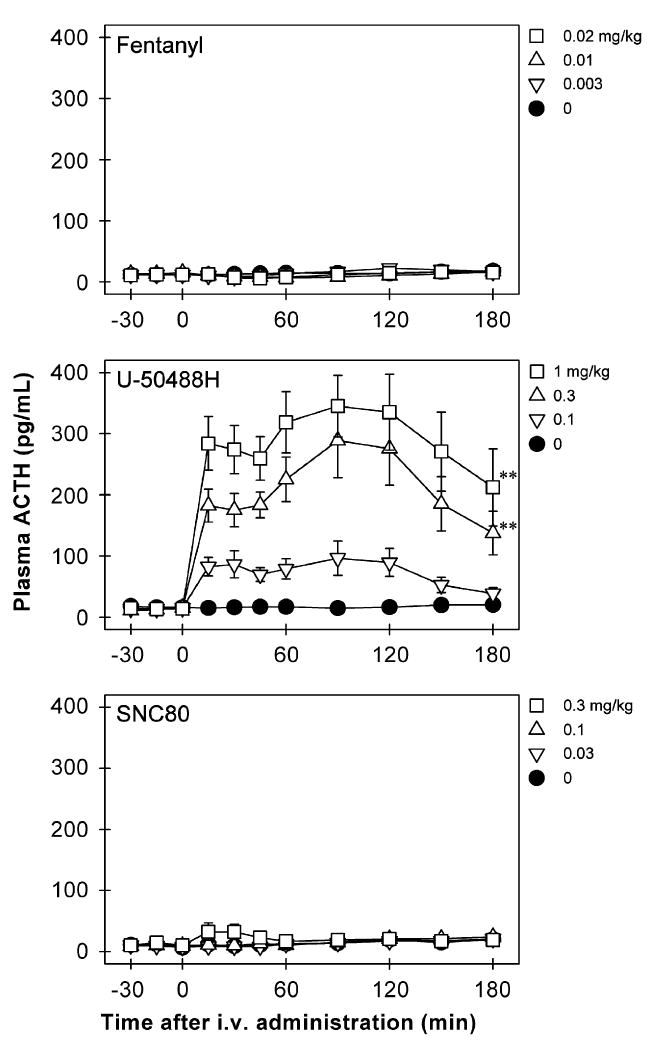
Time course of ACTH (pg/mL) responses to IV administration of opioid receptor agonists, fentanyl, U-50488H, and SNC80. The drug or vehicle solution was given at time 0. Each value represents mean ±SEM (n = 8). The asterisks represent a significant difference from the vehicle condition between time points 15 and 180 min (**p<0.01). The data of lower doses of fentanyl and U-50488H are not shown for sake of clarity. See Figure 2 for other details.
Figure 2.
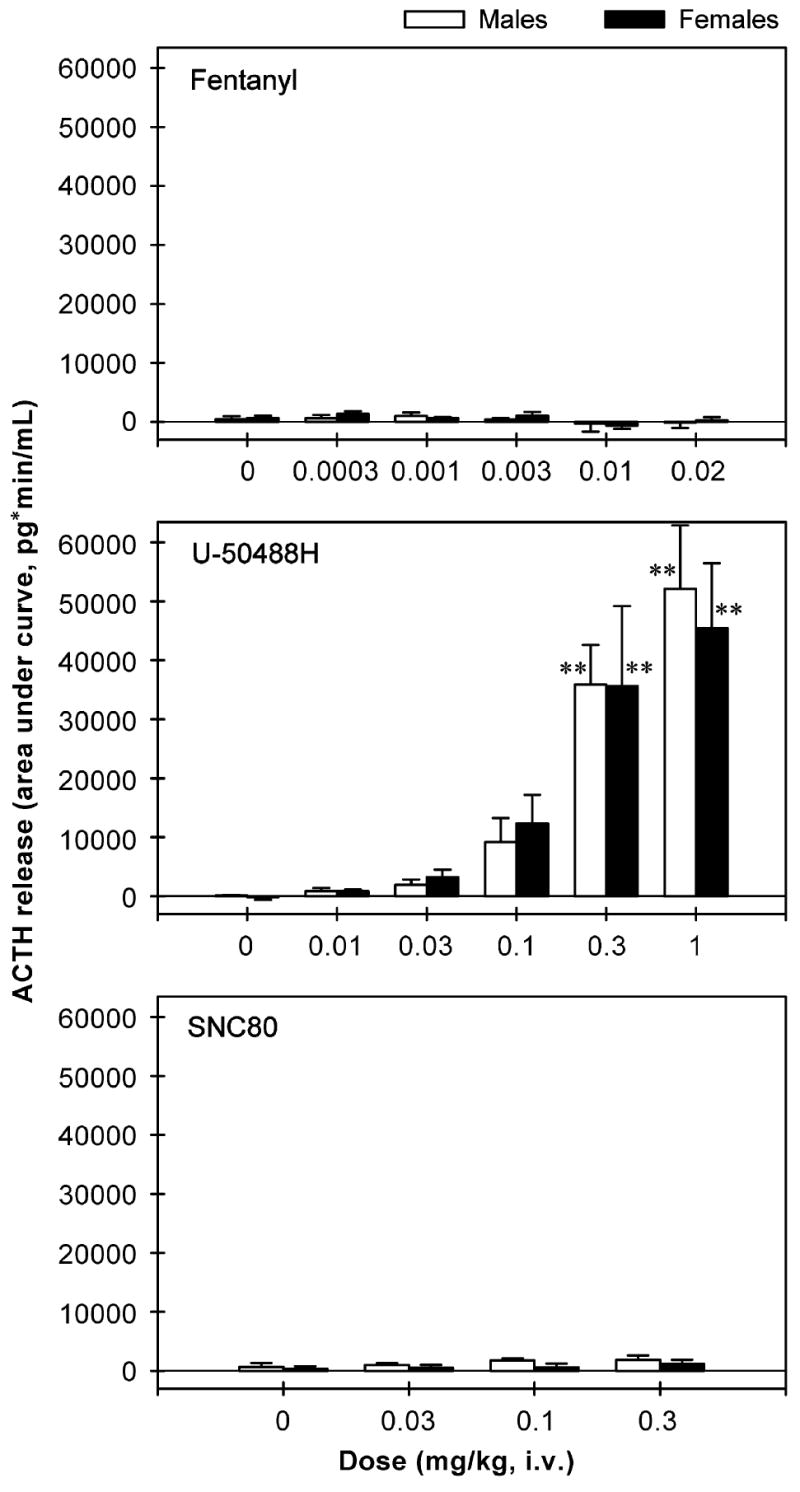
The area under curve (AUC) of ACTH release throughout the entire 3-h session following IV administration of opioid receptor agonists in male and female monkeys. Each AUC value represents mean ±SEM (n = 4). The asterisk represents a significant difference from the vehicle condition (**p<0.01). See Figure 1 for other details.
Figure 3 shows the time course of cortisol release in the plasma of monkeys receiving the opioid receptor agonists. Neither fentanyl [F(5,35) = 0.9, p = 0.5] nor SNC80 [F(3,21) = 0.9, p = 0.4] stimulated cortisol release. In contrast, U-50488H dose-dependently stimulated cortisol release [F(5,35) = 27.3, p<0.01]. Post hoc comparisons indicated that U-50488H at doses between 0.03 and 1 mg/kg significantly increased the release of cortisol (p<0.01). In particular, 0.03 mg/kg of U-50488H increased the release of cortisol between 30 and 90 min following IV administration. All