

Abstract
Treatment of the protected ribose or xylose 5-aldehyde with sulfonyl-stabilized fluorophosphonate gave (fluoro)vinyl sulfones. Stannyldesulfonylation followed by iododestannylation afforded 5,6-dideoxy-6-fluoro-6-iodo-d-ribo or xylo-hex-5-enofuranoses. Coupling of the hexenofuranoses with alkylzinc bromides gave ten-carbon ribosyl- and xylosylhomocysteine analogues incorporating a fluoroalkene. The fluoroalkenyl and alkenyl analogues were evaluated for inhibition of Bacillus subtilis S-ribosylhomocysteinase (LuxS). One of the compounds, 3,5,6-trideoxy-6-fluoro-d-erythro-hex-5-enofuranose, acted as a competitive inhibitor of moderate potency (KI = 96 µM).
Keywords: LuxS enzyme, Negishi coupling, S-ribosylhomocysteine, vinyl fluorides, vinyl stannanes
1. Introduction
S-Adenosyl-L-homocysteine (SAH) is a byproduct of many methyltransferase reactions and a potent inhibitor of the methyltransferases. In eukaryotes and some bacteria, detoxification of SAH is mediated by SAH hydrolase (EC 3.3.1.1), which effects hydrolytic cleavage of SAH to L-homocysteine (Hcy) and adenosine (Figure 1).1 Hcy appears to be a risk factor for coronary artery diseases.2 Alternatively, most bacteria utilize enzyme 5′-methylthioadenosine (MTA)/SAH nucleosidase (EC 3.2.2.9) to irreversibly cleave SAH yielding adenine and S-ribosyl-L-homocysteine (SRH).3 The SRH is then converted to Hcy and 2,4-dihydroxy-2,3-pentadione (DPD) by a metalloenzyme S-ribosylhomocysteinase (LuxS).4 DPD5 spontaneously cyclizes and complexes with borate to form a furanosyl borate diester, which acts as a type 2 autoinducer for bacterial interspecies quorum sensing.6 Since quorum sensing regulates many bacterial behaviors such as virulence and biofilm formation, LuxS and other proteins involved in quorum sensing have been proposed as attractive targets for novel antibacterial drug design.7 Several substrate analogues of SRH (e.g., 1 and 2) showed submicromolar inhibition of LuxS.4e,h
Figure 1.

Reaction pathways for SAH detoxification in eukaryotes (a) and the majority of bacteria (b). The latter is also utilized by the bacteria to produce the type 2 autoinducer.
We have previously observed that SAH hydrolase is capable of the addition of water across 5′,6′ isolated double bond of adenosine analogues 3 and 4 (Figure 2).1c,8 The resulting adduct (or its derivative) caused covalent modification and inactivation8b of the enzyme, a process which required the catalytic activity of the enzyme. Since LuxS catalyzes a similar reaction as SAH hydrolase (i.e., overall elimination of Hcy), we designed analogues of SRH with the vinyl or halovinyl moieties incorporated in place of the carbon-5 and sulfur atoms (e.g., 5). We envisaged that these ribosyl analogues might serve as mechanistic probes to study the mechanism of action of LuxS and evaluate the similarities between SAH hydrolase and LuxS. As mentioned above, LuxS inhibitors may provide a novel class of antibacterial agents. We now describe the syntheses of SRH analogues with the carbon-5 and sulfur atoms replaced by vinyl or (6-fluoro)vinyl motifs and discuss their interactions with LuxS enzyme.
Figure 2.

Inhibitors of LuxS enzyme (1, 2).4e,h 5′-Deoxy-5′-(halomethylene)adenosine analogues (3, 4) which serve as suicide substrates for SAH hydrolase8 and targeted SRH analogues (5) in which the sulfur and C5 atoms are replaced by a vinyl unit.
2. Chemistry
Our initial plan to prepare compound 5 and its congeners is illustrated in Scheme 1. Treatment of the diacetone 3-O-benzoylglucose 6 or allose 7 with periodic acid selectively removed the 5,6-O-isopropylidene group. Subsequent oxidative cleavage of the exposed vicinal diol9a gave the corresponding 5-aldehydes 8 and 9, respectively, in high yields (Scheme 1). Wittig olefination of aldehyde 8 with the ylide derived from commercially available [4-ethoxy-4-oxobutyl)triphenylphoshonium bromide provided a complex mixture of products. Column chromatography yielded protected 5,6,7,8-tetradeoxy-α-D-xylo-non-5(Z)-enofuranuronate 10 (18% yield). The stereochemistry was assigned as Z, based on the magnitude of the coupling constants for olefinic protons (3J5-6 = 11.1 Hz), and literature precedence for the Wittig condensations of aliphatic aldehydes with the nonstabilized ylides.9b Similarly, Wittig-treatment of ribo 5-aldehyde 9 gave 11; a nine-carbon analogue of SRH. Unfortunately, our attempts to add bromine (CH2Cl2/0 °C) across the double bond of 10 or 11 (as well as 16) produced a complex mixture which did not give the desired SRH analogues of type 5 bearing a (6-bromo)vinyl unit when treated with DBU.10
Scheme 1.
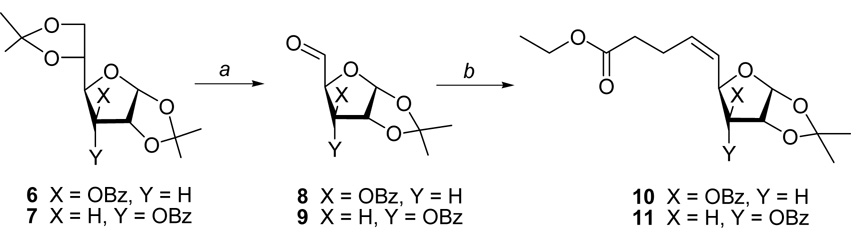
(a) H5IO6/EtOAc; (b) Ph3PCH2CH2CH2CO2Et/HMDS/THF.
In an alternative approach, we attempted a synthesis of 6-bromoalkenyl analogues 5 (X = Br) via Pd-catalyzed monoalkylation11–13 of the readily available (gem-dibromo)vinyl sugar precursors (e.g. 12, 13) with the corresponding alkylzinc reagents. Thus, dibromolefination of xylo 5-aldehyde 8 by the Corey-Fuchs procedure14 gave 5-(dibromomethylene)-5-deoxyxylose 12 (81% from 6; Scheme 2). Analogous treatment of the ribo 5-aldehyde 9 afforded 13.15 Treatment of 12 with 3 equiv. of 4-ethoxy-4-oxobutylzinc bromide in the presence of Pd(PPh3)4 at 55 °C gave monoalkylated 5,6,7,8,9-pentadeoxy-α-D-xylo-dec-5(E)-enofuranuronate 14 (18%, 3J5–6 = 15.4 Hz) and dialkylated 18 (48%) products, but did not yield the desired 6-bromoalkenyl product 15. Analogous Negishi coupling of 5-(dibromomethylene)-5-deoxyribose 13 afforded only dialkylated product 19 (54%). Changing catalyst [(Pd2(dba)3)], solvent (THF), reaction temperature (r.t. to 60 °C) as well as adding additives (CuI, tricyclohexylphosphine) did not lead to the formation of 15 or 17 but instead produced dialkylated byproducts 18 and 19 (3-49%) in agreement with a recent report.13b
Scheme 2.

(a) PPh3/CBr4; (b) BrZn(CH2)3CO2Et/Pd(PPh3)4/benzene/Δ
We next explored stereoselective coupling of the gem-dihalovinyl sugars containing two different halogens. We chose 5-deoxy-5-(fluoroiodomethylene) hexenofuranoses 26 and 27 because the iodo and fluoro substituents are known to have quite different reactivity towards oxidative-addition in Pd-mediated couplings.11b,16,17 The precursors 26 and 27 were prepared employing McCarthy's stannyldesulfonylation methodology.18,19 Thus, treatment of the xylo aldehyde 8 with the enolate generated from the sulfonyl-stabilized fluorophosphonate20 gave (fluoro)vinyl sulphones 20 (E/Z, 7∶3; 76%; Scheme 3). The stereoselective radical-mediated stannyldesulfonylation of 20 with Bu3SnH produced (fluoro)vinyl stannanes 23 (E/Z, 7∶3; 95%). Iododestannylation of 23 with N-iodosuccinimide (NIS) quantitatively afforded 6-fluoro-6-iodo-xylo-hex-5-enofuranoses 26 with retention of the E/Z configuration. The ribo analogue 27 (E/Z, 3∶2; 57% overall yield from 9) was similarly prepared. The isomeric ratio for the fluorinated sugars could be distinguished by the magnitude of the 3JF-H5 in the NMR spectra.
Scheme 3.
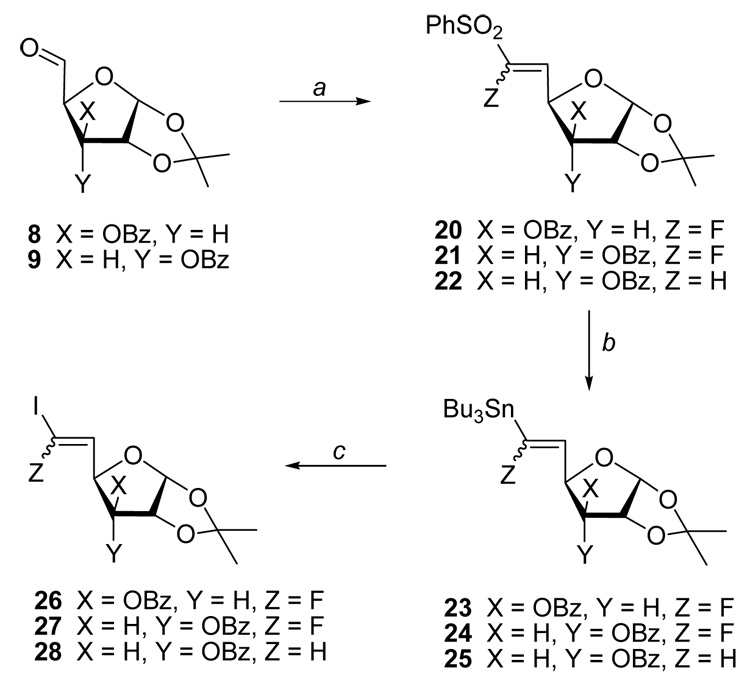
(a) PhSO2CHFPO(OEt)2 or PhSO2CH2PO(OEt)2/LHMDS/THF/-78 °C; (b) Bu3SnH/AIBN/toluene/85 °C; (c) NIS/CH2Cl2
Pd-mediated cross-coupling of the xylo analogue 26 (E/Z, 4∶1) with 2 equiv. of 4-ethoxy-4-oxobutylzinc bromide resulted in selective consumption of (E)-26 to afford (Z)-29 in 61% isolated yield or 76% based on consumption of (E)-26 (Scheme 4). A small amount of (E)-29 was also isolated, although monocoupling with gem-dihalovinyl substrates is considered to be trans selective.12,13b,16 Similar monoalkylation of the ribo analogue 27 (E/Z, 3∶2) with BrZn(CH2)3COOEt yielded (Z)-30 [54%, 90% based on the conversion of (E)-27]21 and (E)-30 [12%, 30% from (Z)-27]. Coupling of the (iodo)vinyl (E)-28, prepared as depicted in Scheme 3 (9 → 22 → 25 → 28), with BrZn(CH2)3COOEt gave the unfluorinated analogue (E)-16 (56%) with the retention of configuration. Treatment of (Z)-29 with NH3/MeOH removed the benzoyl group and converted the ethyl ester into a methyl ester (Z)-31 (74%). Subsequent removal of the isopropylidene group with aqueous trifluoroacetic acid (TFA) at 0 °C gave (Z)-33 (61%; α/β, 1∶1). Successive treatment of (Z)-30 with NH3/MeOH followed by TFA/H2O gave (Z)-34 (52% overall yield; α/β, 3∶7); a ten-carbon 6-fluoroalkenyl analogue of SRH.
Scheme 4.

(a) BrZn(CH2)3CO2Et/Pd(PPh3)4/benzene/Δ; (b) NH3/MeOH; (c) TFA/H2O
The 5,6-dideoxy-6-fluorohex-5-enofuranoses 42 and 43, depurinated analogues of 3 (X = F), were synthesized by protiodestannylation of the (fluoro)vinyl stannanes 23 and 24. Thus, treatment of 23 (E/Z, 7∶3) with NH3/MeOH at 25 °C resulted in the removal of 3-O-benzoyl group to give 36 (Scheme 5). However, prolonged heating of 36 (or 23) with NH3/MeOH at 65 °C for 48 h effected protiodestannylation to yield a separatable mixture of (E)-39 (29%) and (Z)-39 (48%). Treatment of (E)-39 with TFA/H2O at 0 °C gave (E)-42 (α/β, ~1∶1). Analogous debenzoylation and protiodestannylation of 24 (E/Z, 1∶1) with NH3/MeOH yielded (E)-40 (32%) and (Z)-40 (26%). Acid-catalyzed removal of the isopropylidene group in (E)-40 gave 5,6-dideoxy-6-fluoro-D-ribo-hex-5-enofuranose (E)-43 (67%; α/β, ~1∶4). Alternatively, concomitant protiodestannylation and removal of acetone unit in 36 or 37 with TFA also afforded 42 and 43.
Scheme 5.

(a) NH3/MeOH/25 °C; (b) Bu3SnH/AIBN/toluene/85 °C;(c) NH3/MeOH/65 °C or NH3/MeOH/CsF/65 °C; (d) TFA/H2O
The 3,5,6-trideoxy 6-fluorohex-5-enofuranose 44, which lacks a hydroxyl group at C3 and therefore cannot participate in the second enolization step of the LuxS catalyzed reaction,4b was also prepared. Thus, oxidation of the diacetone 3-deoxyglucose22 with H5IO69a and in situ treatment of the rather unstable 3-deoxyribose 5-aldehyde with the enolate generated from the sulfonyl-stabilized fluorophosphonate20 gave the (fluoro)vinyl sulfones 35 (48%; E/Z, 2∶1; ). Subjection of 35 to the stannyldesulfonylation/protiodestannylation18b sequence afforded 3-deoxy (6-fluoro)vinyl sugar 41, which was deprotected to yield 44 (12% from 35). Alternatively, treatment of vinyl stannanes 38 with TFA affected simultaneous protiodestannylation and removal of the acetone unit to give 44 (23% from 35; E/Z ~1∶3, α/β ~1∶4).
