

Crystals of arylmalonate decarboxylase from A. bronchisepticus were obtained which diffracted X-rays to a resolution of at least 3.0 Å.
Keywords: arylmalonate decarboxylase, Alcaligenes bronchisepticus, enantioselectivity
Abstract
Arylmalonate decarboxylase catalyses the enantioselective decarboxylation of α-aryl-α-methylmalonates to produce optically pure α-arylpropionates. The enzyme was crystallized with ammonium sulfate under alkaline pH conditions with the aim of understanding the mechanism of the enantioselective reaction. X-ray diffraction data collected to a resolution of 3.0 Å at cryogenic temperature showed that the crystals belonged to the orthorhombic space group P212121, with unit-cell parameters a = 83.13, b = 99.62, c = 139.64 Å. This suggested that the asymmetric unit would contain between four and six molecules. Small-angle X-ray scattering revealed that the enzyme exists as a monomer in solution. Thus, the assembly of molecules in the asymmetric unit was likely to have been induced during the crystallization process.
1. Introduction
Arylmalonate decarboxylase (AMDase; EC 4.1.1.76) isolated from Alcaligenes bronchisepticus strain KU1201 catalyses the enantioselective decarboxylation of prochiral α-aryl-α-methylmalonates to optically active α-arylpropionates with a high enantiomeric excess in high yield (Fig. 1 ▶ a; Miyamoto & Ohta, 1990 ▶, 1991 ▶; Ohta, 1997 ▶). Unlike other decarboxylases, the enzyme requires no additional cofactors such as metal ions, coenzyme A or ATP (Miyamoto & Ohta, 1992a ▶). AMDase, comprising 240 amino-acid residues (molecular weight 24 737 Da), shows low homology to racemases and isomerases such as glutamate racemases (GluRs; Gallo & Knowles, 1993 ▶; Hwang et al., 1999 ▶), aspartate racemase (AspR; Yohda et al., 1991 ▶; Liu et al., 2002 ▶) and other racemase-related enzymes such as hydanotine racemase (Watabe et al., 1992 ▶) and maleate cis–trans isomerase (Hatakeyama et al., 1997 ▶). GluR and AspR have been classified into the pyridoxal-5′-phosphate-independent racemase family (Tanner, 2002 ▶; Gerlt et al., 2005 ▶). An alignment of the primary sequences of family members (Fig. 1 ▶ b) suggests that Cys188 of AMDase is one of the essential residues forming the active site. This residue is thought to act as a proton donor to the intermediate, the enolate form of α-arylpropionate, from only one side of the enantiomeric face to produce the (R)-product (Matoishi et al., 2004 ▶; Terao et al., 2007 ▶). AMDase becomes inactive on Cys188Ser mutation (Miyazaki et al., 1997 ▶) or in the presence of SH reagents such as iodoacetate, p-chloromercuribenzoate, HgCl2 and Ag(NO3)2 (Miyamoto & Ohta, 1992b ▶), suggesting a crucial role of the cysteine residue in the reaction.
Figure 1.

(a) Scheme showing the enantioselective decarboxylation reaction of phenylmalonate catalyzed by AMDase. (b) An alignment of the amino-acid sequences of AMDase, GluR (Gallo & Knowles, 1993 ▶), AspR (Yohda et al., 1991 ▶), HydR (Watabe et al., 1992 ▶) and MlIR (Hatakeyama et al., 1997 ▶) around the two cysteine residues conserved in the racemases and the racemase-related isomerase. The sequence numbers of the active-site cysteine (or glycine) residues are labelled.