higher doses of U-50488H (i.e., 0.1–1 mg/kg) increased cortisol release between 30 and 180 min following IV administration. In addition, Figure 4 illustrates the AUC of cortisol release across different agonists and doses in both male and female monkeys. Again, neither fentanyl [F(5,42) = 0.8, p = 0.5] nor SNC80 [F(3,28) = 0.8, p = 0.5] increased cortisol-AUC at any dose tested. However, U-50488H dose-dependently increased the values of cortisol-AUC [F(5,42) = 24.3, p<0.01]. Multiple comparisons indicated that U-50488H at doses between 0.1 and 1 mg/kg significantly elevated the cortisol-AUC compared with the vehicle condition (p<0.05). There was no gender difference for the effect of each drug tested herein (fentanyl: F(1,3) = 0.4, p = 0.6; U-50488-H: F(1,3) = 0.1, p = 0.8; SNC80: F(1,3) = 0.9, p = 0.4).
Figure 3.
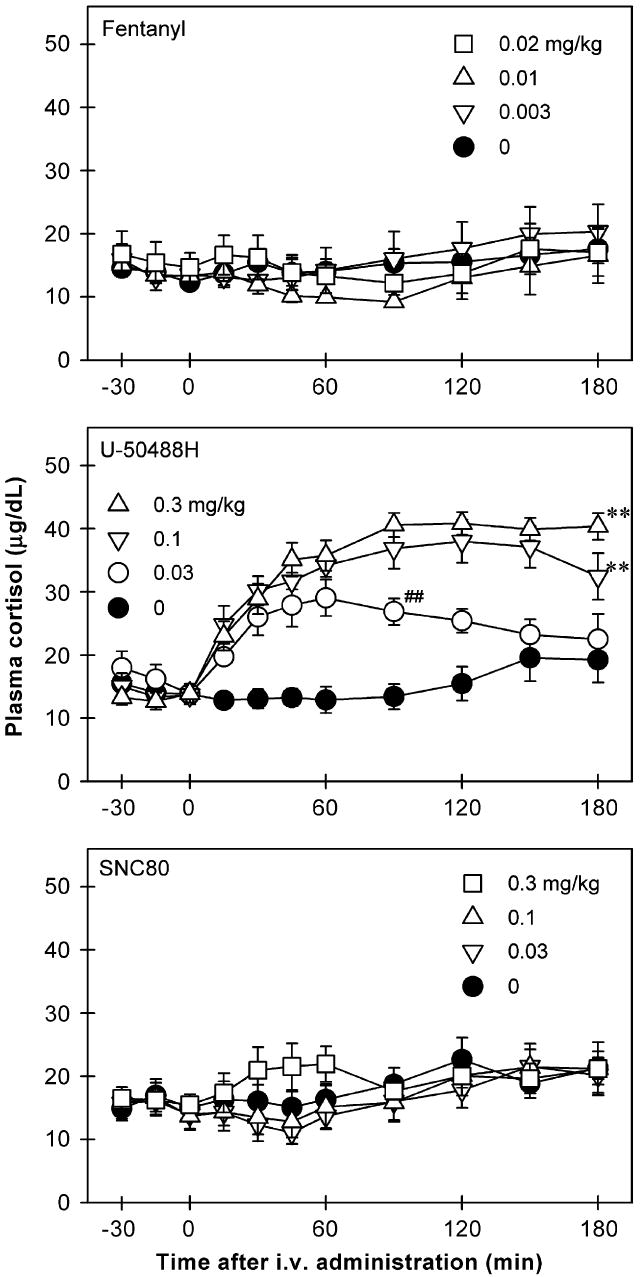
Time course of cortisol (μg/dL) responses to IV administration of opioid receptor agonists, fentanyl, U-50488H, and SNC80. The drug or vehicle solution was given at time 0. Each value represents mean ±SEM (n = 8). The asterisks represent a significant difference from the vehicle condition between time points 30 and 180 min (**p<0.01). The symbol (##) indicates a significant difference from the vehicle condition between time points 30 and 90 min (##p<0.01). The data of other doses of fentanyl and U-50488H are not shown for sake of clarity. See Figure 4 for other details.
