

Single crystals of recombinant S-adenosyl-l-homocysteine hydrolase from L. luteus in complex with adenosine diffract X-rays to 1.17 Å resolution at 100 K. The crystals are tetragonal, space group P43212, and contain one copy of the dimeric enzyme in the asymmetric unit.
Keywords: S-adenosyl-l-homocysteine hydrolase, Lupinus luteus
Abstract
By degrading S-adenosyl-l-homocysteine, which is a byproduct of S-adenosyl-l-methionine-dependent methylation reactions, S-adenosyl-l-homocysteine hydrolase (SAHase) acts as a regulator of cellular methylation processes. S-Adenosyl-l-homocysteine hydrolase from the leguminose plant yellow lupin (Lupinus luteus), LlSAHase, which is composed of 485 amino acids and has a molecular weight of 55 kDa, has been cloned, expressed in Escherichia coli and purified. Crystals of LlSAHase in complex with adenosine were obtained by the hanging-drop vapour-diffusion method using 20%(w/v) PEG 4000 and 10%(v/v) 2-propanol as precipitants in 0.1 M Tris–HCl buffer pH 8.0. The crystals were tetragonal, space group P43212, with unit-cell parameters a = 122.4, c = 126.5 Å and contained two protein molecules in the asymmetric unit, corresponding to the functional dimeric form of the enzyme. Atomic resolution (1.17 Å) X-ray diffraction data have been collected using synchrotron radiation.
1. Introduction
Methylation is one of the most ubiquitous reactions in cell metabolism. The most widely used methyl-group donor is S-adenosyl-l-methionine (SAM; Richards et al., 1978 ▶), which participates in the methylation of many biomolecules ranging from small compounds such as norepinephrine, catecholamines or phospholipids to macromolecules, including proteins, nucleic acids and polysaccharides. During methyl-group transfer, the byproduct S-adenosyl-l-homocysteine (SAH) is formed, which is a strong inhibitor of SAM-dependent methyltransferases. S-Adenosyl-l-homocysteine hydrolase (SAHase; EC 3.3.1.1) catalyzes the reversible breakdown of S-adenosyl-l-homocysteine to adenosine (Ado) and homocysteine (Hcy) (Cantoni & Scarano, 1954 ▶). By removing SAH, SAHase serves as an important regulator of SAM-dependent biological methylation reactions. The equilibrium of the SAHase reaction is shifted far in the direction of SAH synthesis. However, under physiological conditions the removal of Ado and Hcy is rapid and the net result is SAH hydrolysis (Richards et al., 1978 ▶).
All archaeal and some bacterial SAHase sequences are shorter than those from most eukarya and bacteria, which carry an insert of around 40 amino acids. This additional segment is present in all plant SAHases and in some other eukaryotic organisms, such as Entamoebidae, Trichomonadida and Apicomplexa, but is absent in fungal, insect and vertebrate sequences (Stępkowski et al., 2005 ▶).
SAHases are oligomeric enzymes that are typically active as homotetramers. Plant SAHases are exceptional in this respect and have been reported to function as homodimers (Guranowski & Pawełkiewicz, 1977 ▶). All S-adenosyl-l-homocysteine hydrolases require for their activity a tightly but noncovalently bound NAD+ cofactor, which must be present in each subunit.
The current knowledge of the structure of SAHase is based on crystallographic studies of the enzymes from man (Turner et al., 1998 ▶; PDB code 1a7a), rat (Hu et al., 1999 ▶; PDB code 1b3r) and the malaria parasite Plasmodium falciparum (Tanaka et al., 2004 ▶; PDB code 1v8b). The available structures show tetrameric enzymes, which are generally complexed with various inhibitors. The resolution limit of the published structures is 2.01 Å (Yang et al., 2003 ▶; PDB code 1li4).