3. Inhibition of LuxS
The (6-fluoro)vinyl xylo- (42) and ribo-hexofuranoses (43) and their 3-deoxy analogue 44 as well as (6-fluoro)vinyl xylo- and ribo-decofuranoses (33 and 34) were evaluated4h as potential inhibitors of Bacillus subtilis S-ribosylhomocysteinase (LuxS). Compound 44 exhibited competitive inhibition of moderate potency, with a KI value of 96 ± 3 µM (Figure 3). None of the other compounds showed significant inhibition under the assay conditions.
Figure 3.
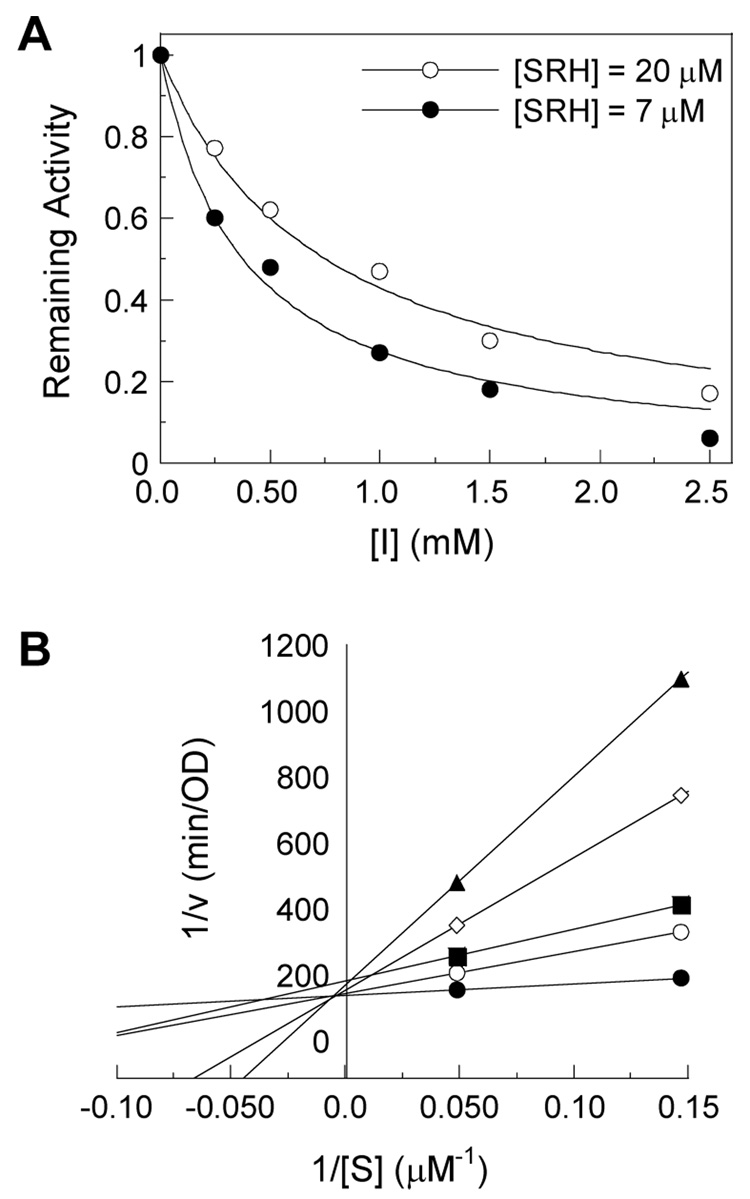
Inhibition of Co2+-substituted B. subtilis LuxS by compound 44. (A) Plot of remaining LuxS activity (relative to that in the absence of inhibitor) as a function of [I]. (B) Lineweaver-Burke plot of data from part A to show the competitive inhibition mode.
4. Summary and Conclusions
We have developed synthesis of six-, nine- and ten-carbon analogues of ribosyl- and xylosylhomocysteines in which the carbon-5 and sulfur atoms are replaced by a vinyl or (fluoro)vinyl unit. These fluoroalkenyl and alkenyl analogues of SRH were synthesized employing either the Wittig reaction or Pd-catalyzed coupling routes. They were evaluated against Bacillus subtilis S-ribosylhomocysteinase (LuxS). Only 3,5,6-trideoxy-6-fluoro-d-erythro-hex-5-enofuranose acted as competitive inhibitor of moderate potency with KI = 96 µM.
5. Experimental Section
1H (Me4Si) NMR spectra were determined with solution in CDCl3 at 400 or 600 MHz, 13C (Me4Si) at 100.6 MHz and 19F (CFCl3) at 376.5 MHz unless otherwise noted. Mass spectra (MS) were obtained by atmospheric pressure chemical ionization (APCI) and HRMS by electron impact techniques unless otherwise noted. Reagent grade chemicals were used as received. Solvents were dried by reflux over and distillation from CaH2 under an argon atmosphere except THF (K/benzophenone). TLC was performed on Merck kieselgel 60-F254 with MeOH/CHCl3 (1∶9) and EtOAc/MeOH (95∶5) as developing systems, and products were detected with 254 nm light or by visualization with Ce(SO4)2/(NH4)6Mo7O24•4H2O/H2SO4/H2O reagent. Merck kieselgel 60 (230-400 mesh) was used for column chromatography. Elemental analyses were determined by Galbraith Laboratories, Knoxville, TN.
Ethyl 3-O-Benzoyl-5,6,7,8-tetradeoxy-1,2-O-isopropylidene-α-D-xylo-non-5(Z)-enofuranuronate (10)
Step (a). H5IO6 (150 mg, 0.66 mmol) was added to a stirred solution of 6 (200 mg, 0.55 mmol) in dried EtOAc at ambient temperature. A precipitate appeared within the first five minutes and the resulting solution was stirred for 90 min. The precipitate was filtered off and was washed with EtOAc (2 × 5 mL). The combined organic layer was washed with NaHCO3/H2O (10 mL), NaCl/H2O (10 mL), dried (Na2SO4) and evaporated to yield 3-O-benzoyl-1,2-O-isopropylidene-α-D-xylo-pentodialdo-1,4-furanose (8; 160 mg, 95%; approx. 90% pure based on 1H NMR): 1H NMR δ 1.35 & 1.48 (2 × s, 2 × 3, 2 × CH3), 4.76 (d, J4-3 = 3.2 Hz, 1, H4), 4.88 (d, J2-1 = 3.1 Hz, 1, H2), 5.77 (d, J1-2 = 3.1 Hz, 1, H1), 6.18 (d, J3-4 = 3.3 Hz, 1, H3), 7.42-8.01 (m, 5, Ar), 9.78 (s, 1, H5). Step (b). LHMDS (1M/THF; 0.69 mL, 0.69 mmol) was added dropwise to a stirred solution of Ph3PCH2CH2CH2CO2Et/Br (314 mg, 0.69 mmol) in anhydrous THF (4 mL) in a flame-dried flask under N2 at ambient temperature. After 15 minutes, a solution of the crude, preferentially freshly prepared, aldehyde 8 (160 mg of the material from step a) in THF (2 mL) was added via syringe and stirring was continued overnight. EtOAc (30 mL) and NaHCO3/H2O (10 mL) was added and the separated organic was washed with NaCl/H2O (10 mL), dried (Na2SO4), and evaporated. Column chromatography (10 → 30% hexanes/EtOAc) gave 10 (39 mg, 18%) as an oil: 1H NMR δ 1.24 (t, J = 7.2 Hz, 3, CH3), 1.37 & 1.62 (2 × s, 2 × 3, 2 × CH3), 2.41 (t, J8-7/7′ = 6.9 Hz, 2, H8/8′), 2.50 ("q", J7-6/8/8′ = 7.3 Hz, 2, H7/7′), 4.15 (q, J = 7.1 Hz, 2, CH2), 4.71 (d, J2-1 = 3.8 Hz, 1, H2), 5.22 (dd, J4-5 = 7.7 Hz, J4-3 = 2.8 Hz, 1, H4), 5.46 (d, J3-4 = 2.8 Hz, H3), 5.58 (dd, J5-6 = 11.1 Hz, J5-4 = 7.9 Hz, 1, H5), 5.68 (dt, J6-5 = 11.1 Hz, J6-7/7′ = 7.1 Hz, 1, H6), 6.05 (d, J1-2 = 3.7 Hz, 1, H1), 7.48-8.04, (m, 5, Ar); 13C NMR δ 14.62 (CH3), 24.08 (C7), 26.62 & 27.19 (CMe2), 34.23 (C8), 60.90 (CH2), 75.54 (C2), 78.59 (C3), 84.20 (C4), 105.02 (C1), 112.47 (CMe2), 123.90 (C6), 128.91 (Bz), 129.78 (Bz), 130.15 (Bz), 133.85 (Bz), 134.39 (C5), 165.64 (Bz), 173.09 (C9); MS m/z 391 (100, MH+). HRMS (AP-ESI) m/z calcd for C21H26O7Li (MLi+) 397.1839; found 397.1833.
Ethyl 3-O-Benzoyl-5,6,7,8-tetradeoxy-1,2-O-isopropylidene-α-D-ribo-non-5(Z)-enofuranuronate (11)
Step (a) Oxidation of 7 (200 mg, 0.55 mmol) with H5IO6 (150 mg, 0.66 mmol), as described for 10, gave 3-O-benzoyl-1,2-O-isopropylidene-α-D-ribo-pentodialdo-1,4-furanose (9; 145 mg, 85%; approx. 90% pure, 1H NMR): 1H NMR δ 1.39 & 1.61 (2 × s, 2 × 3, 2 × CH3), 4.64 (dd, J4-5 = 2.2 Hz, J4-3 = 9.2 Hz, 1, H4), 5.01 (t, J2-1/3 = 4.2 Hz, 1, H2), 5.13 (dd, J3-4 = 9.2 Hz, J3-2 = 4.6 Hz, 1, H3), 6.02 (d, J1-2 = 3.4 Hz, 1, H1), 7.48-8.03 (m, 5, Ar), 9.77 (d, J5-4 = 2.2 Hz, 1, H5). Step (b). Treatment of the crude 9 (145 mg) with Ph3P(CH2)3CO2Et/Br (275 mg, 0.60 mmol) and LHDMS (1M/THF; 0.60 mmol, 0.60 mL), as described for 10, gave 11 (18 mg, 12%): 1H NMR δ 1.26 (t, J = 7.1 Hz, 3, CH3), 1.36 & 1.62 (2 × s, 2 × 3, 2 × CH3), 2.38 (t, J8-7/7′ = 8.2 Hz, 2, H8/8′), 2.46-2.55 (m, 1, H7), 2.55-2.67 (m, 1, H7′), 4.15 (q, J = 7.1 Hz, 2, CH2), 4.74 (dd, J3-4 = 9.1 Hz, J3-2 = 4.8 Hz, 1, H3), 4.96 ("t", J2-1/3 = 4.3 Hz, 1, H2), 5.20 (t, J4-3/5 = 8.7 Hz, 1, H4), 5.50 (ddt, J5-6 = 10.9 Hz, J5-4 = 8.7 Hz, J5-7/7′ = 1.0 Hz, 1, H5), 5.72 (dt, J6-5 = 10.9 Hz, J6-7/7′ = 7.1 Hz, 1, H6), 5.93 (d, J1-2 = 3.8 Hz, 1, H1), 7.48-8.04, (m, 5, Ar); 13C NMR δ 14.30 (CH3), 24.01 (C7), 26.99 & 27.04 (CMe2), 34.53 (C8), 60.89 (CH2), 73.39 (C4), 77.34 (C2), 77.56 (C3), 104.61 (C1), 113.40 (CMe2), 127.26 (C5), 128.90 (Bz), 129.80 (Bz), 130.28 (Bz), 133.80 (Bz), 135.14 (C6), 166.25 (Bz), 173.06 (C9). HRMS (AP-ESI) m/z calcd for C21H26O7Li (MLi+) 397.1839; found 397.1828.
3-O-Benzoyl-5,6-dideoxy-6,6-dibromo-1,2-O-isopropylidene-α-D-xylo-hex-5-enofuranose (12)
(Dibromomethylene)triphenylphosphorane [generated in situ by stirring CBr4 (8.09 g, 24.5 mmol), Ph3P (6.46 g, 24.5 mmol) and activated Zn (dust; 1.60 g, 24.5 mmol) in dried CH2Cl2 (100 mL) at 0 °C (ice-bath) for 30 min followed by stirring at ambient temperature under N2 for 3h] was added to the solution of freshly prepared aldehyde 8 [prepared as described for 10 (step a) from 6 (4.68 g, 12.9 mmol) and dried for 1 h under vacuum prior to use] in CH2Cl2 (75 mL). After stirring for 14 h at ambient temperature, the reaction mixture was partitioned (NaHCO3/H2O//CHCl3), and the organic layer was washed (H2O, brine), dried (MgSO4), and the volatiles were evaporated. Column chromatography (15 → 25% EtOAc/hexane) gave 12 (4.68 g, 81% overall from 6) as a solidifying viscous oil: 1H NMR δ 1.34 & 1.58 (2 × s, 2 × 3, 2 × CH3), 4.68 (d, J2-1 = 3.7 Hz, 1, H2), 5.04 (dd, J4-5 = 7.6 Hz, J4-3 = 3.0 Hz, 1, H4), 5.54 (d, J3-4 = 3.0 Hz, 1, H3), 6.00 (d, J1-2 = 3.7 Hz, 1, H1), 6.60 (d, J5-4 = 7.6 Hz, 1, H5), 7.48 (t, J = 7.6 Hz, 2 Ar), 7.60 (tt, J = 1.3, 7.6 Hz, 1 Ar), 8.02 ("dd", J = 1.4, 7.7 Hz, 2 Ar); 13C NMR δ 24.96 & 25.51 (CMe2), 75.71 (C3), 78.18 (C4), 82.04 (C2), 93.26 (C6), 103.30 (C1), 111.26 (CMe2), 127.31 (Bz), 127.73 (Bz), 128.44 (Bz), 130.49 (C5), 132.37 (Bz), 163.81 (Bz); MS m/z 451 (5, MH+ [81Br2]), 449 (10, MH+ [81/79Br2]), 447 (5, MH+ [79Br2]).