Figure 4.
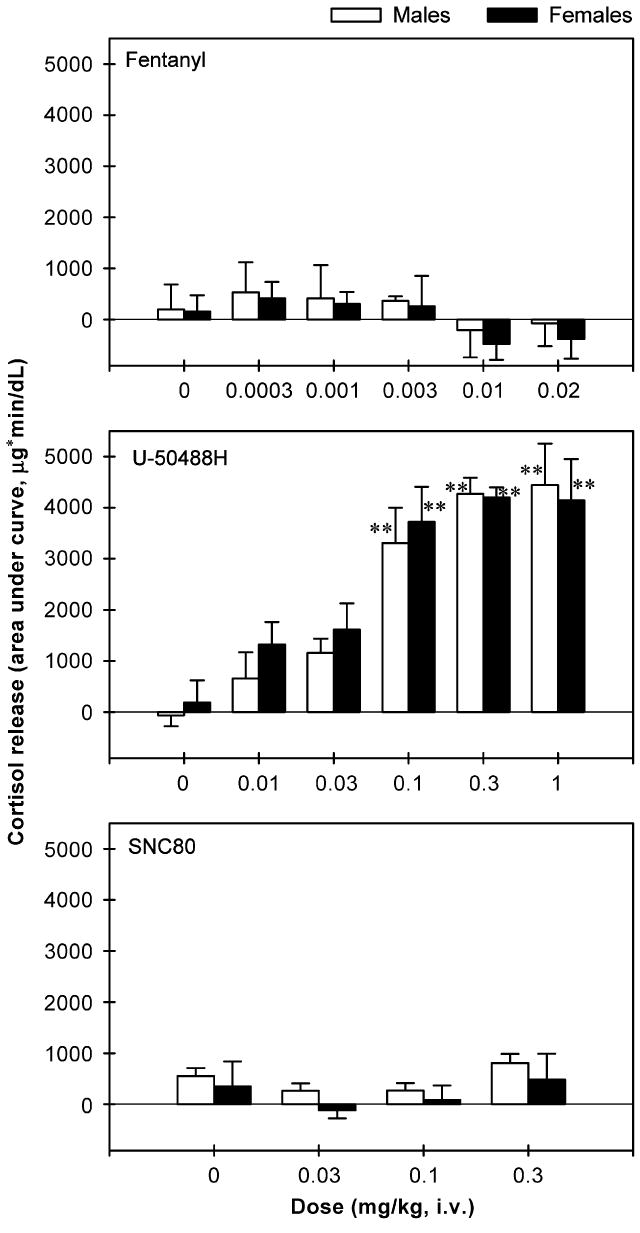
The area under the curve (AUC) of cortisol release throughout the entire 3-h session following IV administration of opioid receptor agonists in male and female monkeys. Each AUC value represents the mean ±SEM (n = 4). The asterisk represents a significant difference from vehicle condition (**p<0.01). See Figure 3 for other details.
Figure 5 shows the time course of the antagonist effect of nor-BNI on U-50488H-stimulated ACTH and cortisol release. The vehicle pretreatment did not change U-50488H-induced ACTH- [F(11,36) = 0.1, p = 0.9] and cortisol-AUC [F(11,36) = 0.5, p = 0.9]. In other words, repeated administration of U-50488H produced similar degrees of ACTH and cortisol release in the vehicle-treated group under this dosing regimen. In contrast, pretreatment with nor-BNI significantly blocked U-50488H-induced ACTH [F(11,36) = 3.5, p<0.01] and cortisol [F(11,36) = 2.3, p<0.05] release (top and bottom panels). Post hoc comparisons indicated that nor-BNI blocked U-50488H-stimulated ACTH-AUC from 3 days to 20 weeks after administration (p<0.01). Nevertheless, nor-BNI only blocked U-50488H-stimulated cortisol-AUC from 3 to 7 days after administration (p<0.05).
Figure 5.