Yellow lupin (Lupinus luteus) SAHase (LlSAHase) was first isolated and characterized by Guranowski & Pawełkiewicz (1977 ▶). Unfortunately, the isolated enzyme could not be crystallized. Subsequent studies showed the presence of at least two SAHase genes in L. luteus cDNA libraries (shh-1 and shh-2; GenBank codes AAD56048 and AAF70071) with 97% identity at the amino-acid level (Brzezinski et al., 2001 ▶). The presence of SAHase isoforms could explain the lack of homogeneity and the difficulties in crystallization using protein samples isolated from plant material.
The present paper describes the purification and crystallization of a recombinant plant SAHase corresponding to the sequence encoded by the L. luteus shh-1 gene. The crystals were obtained in the presence of adenosine, one of the products of the hydrolysis reaction catalyzed by the enzyme. The crystals diffracted X-rays to atomic resolution.
2. Methods
2.1. Cloning, expression and purification
The coding seqence of the shh-1 gene was amplified by PCR from an L. luteus cDNA library. The amplicon was cloned into the pET-15b expression vector (Novagen) using NdeI/BamHI restriction sites (Brzezinski et al., 2001 ▶). The recombinant plasmid was sequenced to confirm the correct sequence of the insert and fusion with an N-terminal His6 tag. The construct was used to transform the BL21-CodonPlus (DE3)-RIPL strain of Escherichia coli.
50 ml TB medium containing 34 µg ml−1 chloramphenicol and 100 µg ml−1 ampicillin was inoculated with a single colony and grown overnight at 310 K. The overnight culture was used for inoculation of 5 l TB medium with appropiate antibiotics and grown to an OD600 of 0.8. The temperature was decreased to 293 K and protein expression was induced by IPTG at a final concentration of 0.35 mM. The cells were harvested 15 h after induction.
The cell pellet was resuspended in buffer A (5 mM imidazole, 500 mM NaCl, 20 mM Tris–HCl pH 7.9, 5 mM β-mercaptoethanol) with the addition of 1 mM PMSF and 20 µg ml−1 lysozyme. Cells were disrupted by sonication on ice and centrifuged to remove cell debris. The supernatant was loaded onto a HiTrap column equilibrated with 0.05 M NiSO4. The protein was eluted in a 0.1–1 M gradient of imidazole in 500 mM NaCl, 20 mM Tris–HCl pH 7.9. SDS–PAGE confirmed the molecular weight of the expressed protein (about 55 kDa). The protein solution was dialyzed against buffer A and the protein concentration was estimated as 2.2 mg ml−1. 3 U thrombin (Novagen) was added per milligram of recombinant SAHase to remove the His tag. After 18 h incubation at 277 K, the reaction was stopped by the addition of 1 mM PMSF. The mixture was loaded onto a HiTrap column equilibrated with 0.05 M NiSO4 and the protein was eluted with buffer A, which was subsequently exchanged for buffer B (50 mM NaCl, 20 mM Tris–HCl pH 8.0, 2.5 mM DTT, 1 mM EDTA). The protein solution was concentrated to 4 mg ml−1 using Amicon Ultra 30 filters. The purified protein contains the LlSAHase sequence extended at the N-terminus by a short peptide (GSA–) which is an artifact of the cloning and purification procedure.
Next, a procedure for cofactor exchange was applied. Firstly, the apo form of LlSAHase was prepared by removal of the enzyme-bound NAD cofactor according to a modification of the procedure of Yuan et al. (1993 ▶). 8 mg recombinant LlSAHase dissolved in 2 ml buffer B was gradually mixed with 5 ml saturated (NH4)2SO4 solution pH 3.3 and then stored for 30 min on ice. The mixture was centrifuged and the precipitate was dissolved in 2 ml buffer B. The enzyme was precipitated again as above and the pellet was washed with 5 ml of a saturated neutral solution of (NH4)2SO4 with 2.5 mM DTT and 1 mM EDTA. The precipitated apo LlSAHase was dissolved in 2 ml buffer B without DTT and subsequently NAD+ was added to a final concentration of 2 mM. After 1 h incubation on ice, the protein solution was loaded onto a Superdex 200 (Pharmacia) gel-filtration column pre-equilibrated with buffer B. The protein was eluted with buffer B as a dimer. Fractions containing LlSAHase were concentrated to 2.3 mg ml−1 using Amicon Ultra 30 filters and the fresh protein solution was used for crystallization experiments. The protein concentration was estimated using the method of Bradford (1976 ▶) with bovine serum albumin as a standard.