Ethyl 3-O-Benzoyl-5,6,7,8,9-pentadeoxy-1,2-O-isopropylidene-α-D-xylo-dec-5(E)-enofuranuronate (14) and Ethyl 3-O-Benzoyl-6-[3-(ethoxycarbonyl)propyl]- 5,6,7,8,9-pentadeoxy-1,2-O-isopropylidene-α-D-xylo-dec-5-enofuranuronate (18)
Pd[P(Ph)3]4 (22 mg, 0.014 mmol) was added to a stirred solution of 12 (42 mg, 0.094 mmol) in anhydrous benzene (3 mL) in a flame dried flask under N2 at ambient temperature. After 2 minutes, 4-ethoxy-4-oxobutylzinc bromide (0.5 M/THF; 0.56 mL, 129 mg, 0.28 mmol) was added and the resulting mixture was heated at 55 °C for 6 h. The reaction mixture was cooled down to ambient temperature and was partitioned between EtOAc (30 mL) and NaHCO3/H2O (10 mL). The separated organic layer was washed with H2O (10 mL), NaCl/H2O (10 mL), dried (Na2SO4), and evaporated. Column chromatography (10 → 30% EtOAc/hexanes) gave recovered 12 (7 mg, 13%), 14 (7 mg, 18%) and 18 (19 mg, 48%). Compound 14 had: 1H NMR δ 1.23 (t, J = 7.1 Hz, 3, CH3), 1.28 & 1.58 (2 × s, 2 × 3, 2 × CH3), 1.68 (quint, J8-7/7'/9/9′ = 7.5 Hz, 2, H8/8′), 2.07 ("q", J7-6/8/8′ = 7.0 Hz, 2, H7/7′), 2.23 (t, J9-8/8′ = 7.4 Hz, 2, H9/9′), 4.15 (q, J = 7.1 Hz, 2, CH2), 4.70 (d, J2-1 = 3.7 Hz, 1, H2), 4.86 (dd, J4-5 = 7.1 Hz, J4-3 = 2.8 Hz, 1, H4), 5.42 (d, J3-4 = 2.7 Hz, 1, H3), 5.56 (dd, J5-6 = 15.4 Hz, J5-4 = 7.3 Hz, 1, H5), 5.92 (dt, J6-5 = 15.4 Hz, J6-7/7′ = 6.9 Hz, 1, H6), 6.03 (d, J1-2 = 3.7 Hz, 1, H1), 7.46-8.05, (m, 5, Ar); 13C NMR δ 14.59 (CH3), 23.34 (C8), 26.69 & 27.16 (CMe2), 30.10 (C7), 34.05 (C9), 60.74 (CH2), 75.90 (C2), 81.58 (C3), 85.44 (C4), 105.00 (C1), 112.52 (CMe2), 128.35 (C5), 130.17 (Bz), 130.65 (Bz), 132.47 (Bz), 133.60 (Bz), 135.50 (C6), 165.89 (Bz), 173.74 (C10); MS m/z 405 (100, MH+). Compound 18 had: 1H NMR δ 1.21 (t, J = 7.1 Hz, 3, CH3), 1.29 (t, J = 7.1 Hz, 3, CH3), 1.36 & 1.61 (2 × s, 2 × 3, 2 × CH3), 1.69 (quint, J = 7.5 Hz, 2H), 1.71-1.80 (m, 2H), 2.05 (t, J = 7.5 Hz, 2H), 2.10-2.26 (m, 4H), 2.33 (t, J = 7.2 Hz, 2H), 4.09 (q, J = 7.1 Hz, 2, CH2), 4.18 (q, J = 7.1 Hz, 2, CH2), 4.70 (d, J2-1 = 3.8 Hz, 1, H2), 5.14 (dd, J4-5 = 8.7 Hz, J4-3 = 2.9 Hz, 1, H4), 5.35 (d, J5-4 = 8.4 Hz, 1, H5), 5.41 (d, J3-4 = 2.8 Hz, 1, H3), 6.02 (d, J1-2 = 3.8 Hz, 1, H1), 7.47-8.06, (m, 5, Ar); 13C NMR δ 14.60 (CH3), 14.65 (CH3), 26.65 & 27.15 (CMe2), 23.32 & 23.84 (C8/8′), 30.53 & 36.22 (C7/7′), 34.04 & 34.08 (C9/9′), 60.59 (CH2), 60.75 (CH2), 75.90 (C4), 78.71 (C3), 84.24 (C2), 104.84 (C1), 112.36 (CMe2), 119.09 (C5), 146.90 (C6), 128.92 (Bz), 128.92 (Bz), 129.81 (Bz), 130.17 (Bz), 133.83 (Bz), 165.70 (Bz), 173.62 & 173.75 (C10/10′); MS m/z 519 (100, MH+).
Ethyl 3-O-Benzoyl-5,6,7,8,9-pentadeoxy-1,2-O-isopropylidene-α-D-ribo-dec-5(E)-enofuranuronate (16)
Treatment (55 °C, 3 h) of 28 (E; 20 mg, 0.048 mmol) with 4-ethoxy-4-oxobutylzinc bromide (0.5 M/THF; 0.19 mL, 65 mg, 0.096 mmol) as described for 14/18 gave 16 (11 mg, 56%): 1H NMR δ 1.24 (t, J = 7.1 Hz, 3, CH3), 1.35 & 1.59 (2 × s, 2 × 3, 2 × CH3), 1.72 (quint, J8-7/7′/9/9′ = 7.4 Hz, 2, H8/8′), 2.13 ("q", J7-6/8/8′ = 7.2 Hz, 2, H7/7′), 2.28 (t, J9-8/8′ = 7.6 Hz, 2, H9/9′), 4.10 (q, J = 7.1 Hz, 2, CH2), 4.65 (dd, J4-5 = 7.6 Hz, J4-3 = 8.9 Hz, 1, H4), 4.74 (dd, J3-4 = 9.2 Hz, J3-2 = 4.6 Hz, 1, H3), 4.96 (t, J2-1/3 = 4.3 Hz, 1, H2), 5.53 (dd, J5-6 = 15.4 Hz, J5-4 = 7.3 Hz, 1, H5), 5.89 (d, J1-2 = 4.0 Hz, 1, H1), 5.91 (dt, J6-5 = 15.8 Hz, J6-7/7′ = 6.8 Hz, 1, H6), 7.46-8.05, (m, 5, Ar); 13C NMR δ 14.62 (CH3), 24.40 (C8), 26.92 & 27.00 (CMe2), 31.99 (C7), 33.89 (C9), 60.65 (CH2), 76.92 (C2), 77.65 (C3), 78.60 (C4), 104.37 (C1), 113.35 (CMe2), 127.04 (C5), 128.85 (Bz), 129.83 (Bz), 130.28 (Bz), 133.76 (Bz), 136.28 (C6), 166.29 (Bz), 173.84 (C10); MS m/z 405 (100, MH+).
Ethyl 3-O-Benzoyl-5,6,7,8,9-pentadeoxy-6-[3-(ethoxycarbonyl)propyl]-1,2-Oisopropylidene-α-D-ribo-dec-5-enofuranuronate (19)
Treatment of 1315 (42 mg, 0.094 mmol) with Pd[P(Ph)3]4 (22 mg, 0.014 mmol) and 4-ethoxy-4-oxobutylzinc bromide (0.56 mL, 129 mg, 0.28 mmol) as described for 14/18 gave 19 (26 mg, 54%): 1H NMR δ 1.22 (t, J = 7.1 Hz, 6, 2 × CH3), 1.37 & 1.62 (2 × s, 2 × 3, 2 × CH3), 1.75 (quint, J = 7.5 Hz, 4H), 2.11 (t, J = 7.0 Hz, 2H), 2.24 (t, J = 9.0 Hz, 2H), 2.26 (t, J = 9.0 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 4.10 (q, J = 7.1 Hz, 2, CH2), 4.16 (q, J = 7.1 Hz, 2, CH2), 4.74 (dd, J3-4 = 9.0 Hz, J3-2 = 4.8 Hz, 1, H3), 4.95 (t, J2-3 = 4.3 Hz, 1, H2), 5.01 (t, J4-5 = 9.0 Hz, 1, H4), 5.26 (d, J5-4 = 8.9 Hz, 1, H5), 5.89 (d, J1-2 = 3.9 Hz, 1, H1), 7.40-8.10 (m, 5, Ar); 13C NMR δ 14.55 (CH3), 14.61 (CH3), 21.48 (C8/8′), 23.11 & 27.03 (CMe2), 30.08 & 30.12 (C7/7′), 32.34 & 34.04 (C9/9′), 60.73 (CH2), 60.83 (CH2), 73.86 (C4), 78.71 (C3), 84.24 (C2), 104.37 (C1), 113.28 (CMe2), 122.65 (C5), 128.92 (Bz), 128.92 (Bz), 129.81 (Bz), 130.17 (Bz), 133.83 (Bz), 143.90 (C6), 165.70 (Bz), 170.57 & 171.62 (C10/10′); MS m/z 519 (100, MH+).
(E/Z)-3-O-Benzoyl-5,6-dideoxy-6-fluoro-1,2-O-isopropylidene-6-phenylsulfonyl-α-D-xylo-hex-5-enofuranose (20)
LHMDS (0.84 mL, 140 mg, 0.84 mmol) was added dropwise to a solution of diethyl fluoro(phenylsulfonyl)methylphosphonate20 (260 mg, 0.84 mmol) in anhydrous THF (8 mL) in a flame dried flask under N2 at −78 °C. After 30 minutes, a solution of 8 (265 mg, 0.82 mmol) in THF (4 mL) was added and stirring was continued for 1.5 h. EtOAc (30 mL) and NH4Cl/H2O (10 mL) were added and reaction mixture was allowed to warm to ambient temperature. The separated organic layer was washed with NaHCO3/H2O (10 mL), NaCl/H2O (10 mL), dried (Na2SO4), and evaporated. Column chromatography (10 → 30% EtOAc/hexanes) gave 20 (166 mg, 76%; E/Z, 7∶3) as inseparable mixture of isomers: HRMS (AP-ESI) m/z: calcd for C22H22FO7S (MH+) 449.1065, found 449.1071; 19F NMR δ −110.25 (d, JF-H5 = 18.8 Hz, 0.30F, Z), −119.30 (d, JF-H5 = 32.1 Hz, 0.70F, E). Compound (E)-20 had: 1H NMR δ 1.35 & 1.56 (2 × s, 2 × 3, 2 × CH3), 4.73 (d, J2-1 = 3.7 Hz, 1, H2), 5.23 (dt, J4-5 = 7.4 Hz, J4-3/F = 2.3 Hz, 1, H4), 5.49 (d, J3-4 = 3.1 Hz, 1, H3), 6.03-6.05 (m, 1, H1), 6.43 (dd, J5-F = 32.4 Hz, J5-4 = 7.2 Hz, 1, H5), 7.48-8.03 (m, 10, Ar); 13C NMR δ 26.55 & 27.10 (CMe2), 73.78 (C4), 78.21 (C3), 83.77 (C2), 105.31 (C1), 113.13 (CMe2), 112.10 (d, 2J5-F = 3.3 Hz, C5), 128.99 (Ph), 129.08 (Bz), 129.13 (Bz), 129.79 (Ph), 129.94 (Ph), 130.12 (Ph), 130.17 (Bz), 134.25 (Bz), 135.02 (Ph), 156.00 (d, 1J6-F = 300.0 Hz, C6), 165.36 (Bz). Compound (Z)-20 had: 1H NMR δ 1.38 & 1.69 (2 × s, 2 × 3, 2 × CH3), 4.76 (d, J2-1 = 3.7 Hz, 1, H2), 5.68 (d, J3-4 = 2.8 Hz, 1, H3), 5.99 (dd, J5-F = 19.3 Hz, J5-4 = 8.6 Hz, 1, H5), 6.05-6.07 (m, 1, H1), 6.07-6.09 (m, 1, H4), 7.48-8.03 (m, 10, Ar); 13C NMR δ 26.98 & 27.32 (CMe2), 73.15 (d, 3J4-F = 10.14 Hz, C4), 79.06 (C3), 83.93 (C2), 105.37 (C1), 113.35 (CMe2), 114.10 (d, 2J5-F = 15.0 Hz, C5), 128.99 (Ph), 129.08 (Bz), 129.13 (Bz), 129.79 (Ph), 129.94 (Ph), 130.12 (Ph), 130.17 (Bz), 134.25 (Bz), 135.02 (Ph), 155.58 (d, 1J6-F = 292.3 Hz, C6), 165.36 (Bz). Note: Freshly prepared aldehyde 8, dried under vacuum for 2 h at ambient temperature prior the use, gave the best results.
(E/Z)-3-O-Benzoyl-5,6-dideoxy-6-fluoro-1,2-O-isopropylidene-6-phenylsulfonyl-α-D-ribo-hex-5-enofuranose (21)
Treatment of 9 (200 mg, 0.68 mmol) with diethyl fluoro(phenylsulfonyl)methylphosphonate20 (212 mg, 0.68 mmol) and LHMDS (0.68 mL, 114 mg, 0.68 mmol) as described for 20 gave 21 (216 mg, 71%; E/Z, 6∶4): HRMS (AP-ESI) m/z: calcd for C22H22FO7S (MH+) 449.1065, found 449.1069; 19F NMR δ −108.98 (d, JF-H5 = 22.6 Hz, 0.40F, Z), −121.25 (d, JF-H5 = 30.1 Hz, 0.60F, E). Compound (E)-21 had: 1H NMR δ 1.28 & 1.38 (2 × s, 2 × 3, 2 × CH3), 4.85 (dd, J3-4 = 9.0 Hz, J3-2 = 4.7 Hz, 1, H3), 5.00 ("t", J2-1/3 = 4.5 Hz, 1, H2), 5.10 (t, J4-3/5 = 8.2 Hz, 1, H4), 5.94 (d, J1-2 = 3.7 Hz, 1, H1), 6.37 (dd, J5-F = 31.3 Hz, J5-4 = 8.3 Hz, 1, H5), 7.44-8.20 (m, 10, Ar); 13C NMR δ 26.86 & 26.93 (CMe2), 71.36 (d, 3J4-F = 2.2 Hz, C4), 76.64 (d, 4J3-F = 1.8 Hz, C3), 77.54 (C2), 104.96 (C1), 114.10 (CMe2), 114.22 (d, 2J5-F = 3.1 Hz, C5), 128.88 (Ph), 129.13 (Bz), 129.18 (Bz), 129.88 (Ph), 130.28 (Ph), 133.98 (Bz), 135.10 (Ph), 136.96 (Ph), 156.91 (d, 1J6-F = 306.0 Hz, C6), 165.96 (Bz). Compound (Z)-21 had: 1H NMR δ 1.28 & 1.38 (2 × s, 2 × 3, 2 × CH3), 4.84 (dd, J3-4 = 9.2 Hz, J3-2 = 4.6 Hz, 1, H3), 5.01 ("t", J2-1/3 = 4.6 Hz, 1, H2), 5.86 (dd, J5-F = 19.8 Hz, J5-4 = 9.9 Hz, H5), 5.96 (d, J1-2 = 3.7 Hz, 1, H1), 6.07 (t, J4-3/5 = 10.4 Hz, 1, H4), 7.44-8.20 (m, 10, Ar); 13C NMR δ 27.17 & 27.39 (CMe2), 70.71 (d, 3J4-F = 8.5 Hz, C4), 77.17 (C3), 77.86 (C2), 104.82 (C1), 114.37 (CMe2), 116.23 (d, 2J5-F = 16.2 Hz, C5), 128.95 (Ph), 129.22 (Bz), 129.46 (Bz), 129.80 (Ph), 130.02 (Ph), 133.61 (Bz), 134.00 (Ph), 135.19 (Ph), 156.62 (d, 1J6-F = 296.3 Hz, C6), 166.30 (Bz).