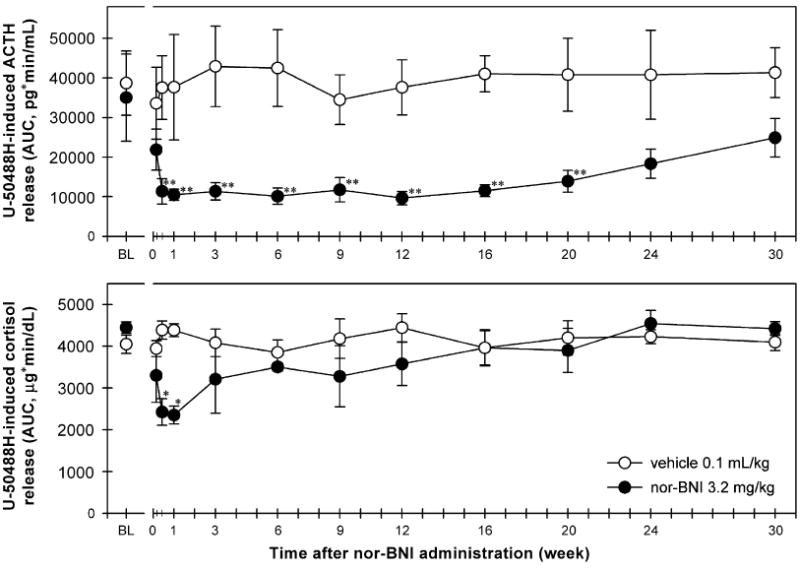
Time course of the antagonist effect of nor-BNI on U-50488H-stimulated ACTH and cortisol release. Each value represents mean ±SEM (n = 4). Top and bottom panels compare the effects of vehicle and nor-BNI pretreatment on the stimulatory effect of U-50488H on ACTH and cortisol levels. The vehicle and nor-BNI groups received IV sterile water 0.1 mL/kg and IV nor-BNI 3.2 mg/kg on day 0, respectively. BL (baseline) represents the effects of IV U-50488H 0.3 mg/kg (i.e., the AUC of ACTH or cortisol release throughout the 3-h session) before each group of monkeys received either vehicle or nor-BNI pretreatment. The effect of U-50488H on the ACTH and cortisol release/AUC was re-determined at different time points as indicated on the abscissas following either vehicle or nor-BNI administration. The asterisk represents a significant difference from each group’s BL condition (*p<0.05; **p<0.01). See Section 2 and Figures 2 and 4 for other details.
4. Discussion
The present study showed that a kappa opioid receptor-selective agonist, U-50488H, dose-dependently increased plasma ACTH and cortisol levels in both male and female monkeys. These results agree with an earlier study, reporting kappa opioid receptor agonist-stimulated cortisol secretion in humans (Ur et al., 1997). It is worth noting that the active doses of U-50488H (0.03–0.3 mg/kg) for both stimulating the HPA activity and inhibiting vasopressin release (i.e., diuresis) are similar, but they are smaller than those (0.3–3 mg/kg) for producing antinociception and sedation in monkeys (Dykstra et al., 1987; Butelman et al., 1993; Ko et al., 1999, 2003). This potency difference indicates that kappa opioid receptor agonists may have therapeutic potential as hormonal regulators, especially for treating patients with underproduction of ACTH and cortisol.
Pretreatment with a kappa opioid receptor-selective antagonist, nor-BNI 3.2 mg/kg, blocked U-50488H-stimulated ACTH and cortisol secretions. There was no development of tolerance to the effects of U-50488H in the vehicle-treated group, indicating that both factors of time and repeated dosing regimen did not affect the measurement of the effects of U-50488H’s in the nor-BNI-treated group under these experimental conditions. These results clearly demonstrate that the kappa opioid receptor mediates the stimulatory effect of U-50488H on the HPA axis in primates. Although the study was not designed to examine the gender difference in the effects of opioid receptor agonists on the HPA activity, future studies with more subjects are needed to determine whether female monkeys have different HPA responses to opioid receptor agonists between their follicular and luteal phases of menstrual cycles.
The duration of nor-BNI-induced blockade of U-50488H-stimulated ACTH versus cortisol release was different (i.e., 20 weeks versus 1 week). There may be several factors contributing to this time course difference. For example, it is likely that there are substantially fewer kappa opioid receptor binding sites in anterior pituitary than in hypothalamus (Simantov and Snyder, 1977; Sim-Selley et al., 1999; Peckys and Landwehrmeyer, 1999). The high density of kappa opioid receptors in the hypothalamus may play an important role in eliciting U-50488H’s stimulatory effect on HPA activity and may be more susceptible to the kappa opioid receptor antagonist effect of nor-BNI. In addition, ACTH acts on the adrenal cortex to release cortisol into blood (Pechnick, 1993; Wand and Schumann, 1998; Miller and O’Callaghan, 2002). Repeated exposures to U-50488H in the presence of nor-BNI may gradually increase the efficiency of ACTH in stimulating cortisol release.