2.2. Crystallization
Protein solution (2.0 mg ml−1) in buffer B was incubated for 12 h with 2 mM adenosine at 277 K. The mixture was used for initial screening for crystallization conditions using a sparse-matrix screen (Jancarik & Kim, 1991 ▶) from Molecular Dimensions (Structure Screen 1). Initial crystals were obtained from 20%(w/v) PEG 4000, 10%(v/v) 2-propanol and 0.1 M HEPES pH 7.5. The crystallization conditions were refined by adjusting the pH as well as the concentration and volume of the protein solution. The best crystals were obtained with 20%(w/v) PEG 4000, 10%(v/v) 2-propanol and 0.1 M Tris–HCl pH 8.0. They were grown using the hanging-drop vapour-diffusion method at 292 K by mixing 3 µl protein–Ado solution with 1 µl precipitating solution on a siliconized coverslide and equilibrating the drop against 1.0 ml precipitant solution. The crystals appeared within two weeks (Fig. 1 ▶).
Figure 1.

A single crystal of S-adenosyl-l-homocysteine hydrolase from L. luteus in complex with adenosine.
2.3. Data collection and processing
Difraction data for the LlSAHase–Ado complex were collected using synchrotron radiation (EMBL/DESY Hamburg, beamline X13, λ = 0.8086 Å) and a 165 mm MAR CCD detector. Since the crystals were very fragile and easily cracked upon transfer between drops, a special cryoprotection technique was implemented. A cryoprotection solution with 50%(v/v) PEG 400 instead of PEG 4000 and with all other ingredients unchanged was prepared. A drop of this cryoprotectant with a matching volume was placed near the crystallization drop containing the selected crystal and the two drops were joined and mixed with a mounting loop. A crystal with dimensions 0.4 × 0.3 × 0.3 mm was fished out from this solution and vitrified at 100 K in a nitrogen-gas stream. The diffraction data were measured in two runs corresponding to low and high resolution. The low-resolution data set consisted of 340 images recorded with 0.4° oscillation at 200 mm crystal-to-detector distance and extended to 2.10 Å resolution. The high-resolution data set consisted of 540 images recorded with 0.25° oscillation at 95 mm crystal-to-detector distance and extended to 1.17 Å resolution (Fig. 2 ▶). In the final scaling, reflections from the ranges 20.0–2.10 Å (low-resolution data set) and 2.7–1.17 Å (high-resolution data set) were used. Indexing and integration of all images was performed in DENZO and scaling was performed in SCALEPACK, both from the HKL program package (Otwinowski & Minor, 1997 ▶). The crystal is tetragonal, space group P43212. Statistics of the final data set are given in Table 1 ▶.
Figure 2.

An X-ray diffraction pattern recorded for a single crystal of LlSAHase in complex with adenosine (0.25° oscillation). The edge of the detector (framed, inset) corresponds to a resolution of 1.15 Å.
Table 1. X-ray data-collection details and processing statistics for yellow lupin SAHase cocrystallized with adenosine.
Values in parentheses correspond to the last resolution shell.
| Space group | P43212 |
| Unit-cell parameters (Å) | a = 122.4, c = 126.5 |
| Temperature (K) | 100 |
| Resolution limits (Å) | 20.0–1.17 (1.21–1.17) |
| No. of observations | 12171512 |
| No. of unique reflections | 319479 |
| Wavelength (Å) | 0.8086 |
| Mosaicity (°) | 0.38 |
| Completeness (%) | 99.9 (99.2) |
| 〈I/σ(I)〉 | 34.3 (2.6) |
| Rmerge† | 0.059 (0.721) |
R
merge = 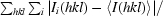
 , where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
2.4. Structure solution
The structure was solved by the molecular-replacement method, as implemented in the program Phaser (McCoy et al., 2007 ▶) from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶), using chain A of P. falciparum SAHase (PDB code 1v8b; Tanaka et al., 2004 ▶) as a model. The correct solution (Z score 50.05) was found in space group P43212 for two protein molecules in the asymmetric unit. Currently, the structure is undergoing anisotropic stereochemically restrained structure-factor refinement.