(E)-3-O-Benzoyl-5,6-dideoxy-1,2-O-isopropylidene-6-phenylsulfonyl-α-D-ribo-hex-5-enofuranose (22)
Treatment of 9 (150 mg, 0.50 mmol) with diethyl (phenylsulfonyl)methylphosphonate20 (146 mg, 0.50 mmol) and LHMDS (0.50 mL, 84 mg, 0.50 mmol) as described for 20 gave 22 (166 mg, 82%): 1H NMR δ 1.33 & 1.55 (2 × s, 2 × 3, 2 × CH3), 4.76 (dd, J3-4 = 9.5 Hz, J3-2 = 4.6 Hz, 1, H3), 4.92 (ddd, J4-5 = 3.7 Hz, J4-6 = 1.7 Hz, J4-3 = 9.5 Hz, 1, H4), 5.01 ("t", J2-3/1 = 4.2 Hz, 1, H2), 5.90 (d, J1-2 = 3.7 Hz, 1, H1), 6.79 (dd, J6-4 = 1.8 Hz, J6-5 = 15.0 Hz, 1, H6), 7.09 (dd, J5-6 = 15.0 Hz, J5-4 = 3.8 Hz, 1, H5), 7.50-8.05 (m, 10, Ar); MS m/z 431 (100, MH+).
(E/Z)-3-O-Benzoyl-5,6-dideoxy-6-fluoro-1,2-O-isopropylidene-6-tributylstannyl-α-D-xylo-hex-5-enofuranose (23)
Bu3SnH (407 mg, 0.38 mL, 1.4 mmol) was added dropwise to a degassed solution of 20 (300 mg, 0.70 mmol; E/Z, 7∶3) in anhydrous toluene (5 mL) in a flame-dried flask under N2 at ambient temperature. After an additional 10 minutes of degassing with N2, AIBN (86 mg, 0.53 mmol) was added and the reaction mixture was refluxed at 110 °C with stirring for 5 h. The volatiles were evaporated and the residue was partitioned between EtOAc (50 mL) and NaHCO3/H2O (30 mL). The organic layer was washed with NaCl/H2O (30 mL), dried (Na2SO4), and evaporated. Column chromatography (hexanes → 10% EtOAc/hexanes) gave 23 (794 mg, 95%; E/Z, 7∶3) as an inseparable mixture: MS m/z 599 (89, MH+, 120Sn), 597 (63, MH+, 118Sn), 595 (33, MH+, 116Sn), 541 (100, M-57, 120Sn), 539 (78, M-57, 118Sn), 537 (42, M-57, 116Sn); 19F NMR δ −87.67 (d, JF-H5 = 34.3 Hz, 84% of 0.30F, Z), −87.67 (dd, JF-Sn = 229.5 Hz, JF-H5 = 34.8 Hz, 16% of 0.30F, Z), −92.73 (d, JF-H5 = 52.7 Hz, 84% of 0.70F, E), −92.73 (ddd, JF-Sn = 213.1 Hz, JF-H5 = 52.7 Hz, JF-H4 = 4.9 Hz, 16% of 0.70F, E). Compound (E)-23 had: 1H NMR δ 0.90-1.60 (m, 27, 3 × Bu), 1.34 & 1.36 (2 × s, 2 × 3, 2 × CH3), 4.71 (d, J2-1 = 3.8 Hz, 1, H2), 5.10 (dd, J5-F = 52.6 Hz, J5-4 = 7.4 Hz, 1, H5), 5.32 (d, J3-4 = 3.0 Hz, 1, H3), 5.47-5.49 (m, 1, H4), 6.02 (d, J1-2 = 3.8 Hz, 1, H1), 7.47-8.06 (m, 5, Ar); 13C NMR δ 10.33 (Bu), 11.15 (Bu), 17.90 (Bu), 27.42 & 27.54 (CMe2), 28.24 (Bu), 70.56 (d, 3J4-F = 17.6 Hz, C4), 77.21 (C3), 77.52 (C2), 104.31 (C1), 113.07 (CMe2), 120.53 (d, 2J5-F = 3.9 Hz, C5), 128.75 (Bz), 129.83 (Bz), 130.25 (Bz), 133.61 (Bz), 166.16 (Bz), 177.14 (d, 2J6-F = 262.0 Hz, C6). Compound (Z)-23 had: 1H NMR δ 0.90-1.60 (m, 27, 3 × Bu), 1.38 & 1.69 (2 × s, 2 × 3, 2 × CH3), 4.69 (d, J1-2 = 3.9 Hz, 1, H2), 4.75 (d, J3-4 = 7.9 Hz, 1, H3), 5.47-5.49 (m, 1, H4), 5.98 (d, J1-2 = 3.8 Hz, 1, H1), 6.02 (dd, J5-F = 34.3 Hz, J5-4 = 9.2 Hz, 1, H5), 7.47-8.06 (m, 5, Ar); 13C NMR δ 10.33 (Bu), 11.15 (Bu), 17.90 (Bu), 27.42 & 27.54 (CMe2), 28.24 (Bu), 74.73 (d, 3J4-F = 22.2 Hz, C4), 77.38 (d, 4J3-F = 1.4 Hz, C3), 77.52 (C2), 104.53 (C1), 113.47 (CMe2), 121.24 (d, 2J5-F = 9.5 Hz, C5), 128.75 (Bz), 129.88 (Bz), 130.34 (Bz), 133.73 (Bz), 166.35 (Bz), 180.03 (d, 2J6-F = 254.3 Hz, C6).
(E/Z)-3-O-Benzoyl-5,6-dideoxy-6-fluoro-1,2-O-isopropylidene-6-tributylstannyl-α-D-ribo-hex-5-enofuranose (24)
Treatment of 21 (300 mg, 0.70 mmol; E/Z, 3∶2) with Bu3SnH (407 mg, 0.38 mL, 1.4 mmol) and AIBN (86 mg, 0.53 mmol) as described for 23 gave 24 (397 mg, 95%; E/Z, 3∶2): MS m/z 599 (89, MH+, 120Sn), 597 (63, MH+, 118Sn), 595 (33, MH+, 116Sn), 541 (100, M-57, 120Sn), 539 (78, M-57, 118Sn), 537 (42, M-57, 116Sn); 19F NMR δ −87.58 (d, JF-H5 = 33.1 Hz, 84% of 0.40F, Z), −87.58 (ddd, JF-Sn = 226.7 Hz, JF-H5 = 32.8 Hz, JF-H4 = 4.1 Hz, 16% of 0.40F), −94.80 (d, JF-H5 = 51.1 Hz, 84% of 0.60F, E), −94.80 (ddd, JF-Sn = 213.9 Hz, JF-H5 = 50.8 Hz, JF-H4 = 4.5 Hz, 16% of 0.60F, E). Compound (E)-24 had: 1H NMR δ 0.70-1.70 (m, 27, 3 × Bu), 1.24 & 1.26 (2 × s, 2 × 3, 2 × CH3), 4.62 (dd, J3-4 = 9.3 Hz, J3-2 = 4.7 Hz, 1, H3), 4.86-4.87 (m, 1, H2), 4.90 (dd, J5-F = 51.0 Hz, J5-4 = 8.4 Hz, 1, H5), 5.28 ("t", J4-3/5 = 8.9 Hz, 1, H4), 5.79 (d, J1-2 = 3.9 Hz, 1, H1), 7.57-8.06 (m, 5, Ar); 13C NMR δ 11.15 (Bu), 14.03 (Bu), 29.96 (Bu), 27.48 & 27.59 (CMe2), 29.23 (Bu), 70.55 (d, 3J4-F = 18.1 Hz, C4), 77.36 (C3), 77.48 (C2), 104.50 (C1), 113.54 (CMe2), 120.59 (d, 2J5-F = 3.8 Hz, C5), 128.77 (Bz), 129.81 (Bz), 130.38 (Bz), 133.69 (Bz), 166.44 (Bz), 176.10 (d, 1J6-F = 260.0 Hz, C6). Compound (Z)-24 had: 1H NMR δ 0.70-1.70 (m, 27, 3 × Bu), 1.28 & 1.30 (2 × s, 2 × 3, 2 × CH3), 4.47 ("t", J4-3/5 = 9.3 Hz, 1, H4), 4.71 (dd, J3-4 = 9.0 Hz, J3-2 = 4.8 Hz, 1, H3), 4.85-4.86 (m, 1, H2), 5.81 (d, J1-2 = 3.8 Hz, 1, H1), 5.83 (dd, J5-F = 33.7 Hz, J5-4 = 9.5 Hz, 1, H5), 7.57-8.06 (m, 5, Ar); 13C NMR δ 11.15 (Bu), 14.03 (Bu), 29.96 (Bu), 27.48 & 27.59 (CMe2), 29.23 (Bu), 74.72 (d, 3J4-F = 22.0 Hz, C4), 77.16 (C3), 77.67 (C2), 104.28 (C1), 113.13 (CMe2), 121.18 (d, 2J5-F = 9.9 Hz, C5), 128.77 (Bz), 129.76 (Bz), 130.28 (Bz), 130.80 (Bz), 166.24 (Bz), 177.00 (d, 1J6-F = 255.0 Hz, C6).
(E)-3-O-Benzoyl-5,6-dideoxy-1,2-O-isopropylidene-6-tributylstannyl-α-D-ribo-hex-5-enofuranose (25)
Treatment of 22 (E; 300 mg, 0.70 mmol) with Bu3SnH (407 mg, 0.38 mL, 1.4 mmol) and AIBN (86 mg, 0.53 mmol) as described for 23 gave 25 (385 mg, 95%): 1H NMR δ 1.51-1.90 (m, 33, 3 × Bu & 2 × CH3), 4.66 (dd, J4-5 = 6.5 Hz, J4-3 = 8.4 Hz, 1, H4), 4.77 (dd, J3-4 = 9.2 Hz, J3-2 = 4.7 Hz, 1, H3), 4.97 (t, J2-1/3 = 3.6 Hz, 1, H2), 5.93 (d, J1-2 = 3.8 Hz, 1, H1), 6.10 (dd, J5-6 = 19.1 Hz, J5-4 = 6.5 Hz 1, H5), 6.50 (dd, J6-5 = 19.1 Hz, J6-4 = 0.9 Hz, 1, H6), 7.57-8.06 (m, 5, Ar); MS m/z 581 (89, MH+, 120Sn), 579 (63, MH+, 118Sn), 577 (33, MH+, 116Sn), 523 (100, M-57, 120Sn), 521 (78, M-57, 118Sn), 519 (42, M-57, 116Sn).
(E/Z)-3-O-Benzoyl-5,6-dideoxy-6-fluoro-6-iodo-1,2-O-isopropylidene-α-D-xylo-hex-5-enofuranose (26)
A solution of NIS (50 mg, 0.23 mmol) in anhydrous CH2Cl2 (3 mL) was added dropwise to a stirred solution of 23 (90 mg, 0.15 mmol; E/Z, 7∶3) in CH2Cl2 (5 mL) under N2 at −20 °C. After 1h, CHCl3 (30 mL) and diluted NaHSO3/H2O (10 mL) were added. The separated organic layer was washed with NaHCO3/H2O (10 mL), NaCl/H2O (10 mL), dried (Na2SO4), and evaporated. Column chromatography (hexanes → 30% EtOAc/hexanes) gave 26 (155 mg, 83%; E/Z, 8∶2) as an inseparable mixture: MS m/z 435 (100, MH+); 19F NMR δ −56.54 (d, JF-H5 = 15.8 Hz, 0.20F, Z), −60.98 (d, JF-H5 = 33.1 Hz, 0.80F, E). Compound (E)-26 had: 1H NMR δ 1.35 & 1.38 (2 × s, 2 × 3, 2 × CH3), 4.69 (d, J2-1 = 3.8 Hz, 1, H2), 5.28 (ddd, J4-5 = 9.8 Hz, J4-3 = 2.9 Hz, J4-6 = 1.6 Hz, 1, H4), 5.45 (d, J3-4 = 3.0 Hz, 1, H3), 5.58 (dd, J5-F = 33.0 Hz, J5-4 = 8.7 Hz, 1, H5), 5.99 (d, J1-2 = 3.7 Hz, 1, H1), 7.47-8.06 (m, 5, Ar); 13C NMR δ 26.60 & 27.12 (CMe2), 74.04 (d, 3J4-F = 4.4 Hz, C4), 77.88 (C3), 83.73 (C2), 104.74 (C1), 107.89 (d, 1J6-F = 338.7 Hz, C6), 112.85 (CMe2), 116.80 (d, 2J5-F = 5.4 Hz, C5), 129.04 (Bz), 129.50 (Bz), 130.18 (Bz), 134.09 (Bz), 165.54 (Bz). Compound (Z)-26 had: 1H NMR δ 1.35 & 1.68 (2 × s, 2 × 3, 2 × CH3), 4.71 (d, J1-2 = 3.7 Hz, 1, H2), 4.84 (dd, J4-5 = 8.7 Hz, J4-3 = 2.7 Hz, 1, H4), 5.48 (d, J3-4 = 3.0 Hz, 1, H3), 5.77 (dd, J5-F = 15.4 Hz, J5-4 = 8.8 Hz, 1, H5), 6.03 (d, J1-2 = 3.7 Hz, 1, H1), 7.47-8.06 (m, 5, Ar); 13C NMR δ 26.78 & 27.27 (CMe2), 77.88 (C3), 80.06 (d, 3J4-F = 8.2 Hz, C4), 83.84 (C2), 105.04 (C1), 112.94 (d, 2J5-F = 16.9 Hz, C5), 112.95 (CMe2), 114.78 (d, 1J6-F = 332.0 Hz, C6), 129.04 (Bz), 129.40 (Bz), 130.18 (Bz), 134.14 (Bz), 165.54 (Bz).