The long duration (i.e., for weeks) of the nor-BNI-induced kappa opioid receptor antagonist effect is well known in the literature (Horan et al., 1992; Butelman et al., 1993; Jewett and Woods, 1995). Nevertheless, this study presents the longest duration (20 weeks) of kappa opioid receptor antagonism in vivo following systemic administration of nor-BNI. As reported previously, nor-BNI itself produced a transient increased secretion of ACTH and cortisol for 2–3 h (Williams et al., 2003). It is possible that nor-BNI displayed different pharmacological actions through kappa opioid receptors by first showing acute partial kappa opioid receptor agonist effects following by long-term kappa opioid receptor antagonist effects. There are other opioid compounds that also display changing pharmacological actions over time. For example, buprenorphine produced acute analgesia (i.e., mu opioid receptor agonist action) followed by long-term mu opioid receptor antagonist effects (Cowan et al., 1977; Walker et al., 1995). Nevertheless, additional study will be needed to verify the role of kappa opioid receptors in the early action of nor-BNI on hormonal regulation. It will be interesting to see whether pretreatment with nor-BNI blocks subsequent administration (i.e., second administration) of nor-BNI-stimulated ACTH and cortisol release.
In a previous study, intracisternal administration of nor-BNI 0.32 mg significantly blocked U-50488H-induced diuresis for 20 weeks in monkeys; this dose of nor-BNI was not an effective antagonist following systemic administration (Ko et al., 2003). These results demonstrated that central kappa opioid receptors in the hypothalamus mediate kappa opioid receptor agonist-induced diuresis in primates. Together with the present findings, these results indicate that nor-BNI produces a long-lasting kappa opioid receptor antagonist effect against kappa opioid receptor agonist-induced hormonal regulation. Although the present study did not investigate the site of action of U-50488H on stimulating ACTH release, other studies have suggested that kappa opioid receptor agonists acts to stimulate the secretion of corticotrophin-releasing factor (CRF) from the hypothalamus (Buckingham and Cooper, 1986; Nikolarakis et al., 1987). Given that the kappa opioid receptor is abundant in the primate hypothalamus (Sim-Selley et al., 1999; Peckys and Landwehrmeyer, 1999), it is important to determine whether CRF blockers can modulate kappa opioid receptor agonist-stimulated ACTH release in primates.
A mu opioid receptor-selective agonist, fentanyl, did not stimulate ACTH and cortisol release in monkeys. The doses of fentanyl used here have been shown to produce antinociceptive and reinforcing effects in monkeys (Ko et al., 1998, 2002; Butelman et al., 2004). This finding differs with the reports of a stimulatory effect of mu opioid receptor agonists on the rodent HPA axis (Iyengar et al., 1987; Nikolarakis et al., 1987; Ignar and Kuhn, 1990; Gonzalvez et al., 1991), but it agrees with the finding of inhibition or no effect of mu opioid receptor agonists on human HPA activity (McDonald et al., 1959; Zis et al., 1984; Hoehe et al., 1988; Pechnick, 1993). On the other hand, opioid receptor antagonists stimulate the HPA axis by increasing plasma levels of ACTH and cortisol in humans, indicating the HPA axis may be tonically inhibited by endogenous opioids (Volavka et al., 1979; Morley, 1983; Jackson et al., 1995). The magnitude of ACTH response to naloxone in mu opioid-dependent subjects exceeds the magnitude of the response in nondependent, healthy subjects given higher doses of naloxone, suggesting the HPA axis in opioid-dependent subjects may also reflect neuroadaptation to the basal signal activity of mu opioid receptors (Volavka et al., 1979; Naber et al., 1981; Rosen et al., 1996).
A delta opioid receptor-selective agonist, SNC80, also did not stimulate ACTH and cortisol secretion in monkeys. The doses of SNC80 used here have been shown to produce antinociceptive and other behavioral effects in monkeys (Negus et al., 1998; Brandt et al., 2001). This finding differs with the stimulatory effects of DOR agonists on the rodent HPA axis (Iyengar et al., 1987; Gonzalvez et al., 1991). Interestingly, the single human study reported that a delta opioid receptor agonist, deltorphin, inhibited ACTH and cortisol responses to insulin-induced hypoglycemia, while it had no effect on ACTH and cortisol responses to CRF (Degli Uberti et al., 1992). Given that anatomical data indicate a sparse density of delta opioid receptor mRNA in the human hypothalamus (Peckys and Landwehrmeyer, 1999), studies using elevated basal levels of the HPA axis to compare the inhibitory effects of mu and delta opioid receptor agonists will help clarify the role of delta opioid receptors in the primate HPA axis.
It is important to note that both mu and kappa opioid receptors are abundant in the primate hypothalamus (Sim-Selley et al., 1999; Peckys and Landwehrmeyer, 1999). Studies using selective mu and kappa opioid receptor agonists have provided a pharmacological basis of endogenous opioids action in the modulation of the primate HPA axis. Given mu opioid receptor’s inhibitory and kappa opioid receptor’s stimulatory roles on the HPA activity, it is expected that when endogenous opioids are released, only dynorphin-like ligands, but not endorphin- or enkephalin-like ligands, may stimulate the HPA axis. Although this study focuses on the stimulatory effects of opioid receptor agonists, future studies of mixed opioids with high affinity for both mu and kappa opioid receptors such as butorphanol (Lee et al., 2007) will facilitate our understanding of whether mu and kappa opioid receptors interact in the primate HPA axis. In addition, the subjective effects such as euphoria and dysphoria may not correlate with the HPA activity. Studies have indicated that drugs with abuse liability in humans have different effects on the HPA activity. For example, cocaine stimulated ACTH and cortisol secretion, but fentanyl inhibited ACTH and cortisol secretion (Broadbear et al., 1999, 2004). Ur et al. (1997) also reported that mild dysphoric effects were noted in some subjects following administration of the kappa opioid receptor agonist, spiradoline, but these did not correlate with the rise of cortisol.