3. Results and discussion
The expression and purification protocol allowed the production of a pure and homogeneous protein preparation. Typical yields were 1 mg pure LlSAHase from 1 l culture. The effects of protein concentration, sample volume and pH have been investigated for crystallization optimization. The best crystals were obtained by mixing 3 µl protein solution at 2.0 mg ml−1, preincubated with adenosine in a 1:100 molar ratio, with 1 µl reservoir solution containg 20%(w/v) PEG 4000, 10%(v/v) 2-propanol and 0.1 M Tris–HCl pH 8.0. Under the optimized conditions, the crystals grew within 14 d.
The crystals were tetragonal, class 422, with unit-cell parameters a = 122.4, c = 126.5 Å. The Matthews coefficient (Matthews, 1968 ▶) was consistent with the presence of two protein molecules in the asymmetric unit (V M = 2.23 Å3 Da−1, solvent content 44.6%).
The initial molecular-replacement solution revealed the presence of a dimeric protein in the asymmetric unit, which is at variance with the tetrameric form of the SAHase structures studied to date but in agreement with early predictions about the quaternary structure of plant SAHases (Guranowski & Pawełkiewicz, 1977 ▶) and with the results of our gel-filtration experiments, which showed that LlSAHase forms dimers in solution. In addition, difference Fourier maps show clear electron density for the cofactor (NAD+) and the ligand (adenosine) in both monomers, confirming successful ternary complex formation.
Acknowledgments
This work was supported in part by a grant from the Ministry of Science and Higher Education No. N N302 4305 34.
References
- Bradford, M. M. (1976). Anal. Biochem.72, 248–254. [DOI] [PubMed]
- Brzezinski, K., Janowski, R., Podkowiński, J. & Jaskolski, M. (2001). Acta Biochim. Pol.48, 477–483. [PubMed]
- Cantoni, G. L. & Scarano, E. (1954). J. Am. Chem. Soc.76, 4744.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Guranowski, A. & Pawełkiewicz, J. (1977). Eur. J. Biochem.80, 517–523. [DOI] [PubMed]
- Hu, Y., Komoto, J., Huang, Y., Gomi, T., Ogawa, H., Takata, Y., Fujioka, M. & Takusagawa, F. (1999). Biochemistry, 38, 8323–8333. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411.
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst.40, 658–674. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Richards, H. H., Chiang, P. K. & Cantoni, G. L. (1978). J. Biol. Chem.253, 4476–4480. [PubMed]
- Stępkowski, T., Brzeziński, K., Legocki, A. B., Jaskolski, M. & Béna, G. (2005). Mol. Phylogenet. Evol.34, 15–28. [DOI] [PubMed]
- Tanaka, N., Nakanishi, M., Kusakabe, Y., Shiraiwa, K., Yabe, S., Ito, Y., Kitade, Y. & Nakamura, K. T. (2004). J. Mol. Biol.343, 1007–1017. [DOI] [PubMed]
- Turner, M. A., Yuan, C. S., Borchardt, R. T., Hershfield, M. S., Smith, G. D. & Howell, P. L. (1998). Nature Struct. Biol.5, 369–376. [DOI] [PubMed]
- Yang, X., Hu, Y., Yin, D. H., Turner, M. A., Wang, M., Borchardt, R. T., Howell, P. L., Kuczera, K. & Schowen, R. L. (2003). Biochemistry, 42, 1900–1909. [DOI] [PubMed]
- Yuan, C. S., Yeh, J., Liu, S. & Borchardt, R. T. (1993). J. Biol. Chem.268, 17030–17037. [PubMed]