(E/Z)-3-O-Benzoyl-5,6-dideoxy-6-fluoro-6-iodo-1,2-O-isopropylidene-α-D-ribo-hex-5-enofuranose (27)
Treatment of 24 (250 mg, 0.42 mmol; E/Z, 3∶2) with NIS (142 mg, 0.63 mmol) as described for 26 gave 27 (155 mg, 85%; E/Z, 3∶2) as an inseparable mixture: HRMS (AP-FAB) m/z: calcd for C16H16FIO5Li (MLi+) 441.0181; found 441.0192; 19F NMR δ −56.42 (d, JF-H5 = 15.1 Hz, 0.40F, Z), −63.30 (d, JF-H5 = 33.5 Hz, 0.60F, E). Compound (E)-27 had: 1H NMR δ 1.36 & 1.60 (2 × s, 2 × 3, 2 × CH3), 4.75 (dd, J3-4 = 9.2 Hz, J3-2 = 4.7 Hz, 1, H3), 4.98 (t, J2-1/3 = 4.5 Hz, 1, H2), 5.16 (t, J4-3/5 = 9.0 Hz, 1, H4), 5.49 (dd, J5-F = 32.7 Hz, J5-4 = 8.7 Hz, 1, H5), 5.89 (d, J1-2 = 3.8 Hz, 1, H1), 7.50-8.10 (m, 5, Ar); 13C NMR δ 26.96 & 27.11 (CMe2), 72.45 (d, 3J4-F = 4.3 Hz, C4), 76.74 (d, 4J3-F = 2.1 Hz, C3), 77.32 (C2), 104.39 (C1), 113.78 (CMe2), 114.96 (d, 1J6-F = 331.5 Hz, C6), 119.90 (d, 2J5-F = 5.5 Hz, C5), 128.92 (Bz), 129.46 (Bz), 130.50 (Bz), 133.94 (Bz), 166.21 (Bz). Compound (Z)-27 had: 1H NMR δ 1.38 & 1.64 (2 × s, 2 × 3, 2 × CH3), 4.72-4.75 (m, 1, H4), 4.84 (dd, J3-4 = 9.2 Hz, J3-2 = 4.6 Hz, 1, H3), 4.98 (t, J2-3/1 = 4.5 Hz, 1, H2), 5.68 (dd, J5-F = 15.3 Hz, J5-4 = 8.9 Hz, 1, H5), 5.91 (d, J1-2 = 3.8 Hz, 1, H1), 7.50-8.14 (m, 5, Ar); 13C NMR δ 26.96 & 27.11 (CMe2), 76.85 (d, 4J3-F = 2.1 Hz, C3), 77.63 (C2), 78.13 (d, 3J4-F = 8.3 Hz, C4), 104.54 (C1), 108.75 (d, 1J6-F = 339.4 Hz, C6), 113.90 (CMe2), 115.77 (d, 2J5-F = 16.2 Hz, C5), 128.91 (Bz), 129.49 (Bz), 130.37 (Bz), 133.95 (Bz), 166.21 (Bz).
(E)-3-O-Benzoyl-5,6-dideoxy-6-iodo-1,2-O-isopropylidene-α-D-ribo-hex-5-enofuranose (28)
Treatment of 25 (150 mg, 0.25 mmol) with NIS (85 mg, 0.38 mmol) as described for 26 gave 28 (93 mg, 87%): 1H NMR δ 1.35 & 1.62 (2 × s, 2 × 3, 2 × CH3), 4.67 (dd, J2-3 = 9.2 Hz, J2-1 = 4.6 Hz, 1, H2), 4.77 (dd, J3-4 = 3.4 Hz, J3-2 = 9.2 Hz, 1, H3), 4.97 (t, J4-3/5 = 4.2 Hz, 1, H4), 5.91 (d, J1-2 = 3.8 Hz, 1, H1), 6.61-6.70 (m, 2, H5/6), 7.49-8.08 (m, 5, Ar); 13C NMR δ 26.95 & 26.96 (CMe2), 76.37 (C4), 77.59 (C3), 79.61 (C2), 81.32 (C6), 104.43 (C1), 113.68 (CMe2), 128.89 (Bz), 129.60 (Bz), 130.94 (Bz), 133.89 (Bz), 141.58 (C5), 166.08 (Bz); MS m/z 417 (100, MH+).
Ethyl 3-O-Benzoyl-5,6,7,8,9-pentadeoxy-6-fluoro-1,2-O-isopropylidene-α-D-xylo-dec-5-(E/Z)-enofuranuronate (29)
Pd[P(Ph)3]4 (5 mg, 0.004 mmol) was added to a stirred solution of 26 (30 mg, 0.07 mmol; E/Z, 4∶1) in anhydrous benzene (3 mL) under N2 at ambient temperature. After 2 minutes, 4-ethoxy-4-oxobutylzinc bromide (0.5M/THF; 0.28 mL, 65 mg, 0.14 mmol) was added and the resulting mixture was heated at 55 °C for 5 h. EtOAc (30 mL) and NaHCO3/H2O (10 mL) were added and the separated organic layer was washed with H2O (10 mL), NaCl/H2O (10 mL), dried (Na2SO4), and then was evaporated. Column chromatography (10 → 30% EtOAc/hexanes) gave (Z)-29 (18 mg, 61%, 76% based on the conversion of the E-isomer), (E)-29 (2 mg, 7%, 35% based on the conversion of the Z-isomer) and more polar byproduct tentatively assigned as 3-O-debenzoylated-(Z)-26 [~5%, TLC; 19F NMR δ −57.21 (JF-H5 = 16.4 Hz)]. Compound (Z)-29 had: 1H NMR δ 1.22 (t, J = 7.1 Hz, 3, CH3), 1.36 & 1.61 (2 × s, 2 × 3, 2 × CH3), 1.81 ("quint", J8-7/7'/9/9' = 7.4 Hz, 2, H8/8′), 2.26 (dt, J7-F = 18.1 Hz, J7-8/8′ = 7.4 Hz, 2, H7/7′), 2.30 (t, J9-8/8′ = 7.4 Hz, 2, H9/9′), 4.09 (q, J = 7.1 Hz, 2, CH2), 4.69 (d, J2-1 = 3.7 Hz, 1, H2), 4.84 (dd, J5-F = 35.8 Hz, J5-4 = 8.4 Hz, H5), 5.33 (dd, J4-5 = 8.5 Hz, J4-3 = 2.8 Hz, 1, H4), 5.45 (d, J3-4 = 2.8 Hz, 1, H3), 6.00 (d, J1-2 = 3.7 Hz, 1, H1), 7.48-8.04, (m, 5, Ar); 13C NMR δ 14.59 (CH3), 21.49 (C8), 26.64 & 27.11 (CMe2), 31.61 (d, 2J7-F = 25.4 Hz, C7), 33.34 (C9), 60.75 (CH2), 73.37 (d, 3J4-F = 6.6 Hz, C4), 77.63 (C2), 78.33 (C3), 100.03 (d, 2J5-F = 10.9 Hz, C5), 104.78 (C1), 112.60 (CMe2), 128.97 (Bz), 129.70 (Bz), 130.16 (Bz), 133.93 (Bz), 162.94 (d, 1J6-F = 260.7 Hz, C6), 165.61 (Bz), 173.32 (C10); 19F NMR δ −99.93 (dt, JF-H5 = 35.8 Hz, J = 18.0 Hz); HRMS (AP-FAB+) m/z calcd for C22H27FO7Li (MLi+) 429.1910; found 429.1900. Compound (E)-29 had: 1H NMR δ 1.28 (t, J = 7.1 Hz, 3, CH3), 1.35 & 1.61 (2 × s, 2 × 3, 2 × CH3), 1.85-1.95 (m, 2, H8/8′), 2.38 (t, J9-8/8′ = 7.2 Hz, 2, H9/9′), 2.39-2.50 (m, 2, H7/7′), 4.15 (q, J = 7.1 Hz, 2, CH2), 4.69 (d, J2-1 = 3.8 Hz, 1, H2), 4.93 ("dt", J4-5 = 9.3 Hz, J4-F/3 = 2.5 Hz, 1, H4), 5.30 (dd, J5-F = 18.6 Hz, J5-4 = 9.4 Hz, H5), 5.38 (d, J3-4 = 2.9 Hz, 1, H3), 6.00 (d, J1-2 = 3.8 Hz, 1, H1), 7.48-8.04 (m, 5, Ar); 19F NMR δ −94.53 ("q", J = 22.1 Hz). HRMS (AP-FAB+) m/z calcd for C22H27FO7Li (MLi+) 429.1910; found 429.1903.
Ethyl 3-O-Benzoyl-5,6,7,8,9-pentadeoxy-6-fluoro-1,2-O-isopropylidene-α-D-ribo-dec-5(E/Z)-enofuranuronate (30)
Treatment of 27 (42 mg, 0.097 mmol; E/Z, 3∶2) with Pd[P(Ph)3]4 (22 mg, 0.01 mmol) and 4-ethoxy-4-oxobutylzinc bromide (0.5M/THF; 0.30 mmol, 0.60 mL) as described for 29 followed by column chromatography (10 → 40% EtOAc/hexanes) gave (Z)-30 (22 mg, 54%; 90% based on the conversion of E-isomer), (E)-30 (5 mg, 12%; 30% based on the conversion of the Z-isomer), and more polar 3-O-debenzoylated-(Z)-27 (3 mg, 10%). Compound (Z)-30 had: 1H NMR δ 1.24 (t, J = 7.1 Hz, 3, CH3), 1.37 & 1.60 (2 × s, 2 × 3, 2 × CH3), 1.84 ("quint", J8-7/7'/9/9' = 7.4 Hz, 2, H8/8′), 2.25 (dt, J7-F = 17.6 Hz, J7-8/8' = 7.5 Hz, 2, H7/7′), 2.32 (t, J9-8/8′ = 7.4 Hz, 2, H9/9′), 4.09 (q, J = 7.1 Hz, 2, CH2), 4.72 (dd, J3-4 = 9.2 Hz, J3-2 = 4.7 Hz, 1, H3), 4.75 (dd, J5-F = 35.0 Hz, J5-4 = 8.9 Hz, 1, H5), 4.95 (t, J2-1/3 = 4.3 Hz, 1, H2), 5.19 (t, J4-3/5 = 9.1 Hz, 1, H4), 5.89 (d, J1-2 = 3.8 Hz, 1, H1), 7.48-8.09 (m, 5, Ar); 13C NMR δ 14.60 (CH3), 21.45 (C8), 26.95 & 27.00 (CMe2), 31.63 (d, 2J7-F = 26.5 Hz, C7), 33.32 (C9), 60.79 (CH2), 71.39 (d, 3J4-F = 6.3 Hz, C4), 77.54 (C2), 73.63 (C3), 103.40 (d, 2J5-F = 11.8 Hz, C5), 104.39 (C1), 113.52 (CMe2), 128.84 (Bz), 129.71 (Bz), 130.33 (Bz), 133.77 (Bz), 164.13 (d, 1J6-F = 272.7 Hz, C6), 165.33 (Bz), 173.30 (C10); 19F NMR δ −102.14 (dt, JF-H5 = 34.1 Hz, JF-H7/7′ = 17.6 Hz). HRMS (AP-ESI) m/z calcd for C22H28FO7 (MH+) 423.1814; found 423.1815. Compound (E)-30 had: 1H NMR δ 1.26 (t, J = 7.1 Hz, 3, CH3), 1.36 & 1.61 (2 × s, 2 × 3, 2 × CH3), 1.90 ("quint", J8-7/7'/9/9' = 6.9 Hz, 2, H8/8′), 2.38 (t, J9-8/8′ = 6.9 Hz, 2, H9/9′), 2.50 (dt, J7-F = 23.0 Hz, J7-8/8' = 7.3 Hz, 2, H7/7′), 4.14 (q, J = 7.1 Hz, 2, CH2), 4.75-4.80 (m, 2, H3/4), 4.95 (t, J2-1/3 = 4.0 Hz, 1, H2), 5.17 (dd, J5-F = 19.2 Hz, J5-4 = 8.7 Hz, 1, H5), 5.88 (d, J1-2 = 3.8 Hz, 1, H1), 7.48-8.09 (m, 5, Ar); 13C NMR δ 14.63 (CH3), 21.99 (C8), 26.92 & 27.00 (CMe2), 28.49 (d, 2J7-F = 27.0 Hz, C7), 33.67 (C9), 60.78 (CH2), 73.65 (d, 3J4-F = 14.7 Hz, C4), 77.50 (C2), 77.61 (C3), 104.58 (d, 2J5-F = 25.8 Hz, C5), 104.37 (C1), 113.39 (CMe2), 128.88 (Bz), 129.64 (Bz), 130.25 (Bz), 133.83 (Bz), 165.15 (d, 1J6-F = 256.5 Hz, C6), 165.99 (Bz), 173.21 (C10); 19F NMR δ - 94.73 ("q", JF-H5/7/7′ = 22.8 Hz). HRMS (AP-ESI) m/z calcd for C22H28FO7 (MH+) 423.1814; found 423.1819. The 3-O-debenzoylated-(Z)-27 had: 1H NMR δ 1.27 & 1.58 (2 × s, 2 × 3, 2 × CH3), 3.82-3.84 (m, 1, H3), 4.18 (t, J4-3/5 = 8.8 Hz, 1, H4), 4.60-4.62 (m, 1, H2), 5.61 (dd, J5-F = 15.1 Hz, J5-4 = 8.9 Hz, 1, H5), 5.83 (d, J1-2 = 3.7 Hz, 1, H1); 13C NMR δ 26.99 & 27.07 (CMe2), 76.63 (C3), 78.56 (C2), 80.47 (d, 3J4-F = 8.0 Hz, C4), 104.16 (C1), 115.85 (d, 1J6-F = 344.1 Hz, C6), 113.70 (CMe2), 115.85 (d, 2J5-F = 15.7 Hz, C5); 19F NMR δ −56.50 (d, JF-H5 = 15.1 Hz); MS m/z 331 (100, MH+).
Methyl 5,6,7,8,9-Pentadeoxy-6-fluoro-1,2-O-isopropylidene-α-D-xylo-dec-5(Z)-enofuranuronate (31)
Compound (Z)-29 (26 mg, 0.062 mmol) was dissolved in MeOH (6 mL) and saturated NH3/MeOH (3 mL) was added at 0 °C (ice bath). The resulting mixture was stirred for 48 h (0 °C → ambient temperature). The volatiles were evaporated and the residue was column chromatographed (15 → 50% EtOAc/hexanes) to give 31 (14 mg, 74%): 1H NMR δ 1.35 & 1.55 (2 × s, 2 × 3, 2 × CH3), 1.90 (quint, J8-7/7'/9/9' = 7.2 Hz, 2, H8/8′), 2.23-2.40 (m, 2, H7/7′), 2.41 (t, J9-8/8′ = 7.2 Hz, 2, H9/9′), 3.70 (s, 3, CH3), 4.17 (d, J3-4 = 2.6 Hz, 1, H3); 4.59 (d, J2-1 = 3.7 Hz, 1, H2), 4.84 (dd, J5-F = 37.6 Hz, J5-4 = 7.7 Hz, 1, H5), 5.08 ("dm", J4-5 = 7.6 Hz, 1, H4), 5.95 (d, J1-2 = 3.7 Hz, 1, H1); 13C NMR δ 21.40 (C8), 26.59 & 27.12 (CMe2), 31.65 (d, 2J7-F = 26.6 Hz, C7), 33.29 (C9), 52.08 (CH3), 75.29 (d, 3J4-F = 4.9 Hz, C4), 76.74 (d, 4J3-F = 1.0 Hz, C3), 85.51 (C2), 101.15 (d, 2J5-F = 11.0 Hz, C5), 104.74 (C1), 112.08 (CMe2), 161.86 (d, 1J6-F = 261.8 Hz, C6), 174.02 (C10); 19F NMR δ −100.23 (dt, JF-H5 = 38.0 Hz, JF-H7 = 18.0 Hz); MS m/z 305 (100, MH+).