Taken together, this study provides new and important information on the primate HPA axis. It demonstrates that acute administration of synthetic kappa, but not mu or delta opioid receptor agonists stimulates the monkey HPA axis. Both mu and delta opioid receptor agonists may be used as analgesics without stimulating HPA activity. Studies of exogenous compounds selective for each opioid receptor subtype facilitate our understanding of the roles of endogenous opioids in the primate HPA activity. Kappa opioid receptor agonists and antagonists may have some utility in the treatment of neuroendocrine disorders (i.e., underproduction or overproduction of ACTH). Given the corresponding anatomical evidence, mu and kappa opioid receptor agonists can be used to test the functional integrity of the primate HPA axis. The study supports the use of the non-human primate model for exploring and predicting effects of opioids on hormones secreted by the hypothalamus and the pituitary in humans (Bowen et al., 2002; Butelman et al., 1999, 2007).
Acknowledgments
This study was supported by US Public Health Service Grants DA-000254 and DA-013685. We thank Amy J. Ensley and Ryan Sherriff for technical assistance. A portion of this work was supported by the Intramural Research Programs of the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism.
Role of the funding source Funding for this study was provided by NIH Grants DA-000254 and DA-013685; the NIDA/NIH had no further role in the study design, in the collection, analysis and interpretation of data, in the writing of the report, and in the decision to submit the paper for publication.
Footnotes
Conflict of interest All authors declare that no conflict of interest, financial, or otherwise, exists with regard to this research.
References
- Bowen CA, Negus SS, Kelly M, Mello NK. The effects of heroin on prolactin levels in male rhesus monkeys: use of cumulative-dosing procedures. Psychoneuroendocrinology. 2002;27:319–336. doi: 10.1016/s0306-4530(01)00053-1. [DOI] [PubMed] [Google Scholar]
- Brandt MR, Furness MS, Mello NK, Rice KC, Negus SS. Antinociceptive effects of δ-opioid agonists in rhesus monkeys: effects on chemically induced thermal hypersensitivity. J Pharmacol Exp Ther. 2001;296:939–946. [PubMed] [Google Scholar]
- Broadbear JH, Winger G, Cicero TJ, Woods JH. Effects of self-administered cocaine on plasma adrenocorticotropic hormone and cortisol in male rhesus monkeys. J Pharmacol Exp Ther. 1999;289:1641–1647. [PubMed] [Google Scholar]
- Broadbear JH, Winger G, Woods JH. Self-administration of fentanyl, cocaine, and ketamine: effects on the pituitary–adrenal axis in rhesus monkeys. Psychopharmacology. 2004;176:398–406. doi: 10.1007/s00213-004-1891-x. [DOI] [PubMed] [Google Scholar]
- Buckingham JC, Cooper TA. Pharmacological characterization of opioid receptors influencing the secretion of corticotrophin releasing factor in the rat. Neuroendocrinology. 1986;44:36–40. doi: 10.1159/000124618. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Negus SS, Ai Y, de Costa BR, Woods JH. Kappa opioid antagonist effects of systemically administered nor-binaltorphimine in a thermal antinociception assay in rhesus monkeys. J Pharmacol Exp Ther. 1993;267:1269–1276. [PubMed] [Google Scholar]
- Butelman ER, Ko MC, Sobczyk-Kojiro K, Mosberg HI, Van Bemmel B, Zernig G, Woods JH. Kappa-Opioid receptor binding populations in rhesus monkey brain: relationship to an assay of thermal antinociception. J Pharmacol Exp Ther. 1998;285:595–601. [PubMed] [Google Scholar]
- Butelman ER, Harris TJ, Perez A, Kreek MJ. Effects of systemically administered dynorphin A(1–17) in rhesus monkeys. J Pharmacol Exp Ther. 1999;290:678–686. [PubMed] [Google Scholar]
- Butelman ER, Harris TJ, Kreek MJ. Antiallodynnic effects of loperamide and fentanyl against topical capsaicin-induced allodynia in unanesthetized primates. J Pharmacol Exp Ther. 2004;311:155–163. doi: 10.1124/jpet.104.068411. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Mandau M, Tidgewell K, Prisinzano TE, Yuferov V, Kreek MJ. Effects of salvinorin A, a kappa-opioid hallucinogen, on a neuroendocrine biomarker assay in nonhuman primates with high kappa-receptor homology to humans. J Pharmacol Exp Ther. 2007;320:300–306. doi: 10.1124/jpet.106.112417. [DOI] [PubMed] [Google Scholar]