Methyl 5,6,7,8,9-Pentadeoxy-6-fluoro-1,2-O-isopropylidene-α-D-ribo-dec-5(Z)-enofuranuronate (32)
Saturated NH3/MeOH (3 mL) was added to a solution of (Z)-30 (26 mg, 0.062 mmol) in MeOH (3 mL) and the mixture was stirred at 0 °C for 48 h to ambient temperature. The volatiles were evaporated and the residue was column chromatographed (15 → 60% EtOAc/hexanes) to give 32 (13 mg, 69%): 1H NMR δ 1.39 & 1.62 (2 × s, 2 × 3, 2 × CH3), 1.90 (quint, J8-7/7'/9/9' = 7.3 Hz, 2, H8/8′), 2.31 (dt, J7-F = 17.6 Hz, J7-8/8′ = 7.4 Hz, 2, H7/7′), 2.40 (t, J9-8/8′ = 6.9 Hz, 2, H9/9′), 3.75 (s, 3, CH3), 4.56-4.72 (m, 4, H2/3/4/5), 5.82 (d, J1-2 = 3.9 Hz, 1, H1); 13C NMR δ 21.44 (C8), 26.81 & 26.94 (CMe2), 31.74 (d, 2J7-F = 26.1 Hz, C7), 33.20 (C9), 52.07 (CH3), 73.94 (d, 3J4-F = 5.1 Hz, C4), 76.80 (C2), 78.62 (C3), 103.63 (d, 2J5-F = 11.6 Hz, C5), 104.00 (C1), 113.07 (CMe2), 163.91 (d, 1J6-F = 262.70 Hz, C6), 173.90 (C10); 19F NMR δ -100.23 (dt, JF-H5 = 37.1 Hz, JF-H7/7′ = 18.1 Hz). HRMS (AP-ESI) m/z calcd for C14H22FO6 (MH+) 305.1395; found 305.1396.
Methyl 5,6,7,8,9-Pentadeoxy-6-fluoro-α/β-D-xylo-dec-5(Z)-enofuranuronate (33)
A solution of 31 (17 mg, 0.056 mmol) in TFA/H2O (9∶1, 3 mL) was stirred for 45 min at 0 °C and was evaporated and coevaporated [toluene (3×), CH3CN (2×)]. The residue was dissolved in H2O and the aqueous layer was extracted with ether (2×). The water layer was evaporated to give 33 (9 mg, 61%; α/β, 1∶1): 1H NMR (MeOH-d4) δ 1.81-1.94 (m, 2, H8/8′), 2.27-2.38 (m, 2, H7/7′), 2.38-2.45 (m, 2, H9/9′), 3.67 (m, 3, CH3), 3.92 (dd, J3-4 = 3.8 Hz, J3-2 = 1.8 Hz, 0.5, H3), 3.97 (dd, J3-4 = 3.9 Hz, J3-2 = 2.6 Hz, 0.5, H3), 4.00-4.04 (m, 1, H2), 4.91 (d, J4-5 = 8.9 Hz, 0.5, H4), 4.99 (d, J4-5 = 9.1 Hz, 0.5, H4), 5.04-5.12 (m, 1, H5), 5.08 (s, 0.5, H1β), 5.37 (d, J1-2 = 4.0 Hz, 0.5, H1α); 19F NMR δ −106.29 (dt, JF-H5 = 37.2 Hz, JF-H7 = 17.6 Hz, 0.5F), −106.87 (dt, JF-H5 = 37.8 Hz, JF-H7 = 18.2 Hz, 0.5F). HRMS (AP-ESI) m/z calcd for C11H18FO6 (MH+) 265.1082; found 265.1088.
Methyl 5,6,7,8,9-Pentadeoxy-6-fluoro-α/β-D-ribo-dec-5(Z)-enofuranuronate (34)
A solution of 32 (12 mg, 0.04 mmol) in TFA/H2O (9∶1, 3 mL) was stirred for 30 min at 0 °C and was evaporated and coevaporated [toluene (3×)]. The residue was dissolved in H2O and the aqueous layer was extracted with ether (2×). The water layer was evaporated to give 34 (8 mg, 76%; α/β, 3∶7): 1H NMR (D2O) δ 1.72-1.76 (m, 2, H8/8′), 2.20 (dt, J7-F = 18.1 Hz, J7-8/8′ = 8.4 Hz, 2, H7/7′), 2.31-2.38 (m, 2, H9/9′), 3.59 (d, J2-3 = 2.4 Hz, 0.7, H2), 3.82-3.85 (m, 0.3, H2), 3.90-3.93 (s, 0.7, H3), 3.90-4.06 (m, 4.3, H3/H4/CH3), 4.64-4.77 (m, 1, H5), 5.12 (s, 0.7, H1), 5.28 (d, J1-2 = 3.7 Hz, 0.3, H1); 19F NMR δ −104.06 (dt, JF-H5 = 36.4 Hz, JF-H7/7′ = 17.8 Hz, 0.3, α); −105.04 (dt, JF-H5 = 35.8 Hz, JF-H7/7′ = 18.8 Hz, 0.7F, β). HRMS (AP-ESI) m/z calcd for C11H18FO6 (MH+) 265.1082; found 265.1090.
(E/Z)-3,5,6-Trideoxy-6-fluoro-1,2-O-isopropylidene-6-phenylsulfonyl-α-D-erythro-hex-5-enofuranose (35)
Step (a). Treatment of diacetone 3-deoxyglucose22 (204 mg, 0.83 mmol) with H5IO6 (228 mg, 1.00 mmol), as described for 10 (Step a, except no aqueous workup was performed) gave 3-deoxy-1,2-O-isopropylidene-α-D-erythro-pentdialdo-1,4-furanose [~85% pure; 1H NMR δ 9.75 (d, J5-4 = 4.8 Hz, H5)] which was directly used in the next step. Step (b) Treatment of the crude aldehyde with diethyl fluoro(phenylsulfonyl)methylphosphonate20 (297 mg, 0.96 mmol) and LHMDS (0.96 mL, 0.96 mmol), as described for 20, gave 35 (150 mg, 48%; E/Z, 2∶1) as an inseparable mixture of isomers. Compound (E)-35 had: 1H NMR δ 1.30 & 1.47 (2 × s, 2 × 3, 2 × CH3), 1.71 (ddd, J3-4 = 10.9 Hz, J3-3′ = 15.5 Hz, J3-2 = 4.6 Hz, 1, H3), 2.28 (dd, J3′-4 = 4.5 Hz, J3′-3 = 13.4 Hz, 1, H3′), 4.77 (t, J2-1/3 = 4.0 Hz, 1, H2), 4.93-4.97 (m, 1, H4), 5.85 (d, J1-2 = 3.6 Hz, 1, H1), 6.31 (dd, J5-F = 32.4 Hz, J5-4 = 7.5 Hz, 1, H5), 7.56-7.97 (m, 5, Ar); 13C NMR δ 26.42 & 27.00 (CMe2), 39.25 (d, 4J3-F = 2.0 Hz, C3), 71.26 (d, 3J4-F = 2.4 Hz, C4), 80.69 (C2), 105.90 (C1), 112.01 (CMe2), 116.77 (d, 2J5-F = 3.7 Hz, C5), 129.17 (Ph), 129.91 (ph), 135.08 (Ph), 137.22 (Ph), 155.54 (d, 1J6-F = 301.9 Hz, C6); 19F NMR δ −122.72 (d, JF-H5 = 32.5 Hz, 0.66). Compound (Z)-35 had: 1H NMR δ 1.34 & 1.60 (2 × s, 2 × 3, 2 × CH3), 1.68 (ddd, J3-4 = 10.5 Hz, J3-3′ = 15.2 Hz, J3-2 = 4.7 Hz, 1, H3), 2.46 (ddd, J3′-4 = 4.6 Hz, J3′-3 = 13.2 Hz, 1, H3′), 4.79 (t, J2-1/3 = 3.9 Hz, 1, H2), 5.71 (ddd, J4-5 = 8.7 Hz, J4-3 = 10.6 Hz, J4-3′ = 4.5 Hz, 1, H4), 5.84-5.85 (m, 1, H1), 5.86 (dd, J5-F = 20.1 Hz, J5-4 = 8.6 Hz, H5), 7.56-7.97 (m, 5, Ar); 19F NMR δ −114.04 (d, JF-H5 = 20.0 Hz, 0.33); MS m/z 329 (100, MH+).
(E/Z)-3,5,6-Trideoxy-6-fluoro-1,2-O-isopropylidene-6-tributylstannyl-α-D-erythro-hex-5-enofuranose (38)
Treatment of 35 (128 mg, 0.39 mmol) with Bu3SnH (0.21 mL, 228 mg, 0.78 mmol,) and AIBN (481 mg, 0.29 mmol), as described for 23, gave 38 (83 mg, 44%; E/Z, 1∶1): 19F NMR δ −92.83 (d, JF-H5 = 33.9 Hz, 84% of 0.50F, Z), −92.83 (ddd, JF-Sn = 230.4 Hz, JF-H5 = 34.7 Hz, JF-H4 = 5.2 Hz 16% of 0.50F, Z), −96.75 (d, JF-H5 = 52.7 Hz, 84% of 0.50F, E), −96.75 (ddd, JF-Sn = 221.1 Hz, JF-H5 = 52.7 Hz, JF-H4 = 4.9 Hz, 16% of 0.50F, E); MS m/z 479 (100, MH+, 120Sn), 477 (73, MH+, 118Sn), 475 (48, MH+, 116Sn). Compound (E)-38 had: 1H NMR δ 0.98-1.70 (m, 34, 3 × Bu/2 × CH3/H3), 2.26 (dd, J3′-4 = 4.3 Hz, J3′-3 = 13.4 Hz, 1, H3), 4.45-4.55 (m, 1, H4), 4.75 (t, J2-1/3 = 4.2 Hz, 1, H2), 4.96 (dd, J5-F = 52.9 Hz, J5-4 = 7.5 Hz, 1, H5), 5.83 (d, J1-2 = 3.8 Hz, 1, H1); 13C NMR δ 10.31 (Bu), 11.00 (Bu), 13.97 (Bu), 27.04 & 27.08 (CMe2), 27.50 (Bu), 40.19 (d, 2J3-F = 1.6 Hz, C3), 71.25 (d, 3J4-F = 17.3 Hz, C4), 81.06 (C2), 105.47 (C1), 111.41 (CMe2), 123.42 (d, 2J5-F = 3.7 Hz, C5), 174.29 (d, 1J5-F = 323.5 Hz, C6). Compound (Z)-38 had: 1H NMR δ 0.98-1.70 (m, 34, 3 × Bu/2 × CH3/H3), 2.11 (dd, J3′-4 = 4.3 Hz, J3′-3 = 13.4 Hz, 1, H3′), 4.73 (t, J2-1/3 = 4.2 Hz, 1, H2), 5.21 (ddd, J4-5 = 7.5 Hz, J4-3 = 4.4 Hz, J4-3′ = 15.2 Hz, 1, H4), 5.81 (d, J1-2 = 3.7 Hz, 1, H1), 5.84 (dd, J5-F = 34.2 Hz, J5-4 = 9.2 Hz, 1, H5); 13C NMR δ 10.31 (Bu), 11.00 (Bu), 13.97 (Bu), 27.04 & 27.08 (CMe2), 27.50 (Bu), 41.00 (d, 2J3-F = 1.8 Hz, C3), 74.73 (d, 3J4-F = 21.7 Hz, C4), 80.83 (C2), 105.69 (C1), 111.17 (CMe2), 123.26 (d, 2J5-F = 8.1 Hz, C5), 177.16 (d, 1J6-F = 316.5 Hz, C6).
(E/Z)-5,6-Dideoxy-6-fluoro-1,2-O-isopropylidene-α-D-xylo-hex-5-enofuranose (39)
Step (a). Compound 23 (200 mg, 0.34 mmol; E/Z, 7∶3) was dissolved in saturated NH3/MeOH (20 mL) and the resulting solution was stirred overnight at ambient temperature. The volatiles were evaporated to give 36 in quantitative yield of sufficient purity to use in the subsequent reaction. Step (b). Compound 36 (crude from step a, 0.34 mmol) was dissolved in NH3/MeOH (20 mL) and the resulting mixture was heated in a pressure Ace tube at 65 °C for 18 h. The volatiles were evaporated and the residue was column chromatographed (hexanes/EtOAc, 8∶2 → 3∶7) to give (E)-39 (20 mg, 29% from 23) and (Z)-39 (33 mg, 48% from 23). Compound (E)-39 had: 1H NMR δ 1.35 & 1.58 (2 × s, 2 × 3, 2 × CH3), 1.78 (br s, 1, OH3), 4.32 (d, J3-4 = 2.5 Hz, 1, H3), 4.59 (d, J2-1 = 3.7 Hz, 1, H2), 4.69 ("dm", J4-5 = 7.0 Hz, 1, H4), 5.53 (ddd, J5-F = 18.1 Hz, J5-6 = 11.2 Hz, J5-4 = 7.1 Hz, 1, H5), 5.94 (d, J1-2 = 3.7 Hz, 1, H1), 6.86 (ddd, J6-F = 82.9 Hz, J6-5 = 11.2 Hz, J6-4 = 1.0 Hz, 1, H6); 13C NMR δ 26.47 & 27.06 (CMe2), 76.48 (d, 4J3-F = 2.0 Hz, C3), 76.80 (d, 3J4-F = 12.6 Hz, C4), 85.31 (C2), 104.87 (C1), 106.20 (d, 2J5-F = 13.7 Hz, C5), 112.25 (CMe2), 153.79 (d, 1J6-F = 262.6 Hz, C6); 19F NMR δ −122.18 (dd, JF-H5 = 17.8 Hz, JF-H6 = 82.9 Hz). Compound (Z)-39 had: 1.35 & 1.54 (2 × s, 2 × 3, 2 × CH3), 1.81 (br s, 1, OH3), 4.22 (br s, 1, H3), 4.58 (d, J2-1 = 3.7 Hz, 1, H2), 5.07 (ddd, J5-F = 40.1 Hz, J5-6 = 4.9 Hz, J5-4 = 7.5 Hz, 1, H5), 5.12-5.15 (m, 1, H4), 5.96 (d, J1-2 = 3.7 Hz, 1, H1), 6.63 (dd, J6-F = 82.7 Hz, J6-5 = 4.8 Hz, J6-4 = 1.2 Hz, 1, H6); 13C NMR δ 26.57 & 27.10 (CMe2), 74.49 (d, 3J4-F = 5.1 Hz, C4), 76.74 (d, 4J3-F = 1.9 Hz, C3), 85.47 (C2), 104.80 (C1), 106.24 (C5), 112.24 (CMe2), 150.20 (d, 1J6-F = 265.2 Hz, C6); 19F NMR δ −121.02 (dd, JF-H5 = 41.1 Hz, JF-H6 = 83.3 Hz). MS (APCI+) m/z 205 (100, MH+). Anal. Calcd for C9H13FO4 (204.19): C, 52.94; H, 6.42. Found: C, 53.19; H, 6.63.