- Cowan A, Lewis JW, MacFarlane I. Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br J Pharmacol. 1977;60:537–545. doi: 10.1111/j.1476-5381.1977.tb07532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli Uberti EC, Salvadori S, Trasforini G, Margutti A, Ambrosio MR, Rossi R, Portaluppi F, Pansini R. Effect of deltorphin on pituitary–adrenal response to insulin-induced hypoglycemia and ovine corticotrophin-releasing hormone in healthy man. J Clin Endocrinol Metab. 1992;75:370–374. doi: 10.1210/jcem.75.2.1322422. [DOI] [PubMed] [Google Scholar]
- Dykstra LA, Gmerek DE, Winger G, Woods JH. Kappa opioids in rhesus monkeys. I. Diuresis, sedation, analgesia and discriminative stimulus effects. J Pharmacol Exp Ther. 1987;242:413–420. [PubMed] [Google Scholar]
- Endoh T, Matsuura H, Tanaka C, Nagase H. Nor-binaltorphimine: a potent and selective κ-opioid receptor antagonist with long-lasting activity in vivo. Arch Int Pharmacodyn. 1992;316:30–42. [PubMed] [Google Scholar]
- Gonzalvez ML, Milaneés MV, Vargas ML. Effects of acute and chronic administration of μ- and δ-opioid agonists on the hypothalamic–pituitary–adrenocortical (HPA) axis in the rat. Eur J Pharmacol. 1991;200:155–158. doi: 10.1016/0014-2999(91)90678-j. [DOI] [PubMed] [Google Scholar]
- Hoehe M, Duka T, Doenicke A. Human studies on the μ opidate receptor agonist fentanyl: neuroendocrine and behavioral responses. Psychoneuroendocrinology. 1988;13:397–408. doi: 10.1016/0306-4530(88)90046-7. [DOI] [PubMed] [Google Scholar]
- Horan P, Taylor J, Yamamura HI, Porreca F. Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther. 1992;260:1237–1243. [PubMed] [Google Scholar]
- Ignar DM, Kuhn CM. Effects of specific mu and kappa opiate tolerance and abstinence on hypothalamo–pituitary–adrenal axis secretion in the rat. J Pharmacol Exp Ther. 1990;255:1287–1295. [PubMed] [Google Scholar]
- Iyengar S, Kim HS, Wood PL. Mu-, delta-, kappa- and epsilon-opioid receptor modulation of the hypothalamic–pituitary–adrenocortical (HPA) axis: subchronic tolerance studies of endogenous opioid peptides. Brain Res. 1987;435:220–226. doi: 10.1016/0006-8993(87)91604-0. [DOI] [PubMed] [Google Scholar]
- Jackson RV, Grice JE, Hockings GI, Torpy DJ. Naloxone-induced ACTH release: mechanism of action in humans. Clin Endocrinol. 1995;43:423–424. doi: 10.1111/j.1365-2265.1995.tb02612.x. [DOI] [PubMed] [Google Scholar]
- Jewett DC, Woods JH. Nor-binaltorphimine: an ultra-long acting kappa-opioid antagonist in pigeons. Behav Pharmacol. 1995;6:815–820. [PubMed] [Google Scholar]
- Ko MC, Butelman ER, Woods JH. The role of peripheral Mu opioid receptors in the modulation of capsaicin-induced thermal nociception in rhesus monkeys. J Pharmacol Exp Ther. 1998;286:150–156. [PMC free article] [PubMed] [Google Scholar]
- Ko MCH, Johnson MD, Butelman ER, Willmont KJ, Mosberg HI, Woods JH. Intracisternal nor-binaltorphimine distinguishes central and peripheral κ-opioid antinociception in rhesus monkeys. J Pharmacol Exp Ther. 1999;291:1113–1120. [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Terner J, Hursh S, Woods JH, Winger G. Relative reinforcing effects of three opioids with different durations of action. J Pharmacol Exp Ther. 2002;301:698–704. doi: 10.1124/jpet.301.2.698. [DOI] [PubMed] [Google Scholar]
- Ko MCH, Willmont KJ, Lee H, Flory GS, Woods JH. Ultra-long antagonism of kappa opioid agonist-induced diuresis by intracisternal nor-binaltorphimine in monkeys. Brain Res. 2003;982:38–44. doi: 10.1016/s0006-8993(03)02938-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Naughton NN, Woods JH, Ko MC. Effects of butorphanol on morphine-induced itch and analgesia in primates. Anesthesiology. 2007;107:478–485. doi: 10.1097/01.anes.0000278876.20263.a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TJ, Eisenach JC. Pharmacology of opioid and nonopioid analgesics in chronic pain states. J Pharmacol Exp Ther. 2001;299:811–817. [PubMed] [Google Scholar]
- McDonald RK, Evans FT, Weise VK, Patrick RW. Effect of morphine and nalorphine on plasma hydrocortisone levels in man. J Pharmacol Exp Ther. 1959;125:241–247. [PubMed] [Google Scholar]
- Mellon RD, Bayer BM. Evidence for central opioid receptors in the immunomodulatory effects of morphine: review of potential mechanism(s) of action. J Neuroimmunol. 1998;83:19–28. doi: 10.1016/s0165-5728(97)00217-8. [DOI] [PubMed] [Google Scholar]
- Miller DB, O’Callaghan JP. Neuroendocrine aspects of the response to stress. Metabolism. 2002;51:5–10. doi: 10.1053/meta.2002.33184. [DOI] [PubMed] [Google Scholar]
- Morley J. Neuroendocrine effects of endogenous opioid peptides in human subjects: a review. Psychoneuroendocrinology. 1983;8:361–379. doi: 10.1016/0306-4530(83)90016-1. [DOI] [PubMed] [Google Scholar]
- Naber D, Pickar D, Davis GC, Cohen RM, Jimerson DC, Elchisak MA, Defraites EG, Kalin NH, Risch SC, Buchsbaum MS. Naloxone effects on beta-endorphin, cortisol, prolactin, growth hormone, HVA, MHPG in plasma of normal volunteers. Psychopharmacology. 1981;74:125–128. doi: 10.1007/BF00432677. [DOI] [PubMed] [Google Scholar]
- Negus SS, Gatch MB, Mello NK, Zhang X, Rice K. Behavioral effects of the delta-selective opioid agonist SNC80 and related compounds in rhesus monkeys. J Pharmacol Exp Ther. 1998;286:362–375. [PubMed] [Google Scholar]
- Nikolarakis K, Pfeiffer A, Stalla GK, Herz A. The role of CRF in the release of ACTH by opiate agonists and antagonists in rats. Brain Res. 1987;421:373–376. doi: 10.1016/0006-8993(87)91311-4. [DOI] [PubMed] [Google Scholar]
- Pechnick RN. Effects of opioids on the hypothalamo–pituitary–adrenal axis. Annu Rev Pharmacol Toxicol. 1993;32:353–382. doi: 10.1146/annurev.pa.33.040193.002033. [DOI] [PubMed] [Google Scholar]
- Peckys D, Landwehrmeyer GB. Expression of mu, kappa, and delta opioid receptor messenger RNA in the human CNS: a 33P in situ hybridization study. Neuroscience. 1999;88:1093–1135. doi: 10.1016/s0306-4522(98)00251-6. [DOI] [PubMed] [Google Scholar]
- Pende A, Musso NR, Montaldi ML, Pastorino G, Arzese M, Devilla L. Evaluation of the effects induced by four opiate drugs, with different affinities to opioid receptor subtypes, on anterior pituitary LH, TSH, PRL and GH secretion and on cortisol secretion in normal men. Biomed Pharmacother. 1986;40:178–182. [PubMed] [Google Scholar]
- Peng PW, Sandler AN. A review of the use of fentanyl analgesia in the management of acute pain in adults. Anesthesiology. 1999;90:576–599. doi: 10.1097/00000542-199902000-00034. [DOI] [PubMed] [Google Scholar]
- Rosen MI, McMahon TJ, Hameedi FA, Pearsall HR, Woods SW, Kreek MJ, Kosten TR. Effect of clonidine pretreatment on naloxone-precipitated opiate withdrawal. J Pharmacol Exp Ther. 1996;276:1128–1135. [PubMed] [Google Scholar]
- Simantov R, Snyder SH. Opiate receptor binding in the pituitary gland. Brain Res. 1977;124:178–184. doi: 10.1016/0006-8993(77)90877-0. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ, Daunais JB, Porrino LJ, Childers SR. Mu and kappa-1 opioid-stimulated [35S]Guanylyl-5′-O-(γ-Thio)-triphosphate binding in cynomolgus monkey brain. Neuroscience. 1999;94:651–662. doi: 10.1016/s0306-4522(99)00344-9. [DOI] [PubMed] [Google Scholar]
- Ur E, Wright DM, Bouloux PMG, Grossman A. The effects of spiradoline (U-62066E), a κ-opioid receptor agonist, on neuroendocrine function in man. Br J Pharmacol. 1997;120:781–784. doi: 10.1038/sj.bjp.0700971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volavka J, Cho D, Mallya A. Naloxone increases ACTH and cortisol levels in man. N Engl J Med. 1979;300:1056–1057. doi: 10.1056/nejm197905033001817. [DOI] [PubMed] [Google Scholar]
- Walker EA, Zernig G, Woods JH. Buprenorphine antagonism of mu opioids in the rhesus monkey tail-withdrawal procedure. J Pharmacol Exp Ther. 1995;273:1345–1352. [PubMed] [Google Scholar]
- Wand GS, Schumann H. Relationship between plasma adrenocorticotropin, hypothalamic opioid tone, and plasma leptin. J Clin Endocrinol Metab. 1998;83:2138–2142. doi: 10.1210/jcem.83.6.4900. [DOI] [PubMed] [Google Scholar]
- Williams KL, Ko MCH, Rice KC, Woods JH. Effect of opioid receptor antagonists on hypothalamic–pituitary–adrenal activity in rhesus monkeys. Psychoneuroendocrinology. 2003;28:513–528. doi: 10.1016/s0306-4530(02)00037-9. [DOI] [PubMed] [Google Scholar]
- Winger G, Stitzer ML, Woods JH. Barbiturate-reinforced responding in rhesus monkeys: comparisons of drugs with different durations of action. J Pharmacol Exp Ther. 1975;195:505–514. [PubMed] [Google Scholar]
- Zis AP, Haskett RF, Albala AA, Carroll BJ. Morphine inhibits cortisol and stimulates prolactin secretion in man. Psychoneuroendocrinology. 1984;9:423–427. doi: 10.1016/0306-4530(84)90050-7. [DOI] [PubMed] [Google Scholar]