(E/Z)-5,6-Dideoxy-6-fluoro-1,2-O-isopropylidene-α-D-ribo-hex-5-enofuranose (40)
Step (a). Compound 24 (200 mg, 0.34 mmol; E/Z, 1∶1) was dissolved in NH3/MeOH (20 mL) and stirred overnight at ambient temperature. The volatiles were evaporated to give 37 in quanitative yield of sufficient purity to use in the subsequent step. Step (b). Treatment of 37 (crude, 0.34 mmol) with NH3/MeOH (20 mL) at 65 °C, as described for 39, gave unchanged 37 (17 mg, 10% from 24; E/Z, 2∶3) and 40 as separable isomers (E; 22 mg, 32% from 24) and (Z; 18 mg, 26% from 24). Compound (E)-40 had: 1H NMR δ 1.40 & 1.60 (2 × s, 2 × 3, 2 × CH3), 2.37 (d, JOH3-3 = 10.0 Hz, 1, OH3), 3.70-3.73 (m, 1, H3), 4.12 (t, J4-5/3 = 8.3 Hz, 1, H4), 4.60 (t, J2-1/3 = 4.6 Hz, 1, H2), 5.53 (ddd, J5-F = 17.1 Hz, J5-6 = 11.2 Hz, J5-4 = 7.8 Hz, 1, H5), 5.84 (d, J1-2 = 3.9 Hz, 1, H1), 6.82 (ddd, J6-F = 82.4 Hz, J6-5 = 11.1 Hz, J6-4 = 0.7 Hz, 1, H6); 13C NMR δ 26.75 & 26.84 (CMe2), 76.55 (d, 3J4-F = 17.0 Hz, C4), 76.49 (C3), 78.57 (C2), 104.08 (C1), 109.44 (d, 2J5-F = 12.9 Hz, C5), 113.11 (CMe2), 152.81 (d, 1J6-F = 262.1 Hz, C6); 19F NMR δ −123.67 (dd, JF-H5 = 17.1 Hz, JF-H6 = 82.5 Hz). Compound (Z)-40 had: 1.40 & 1.60 (2 × s, 2 × 3, 2 × CH3), 2.39 (d, JOH3-3 = 11.0 Hz, 1, OH3), 3.74 (ddd, J3-OH3 = 10.9 Hz, J3-4 = 8.9 Hz, J3-2 = 5.1 Hz, 1, H3), 4.60 (t, J2-1/3 = 4.5 Hz, 1, H2), 4.70 (t, J4-3/5 = 8.8 Hz, 1, H4), 4.89 (ddd, J5-F = 40.0 Hz, J5-6 = 4.9 Hz, J5-4 = 8.9 Hz, 1, H5), 5.84 (d, J1-2 = 3.9 Hz, 1, H1), 6.69 (ddd, J6-F = 82.7 Hz, J6-5 = 4.9 Hz, J6-4 = 0.8 Hz, 1, H6); 13C NMR δ 26.79 & 26.92 (CMe2), 73.18 (d, 3J4-F = 5.1 Hz, C4), 76.74 (d, 4J3-F = 2.0 Hz, C3), 78.58 (C2), 104.20 (C1), 108.69 (2J5-F = 1.9 Hz, C5), 113.17 (CMe2), 153.71 (d, 1J6-F = 265.8Hz, C6); 19F NMR δ −123.90 (dd, JF-H5 = 40.1 Hz, JF-H6 = 82.6 Hz): MS (APCI+) m/z 205 (100, MH+). Anal. Calcd for C9H13FO4 (204.19): C, 52.94; H, 6.42. Found: C, 53.07; H, 6.67.
(E/Z)-3,5,6-Trideoxy-6-fluoro-1,2-O-isopropylidene-α-D-erythro-hex-5-enofuranose (41)
Treatment of 38 (100 mg, 0.21 mmol; E/Z, 1∶1) with NH3/MeOH (15 mL) and CsF (51 mg, 0.33 mmol) at 65 °C for 4 h, as described for 39 (Step b), gave 41 (16 mg, 40%; E/Z, ~45∶55): 19F NMR δ −124.79 (dd, JF-H5 = 41.8 Hz, JF-H6 = 83.0 Hz, 0.55F), −125.95 (dd, JF-H5 = 16.7 Hz, JF-H6 = 82.9 Hz, 0.45F); MS (APCI+) m/z 189 (100, MH+). Compound (E)-41 had: 1H NMR δ 1.35 & 1.55 (2 × s, 2 × 3, 2 × CH3), 1.55-1.68 (m, 1, H3), 2.20 (dd, J3′-3 = 13.5 Hz, J3′-4 = 4.3 Hz, 1, H3′), 4.62 (ddd, J4-3 = 11.5 Hz, J4-5 = 8.2 Hz, J4-3′ = 4.3 Hz, 1, H4), 4.75-4.79 (m, 1, H2), 5.42 (ddd, J5-F = 16.8 Hz, J5-6 = 11.2 Hz, J5-4 = 8.3 Hz, 1, H5), 5.82-5.84 (m, 1, H1), 6.80 (dd, J6-F = 82.7 Hz, J6-5 = 11.2 Hz, 1, H6). Compound (Z)-41 had: 1.28 & 1.57 (2 × s, 2 × 3, 2 × CH3), 1.55-1.68 (m, 1, H3), 2.28 (dd, J3′-3 = 13.5 Hz, J3′-4 = 4.3 Hz, 1, H3′), 4.75-4.79 (m, 1, H2), 4.92 (ddd, J5-F = 41.3 Hz, J5-4 = 8.1 Hz, J5-6 = 4.8 Hz, 1, H5), 5.13 (ddd, J4-3 = 11.5 Hz, J4-5 = 8.0 Hz, J4-3' = 4.1 Hz, 1, H4), 5.82-5.84 (m, 1, H1), 6.53 (dd, J6-F = 82.8 Hz, J6-5 = 4.8 Hz, 1, H6).
(E)-5,6-Dideoxy-6-fluoro-α/β-D-xylo-hex-5-enofuranose (42)
A solution of (E)-39 (13 mg, 0.064 mmol) in TFA/H2O (9∶1; 3 mL) was stirred for 50 min at 0 °C (ice bath). The volatiles were evaporated, coevaporated [toluene (3×) and MeCN (2×)], and the residue was flash column chromatographed (50 → 95% EtOAc/hexanes) to give 42 (4 mg, 38%; α/β, 1∶1): 1H NMR (MeOH-d4) δ 3.91-4.03 (m, 2 H2/3), 4.56-4.63 (m, 1, H4), 5.07 (s, 0.5, H1β), 5.37 (d, J1-2 = 4.0 Hz, 0.5, H1α), 5.51 (ddd, J5-F = 17.8 Hz, J5-6 = 11.2 Hz, J5-4 = 8.9 Hz, 0.5, H5), 5.65 (ddd, J5-F = 17.9 Hz, J5-6 = 11.1 Hz, J5-4 = 8.9 Hz, 0.5, H5), 6.87 (dd, J6-F = 84.0 Hz, J6-5 = 11.0 Hz, 0.5, H6), 6.90 (dd, J6-F = 83.7 Hz, J6-5 = 11.0 Hz, 0.5, H6); 13C NMR (MeOH-d4) δ 75.29 (d, 3J4-F = 13.8 Hz, C4), 77.00 & 77.20 (C3), 77.73 (C2), 78.17 (d, 3J4-F = 13.7 Hz, C4), 81.26 (C2), 96.72 (C1α), 103.17 (C1β), 108.71 (d, 2J5-F = 12.0 Hz, C5), 109.32 (d, 2J5-F = 11.7 Hz, C5), 152.14 (d, 1J6-F = 258.7 Hz, C6), 152.24 (d, 1J6-F = 259.2 Hz, C6); 19F NMR (MeOH-d4) δ - 126.55 (dd, JF-H5 = 17.8 Hz, JF-6 = 83.9 Hz, 0.5F), −126.85 (dd, JF-H5 = 18.1 Hz, JF-H6 = 84.0 Hz, 0.5F); MS (APCI−) m/z 163 (100, MH−).
Analogous treatment of 39 (E/Z, 1∶1; 20 mg, 0.040 mmol) gave 42 (5 mg, 76%) as a mixture (E/Z, ~1∶1; α/β, ~1∶1). Compound (E/Z)-37 had: 19F NMR (MeOH-d4) δ −126.55 (dd, JF-H5 = 17.3 Hz, JF-H6 = 84.0 Hz; E, 0.25, β), −126.85 (dd, 3JF-H5 = 17.5 Hz, 2JF-H6 = 84.0 Hz; E, 0.25, β), −127.66 (dd, JF-H5 = 41.5 Hz, JF-H6 = 84.9 Hz; Z, 0.25, β), −128.34 (dd, JF-H5 = 42.2 Hz, JF-H6 = 84.8 Hz; Z, 0.25, α); MS(APCI−) m/z 163 (100, MH−).
Treatment of the crude 36 [from step (a) for the preparation of 39] with TFA/H2O (1 h, 0 °C) followed by evaporation, coevaporation [toluene (2×) and MeCN (1×)], partition (H2O/ethyl ether), and evaporation of the aqueous layer also gave 42 (55% from 23, α/β, 1∶1).
(E)-5,6-Dideoxy-6-fluoro-α/β-D-ribo-hex-5-enofuranose (43)
Treatment of (E)-40 (13 mg, 0.064 mmol) with TFA/H2O (9∶1, 3 mL), as described for 42, gave 43 (7 mg, 67%; α/β, 1∶4): 1H NMR (MeOH-d4) δ 3.77 (t, J3-2/4 = 6.1 Hz, 0.2, H3), 3.87 (d, J2-1 = 4.5 Hz, 0.8, H2), 4.01-4.05 (m, 1, H2α & H3β), 4.20 (t, J4-3/5 = 8.0 Hz, 0.8, H4), 4.30 (dd, J4-5 = 8.2 Hz, J4-3 = 6.4 Hz, 0.2, H4), 5.12 (br s, 0.8, H1), 5.28 (d, J1-2 = 4.1 Hz, 0.2, H1), 5.42 (ddd, J5-F = 17.5 Hz, J5-6 = 11.1 Hz, J5-4 = 8.3 Hz, 0.2, H5), 5.49 (ddd, J5-F = 17.6 Hz, J5-6 = 11.1 Hz, J5-4 = 8.5 Hz, 0.8, H5), 6.86 (dd, J6-F = 83.9 Hz, J6-5 = 11.1 Hz, 0.8, H6), 6.87 (dd, J6-F = 83.7 Hz, J6-5 = 11.0 Hz, 0.2, H6); 13C NMR (MeOH-d4) δ 70.90 (C2α), 75.32 (d, 4J3-F = 2.6 Hz, C3α), 75.5 (d, 4J3-F = 2.5 Hz, C3β), 76.13 (C2β), 77.55 (d, 3J4-F = 13.6 Hz, C4α), 77.88 (d, 3J4-F = 13.7 Hz, C4β), 96.63 (C1α), 102.09 (C1β), 111.19 (d, 2J5-F = 11.4 Hz, C5α), 112.65 (d, 2J5-F = 10.6 Hz, C5β), 151.98 (d, 1J6-F = 258.6 Hz, C6β), 152.17 (d, 1J6-F = 258.7 Hz, C6α); 19F NMR (MeOH-d4) δ −128.55 (dd, JF-H5 = 17.4 Hz, JF-H6 = 83.5 Hz, 0.2F, α), −129.00 (dd, JF-H5 = 17.3 Hz, JF-H6 = 83.7 Hz, 0.8F, β); MS (APCI−) m/z 163 (100, MH−).
Analogous treatment of 40 (E/Z, 1∶1; 16 mg, 0.032 mmol) gave 43 (3 mg, 57%) as a mixture (E/Z, ~3∶1; α/β, ~1∶4 for E isomer and α/β, ~1∶15 for Z isomer): 19F NMR (MeOH-d4) δ −128.01 (dd, JF-H5 = 41.3 Hz, JF-H6 = 83.4 Hz; Z, 0.02F, α) −128.55 (dd, JF-H5 = 17.6 Hz, JF-H6 = 84.2 Hz; E, 0.14F, α), −129.00 (dd, JF-H5 = 17.5 Hz, JF-H6 = 83.8 Hz; E, 0.60F, β), −129.69 (dd, JF-H5 = 40.8 Hz, JF-H6 = 84.4 Hz; Z, 0.24F, β); MS(APCI−) m/z 163 (100, MH−).
Treatment of the crude 37 [from step (a) for the preparation of 40] with TFA/H2O (1 h, 0 °C) followed by evaporation, coevaporation [toluene (2×) and MeCN (1×)], partition (H2O/ethyl ether), and evaporation of the aqueous layer also gave 43 (45% from 24, α/β, 1∶3).
(E/Z)-3,5,6-Trideoxy-6-fluoro-α-D-erythro-hex-5-enofuranose (44)
Treatment of 38 (62 mg, 0.13 mmol; E/Z, 3∶2) with TFA/H2O (9∶1, 1mL; 1 h, 0 °C) followed by evaporation, coevaporation [toluene (2×) and MeCN (1×)], partition (H2O/ethyl ether), and evaporation of the aqueous layer gave 44 (10 mg, 52%; E/Z ~1∶3, α/β, ~1∶4): 1H NMR (D2O) δ 1.85-2.10 (m, 2, H3,3'), 4.05-4.25 (m, 1, H2), 4.58-4.75 (m, 1, H4), 4.86 (ddd, J5-F = 41.7 Hz, J5-4 = 8.9 Hz, J5-6 = 4.7, Hz, 0.15, H5), 4.92 (ddd, J5-F = 41.6 Hz, J5-4 = 8.9 Hz, J5-6 = 4.7 Hz, 0.6, H5), 5.12 (s, 0.15, H1β), 5.13 (s, 0.6, H1β), 5.24 (d, J1-2 = 3.5 Hz, 0.05, H1α), 5.25 (d, J1-2 = 3.8 Hz, 0.2, H1α), 5.39 (ddd, J5-F = 17.4 Hz, J5-6 = 11.2 Hz, J5-4 = 9.3 Hz, 0.2, H5), 5.45 (ddd, J5-F = 17.6 Hz, J5-6 = 11.1 Hz, J5-4 = 9.1 Hz, 0.05, H5), 6.52 (ddd, J6-F = 83.7 Hz, J6-5 = 4.7 Hz, J6-4 = 1.0 Hz, 0.15, H6), 6.55 (dd, J6-F = 83.7 Hz, J6-5 = 4.7 Hz, J6-4 = 1.0, 0.60, H6), 6.77 (dd, J6-F = 83.9 Hz, J6-5 = 10.7 Hz, 0.05, H6), 6.78 (dd, J6-F = 83.9 Hz, J6-5 = 11.0 Hz, 0.20, H6); 19F NMR (D2O) δ −126.35 (dd, JF-H5 = 17.3 Hz, JF-H6 = 83.6 Hz; E, 0.20F, β), −126.45 (dd, JF-H5 = 17.2 Hz, JF-H6 = 82.8 Hz; E, 0.05F, α), −126.81 (dd, JF-H5 = 42.0 Hz, JF-H6 = 83.7 Hz; Z, 0.15F, α), −127.55 (dd, JF-H5 = 42.0 Hz, JF-H6 = 83.8 Hz; Z, 0.60F, β); HRMS (LCT-ESI) m/z: calcd for C6H9FO3 [M + Na]+ 171.0433; found 171.0434.
Analogous treatment of 41 (10 mg, 0.053 mmol; E/Z, ~45∶55) with TFA/H2O gave 44 (5 mg, 64%; E/Z ~1∶2, α/β ~1∶3).
Enzymatic assay
Inhibition assays were performed in a buffer containing 50 mM HEPES (pH 7.0), 150 mM NaCl, 150 µM 5,5′-dithio-bis-(2-nitrobenzoic acid),23 and various concentrations of SRH (0–55 µM) and inhibitors (0–1 mM). The reactions were initiated by the addition of Co2+-substituted LuxS from Bacillus subtilis (final concentration 0.4–0.5 µM) and monitored continuously at 412 nm (ε = 14150 M−1 cm−1) in a Perkin-Elmer λ25 UV-VIS spectrophotometer at room temperature. The initial rates recorded from the early regions of the progress curves were fitted into the Lineweaver-Burk equation 1/V = KM'/(kcat [E]0) × 1/[S] +1/(kcat [E]0) and the Michaelis-Menten equation V = kcat [E]0 [S]/(KM' + [S]) using KaleidaGraph 3.5 to determine the inhibition pattern. KI values were calculated from the equation KM' = KM × (1 + [I]/KI), where KM = 2.2 µM.
Acknowledgements
This work was partially supported by grants from the National Institutes of Health (S06GM08205 and R01AI062901). C.A.G and J.R were sponsored by MBRS RISE program (NIH/NIGMS; R25GM61347). C.A.G is also thankful to R. E. McNair Program for summer support. The support of US Army Research Office (W911NF-04-1-0022) in the purchase of 600 MHz NMR spectrometer is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.(a) Yuan C-S, Liu S, Wnuk SF, Robins MJ, Borchardt RT. In: Advances in Antiviral Drug Design. De Clercq E, editor. Vol. 2. Greenwich: JAI Press; 1996. pp. 41–88. [Google Scholar]; (b) Turner MA, Yang X, Yin D, Kuczera K, Borchardt RT, Howell PL. Cell Biochem. Biophys. 2000;33:101. doi: 10.1385/CBB:33:2:101. [DOI] [PubMed] [Google Scholar]; (c) Wnuk SF. Mini-Rev. Med. Chem. 2001;1:307. doi: 10.2174/1389557013406918. [DOI] [PubMed] [Google Scholar]
- 2.(a) Schneyder G, Roffi M, Pin R, Flammer Y, Lange H, Eberli FR, Meier B, Turi ZG, Hess OM. N. Engl. J. Med. 2001;345:1593. doi: 10.1056/NEJMoa011364. [DOI] [PubMed] [Google Scholar]; (b) Langheinrich AC, Braun-Dullaeus RC, Walker G, Jeide I, Schiling R, Tammoscheit K, Dreyer T, Fink L, Bohle RM, Haberbosch W. Atherosclerosis. 2003;171:181. doi: 10.1016/j.atherosclerosis.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 3.(a) Lee JE, Cornel KA, Riscoe, Howell PL. Structure. 2001;9:941. doi: 10.1016/s0969-2126(01)00656-6. [DOI] [PubMed] [Google Scholar]; (b) Lee JE, Cornel KA, Riscoe MK, Howell PL. J. Biol. Chem. 2003;278:8761. doi: 10.1074/jbc.M210836200. [DOI] [PubMed] [Google Scholar]
- 4.(a) Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM. Nature. 2002;415:545. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]; (b) Zhu J, Hu X, Dizin E, Pei D. J. Am. Chem. Soc. 2003;125:13379. doi: 10.1021/ja0369663. [DOI] [PubMed] [Google Scholar]; (c) Zhu J, Dizin E, Hu X, Wavreille AS, Park J, Pei D. Biochemistry. 2003;42:4717. doi: 10.1021/bi034289j. [DOI] [PubMed] [Google Scholar]; (d) Zhu J, Patel R, Pei D. Biochemistry. 2004;43:10166. doi: 10.1021/bi0491088. [DOI] [PubMed] [Google Scholar]; (e) Alfaro JF, Zhang T, Wynn DP, Karschner EL, Zhou ZS. Org. Lett. 2004;6:3043. doi: 10.1021/ol049182i. [DOI] [PubMed] [Google Scholar]; (f) Pei D, Zhu J. Curr. Opin. Chem. Biol. 2004;8:492. doi: 10.1016/j.cbpa.2004.08.003. [DOI] [PubMed] [Google Scholar]; (g) Rajan R, Zhu J, Hu X, Pei D, Bell CE. Biochemistry. 2005;44:3745. doi: 10.1021/bi0477384. [DOI] [PubMed] [Google Scholar]; (h) Shen G, Rajan R, Zhu J, Bell CE, Pei D. J. Med. Chem. 2006;49:3003. doi: 10.1021/jm060047g. [DOI] [PubMed] [Google Scholar]
- 5.(a) Meijler MM, Hom LG, Kaufmann GF, McKenzie KM, Sun C, Moss JA, Matsushita M, Janda KD. Angew. Chem. Int. Ed. 2004;43:2106. doi: 10.1002/anie.200353150. [DOI] [PubMed] [Google Scholar]; (b) Semmelhack MF, Campagna SR, Federle MJ, Bassler BL. Org. Lett. 2005;7:569. doi: 10.1021/ol047695j. [DOI] [PubMed] [Google Scholar]; (c) De Keersmaecker SCJ, Varszegi C, van Boxel N, Habel LW, Metzger K, Daniels R, Marchal K, De Vos D, Vanderleyden J. J. Biol. Chem. 2005;280:19563. doi: 10.1074/jbc.M412660200. [DOI] [PubMed] [Google Scholar]; (d) Frezza M, Soulere L, Balestrino D, Gohar M, Deshayes C, Queneau Y, Forestier C, Doutheau A. Bioorg. Med. Chem. Lett. 2007;17:1428. doi: 10.1016/j.bmcl.2006.11.076. [DOI] [PubMed] [Google Scholar]
- 6.(a) Waters CM, Bassler BL. Annu. Rev. Cell Dev. Biol. 2005;21:319. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]; (b) Bassler BL, Losick R. Cell. 2006;125:237. doi: 10.1016/j.cell.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 7.(a) Geske GD, Wezeman RJ, Siegel AP, Blackwell HE. J. Am. Chem. Soc. 2005;127:12762. doi: 10.1021/ja0530321. [DOI] [PubMed] [Google Scholar]; (b) Pomianek ME, Semmelhack MF. ACS Chem. Biol. 2007;2:293. doi: 10.1021/cb700098c. [DOI] [PubMed] [Google Scholar]; (c) Higgins DA, Pomianek ME, Kraml CM, Taylor RK, Semmelhack MF, Bassler BL. Nature. 2007;450:883. doi: 10.1038/nature06284. [DOI] [PubMed] [Google Scholar]
- 8.(a) Wnuk SF, Yuan C-S, Borchardt RT, Balzarini J, De Clercq E, Robins MJ. J. Med. Chem. 1994;37:3579. doi: 10.1021/jm00047a015. [DOI] [PubMed] [Google Scholar]; (b) Yuan C-S, Wnuk SF, Robins MJ, Borchardt RT. J. Biol. Chem. 1998;273:18191. doi: 10.1074/jbc.273.29.18191. [DOI] [PubMed] [Google Scholar]; (c) Wnukx SF, Mao Y, Yuan CS, Borchardt RT, Andrei J, Balzarini J, DeClercq E, Robins MJ. J. Med. Chem. 1998;41:3078. doi: 10.1021/jm9801410. [DOI] [PubMed] [Google Scholar]
- 9.(a) Xie M, Berges DA, Robins MJJ. Org. Chem. 1996;61:5178. [Google Scholar]; (b) Maryanoff BE, Reitz AB. Chem. Rev. 1989;89:863. [Google Scholar]
- 10.Analogous bromination-dehydrobromination strategy applied to the adenosine derivatives bearing a C5′–C6′ double bond produced a single (5′-bromo)vinyl SAH analogue: Andrei D, Wnuk SF. Org. Lett. 2006;8:5093. doi: 10.1021/ol062026m.
- 11.(a) Although Pd-catalyzed cross-coupling reactions are powerful methods for the formation of new carbon-carbon bonds,11b monocross-coupling reactions of 1,1-dihalovinyl electrophiles with Csp2 and Csp nucleophiles are less common12 and monocouplings between 1,1-dihalovinyl electrophiles and Csp3 nucleophiles are scarce.13 de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Weinheim: Wiley-VCH; 2004.
- 12.Negishi E-I, Hu Q, Huang Z, Qian M, Wang G. Aldrichim. Acta. 2005;38:71.; references cited therein.
- 13.For successful Pd-catalyzed monoalkylation of vinyl dihalides via trans-selective monobutylation of 1,1-dichloro-2-phenylethene with n-C4H9ZnCl in 81% yield see: Minato A, Suzuki K, Tamao K. J. Am. Chem. Soc. 1987;109:1257. For studies on differentiation of two chlorine atoms in stepwise double alkylation reactions of 1,1-dichloroalkenes see: Tan Z, Negishi E-i. Angew. Chem. Int. Ed. 2006;45:762. doi: 10.1002/anie.200503519.For Suzuki-Miyaura protocol to the selective monocoupling of 1,1-dichloroalkenes with 9-alkyl-9-BBN see: Liron F, Fosse C, Pernolet A, Roulland E. J. Org. Chem. 2007;72:2220. doi: 10.1021/jo061908w.
- 14.(a) Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;36:3769. [Google Scholar]; (b) Tronchet JMJ, Bonenfant AP, Perret F, Gonzalez A, Zumwald JB, Martinez EM, Baehler B. Helv. Chim. Acta. 1980;63:1181. [Google Scholar]
- 15.Robins MJ, Wnuk SF, Yang X, Yuan C-S, Borchardt RT, Balzarini J, De Clercq E. J. Med. Chem. 1998;41:3857. doi: 10.1021/jm980163m. [DOI] [PubMed] [Google Scholar]
- 16.Pd-catalyzed Negishi monoalkylation of 1-fluoro-1-(iodo, or bromo, or chloro)alkenes with alkylzincs give stereoselective access to the internal fluoroalkenes: Andrei D, Wnuk SF. J. Org. Chem. 2006;71:405. doi: 10.1021/jo051980e.
- 17.For Pd-catalyzed couplings of 1-fluoro-1-haloalkenes with Csp2 and Csp nucleophiles see: Chen C, Wilcoxen K, Huang CQ, Strack N, McCarthy JR. J. Fluorine Chem. 2000;101:285.Xu J, Burton DJ. Tetrahedron Lett. 2002;43:2877–2879.Xin Z, Burton DJ. J. Fluorine Chem. 2001;112:317.
- 18.(a) McCarthy JR, Huber EW, Le T-B, Laskovics M, Matthews DP. Tetrahedron. 1996;52:45. [Google Scholar]; (b) McCarthy JR, Matthews DP, Stemerick DM, Bey P, Lippert BJ, Snyder RD, Sunkara PS. J. Am. Chem. Soc. 1991;113:7439. [Google Scholar]
- 19.For other examples of synthetic sequences involving introduction of alkenes substituted with F/SO2Ph, F/SnBu3, or F/H into amino acids or nucleoside frames see: Berkowitz DB, De la Salud-Bea R, Jahng W-J. Org. Lett. 2004;6:1821. doi: 10.1021/ol049422u.Karukurichi KR, De la Salud-Bea R, Jahng W-J, Berkowitz DB. J. Am. Chem. Soc. 2007;129:258. doi: 10.1021/ja067240k.Pan Y, Calvert K, Silverman RB. Bioorg. Med. Chem. 2004;12:5719. doi: 10.1016/j.bmc.2004.07.065.Shen Y. J. Organomet. Chem. 2006;691:1452.Rapp M, Haubrich TA, Perrault J, Mackey ZB, McKerrow JH, Chiang PK, Wnuk SF. J. Med. Chem. 2006;49:2096. doi: 10.1021/jm0511379.
- 20.(a) Appel RB. Synth. Commun. 1995;25:3593. [Google Scholar]; (b) Wnuk SF, Bergolla LA, Garcia PI., Jr J. Org. Chem. 2002;67:3065. doi: 10.1021/jo0111560. [DOI] [PubMed] [Google Scholar]; (c) Wnuk SF, Garcia PI, Jr, Wang Z. Org. Lett. 2004;6:2047. doi: 10.1021/ol049312n. [DOI] [PubMed] [Google Scholar]
- 21.Wnuk SF, Lalama J, Robert J, Garmendia CA. Nucleosides Nucleotides Nucleic Acids. 2007;26:1051. doi: 10.1080/15257770701513190. [DOI] [PubMed] [Google Scholar]
- 22.Robins MJ, Wilson JS, Hansske F. J. Am. Chem. Soc. 1983;105:4059. [Google Scholar]
- 23.Ellman GL. Arch. Biochem. Biophys. 1959;82:70. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]