In the crystal structures of GluR from Aquifex pyrophilus (Hwang et al., 1999 ▶) and AspR from Pyrococcus horikoshii (Liu et al., 2002 ▶), two cysteine residues are symmetrically arranged in the active-site cleft and provide a reaction field that enables a two-base mechanism for their racemization reaction (Tanner, 2002 ▶; Gallo et al., 1993 ▶; Glavas & Tanner, 1999 ▶, 2001 ▶). In AMDase, Cys188 corresponds to one of the cysteine residues of the racemases and Gly74 takes the place of the other. This difference probably causes an asymmetric structure of the active site, enabling the enantioselective reaction. In fact, a Gly74Cys mutation converts AMDase to an enzyme that catalyses the racemization of α-arylpropionate (Terao et al., 2006a ▶) and Gly74Cys/Cys188Ser-mutated AMDase produces arylpropionate of the opposite enantiomorph to that of the wild-type enzyme, although the enzymatic reaction proceeds with a slower rate than that of the wild type (Ijima et al., 2005 ▶; Miyamoto et al., 2007 ▶). Thus, AMDase is of particular interest with regard to this conversion of the function by simple mutations and the three-dimensional structure of AMDase is indispensable in order to understand the reaction mechanism and for the design and development of novel enzymes (Terao et al., 2006b ▶).
Here, we describe the crystallization of AMDase and preliminary X-ray diffraction experiments at cryogenic temperature. The X-ray diffraction data suggested that 4–6 AMDase molecules occupy the crystallographic asymmetric unit. In addition, the quaternary structure of AMDase was studied by small-angle X-ray scattering (SAXS) measurements in order to determine the functional unit in solution.
2. Methods
2.1. Protein purification and crystallization
Untagged AMDase was overexpressed in Escherichia coli strain DH5α-MCR. The strain was transformed with the plasmid vector pAMD101 coding for the cloned AMDase gene from Al. bronchisepticus strain KU1201 (Miyamoto & Ohta, 1992b ▶). The cells were grown in LB medium at 303 K for 2 h, induced with 1 mM IPTG and harvested 12 h after induction. Harvested cells were suspended in 0.1 M phosphate buffer containing 5 mM EDTA and 5 mM β-mercaptoethanol pH 7.5 and lysed by sonication. Subsequent purification was carried out by ammonium sulfate precipitation and column chromatography using Toyopearl DEAE-650M and Butyl-650M columns (Tohso, Japan) according to the procedure described previously (Terao et al., 2006b ▶). The purity of the eluted enzyme solution after column chromatography was examined by SDS–PAGE. The molecular weight of the purified enzyme was determined to be 24 744 Da by electrospray ion mass spectrometry (Matoishi et al., 2004 ▶). The purified enzyme was suspended at a concentration of 10 mg ml−1 in 10 mM Tris–HCl buffer containing 0.5 mM EDTA and 5 mM β-mercaptoethanol pH 7.0.
Crystallization of AMDase was carried out by the hanging-drop vapour-diffusion method at 293 K. Crystal Screens I and II (Hampton Research, USA) were used in the initial survey of crystallization conditions. 2 µl AMDase solution mixed with 2 µl precipitant solution was equilibrated against 1 ml precipitant solution. Crystallization was set up within a few days of purification.
2.2. X-ray diffraction experiment
X-ray diffraction data were collected in our laboratory by the oscillation method using Cu Kα radiation (λ = 1.5418 Å) from an Ultrax-18 X-ray generator (Rigaku, Japan) operated at 45 kV and 90 mA, Pt-coated double-focusing optics (Rigaku, Japan) and an R-AXIS IV detector (Rigaku, Japan). The crystal-to-detector distance was 200 mm. Crystals as large as 0.2 × 0.1 × 0.1 mm were used in cryogenic X-ray diffraction experiments at 110 K. Indexing, integration, scaling and post-refinement were carried out using the HKL-2000 suite (Otwinowski & Minor, 1997 ▶).
2.3. Small-angle X-ray scattering measurements
The functional unit of AMDase in solution was studied by the small-angle X-ray scattering (SAXS) method. SAXS of wild-type and Gly74Cys/Cys188Ser-mutated (Ijima et al., 2005 ▶) AMDase were collected on BL45SX of SPring-8 (Fujisawa et al., 2000 ▶) using an R-AXIS IV++ detector (Rigaku, Japan). The X-ray wavelength was 1.000 Å and the camera distance was set to 2000 mm. An aluminium attenuator was used for the incident X-ray beam in order to reduce radiation damage to the solution samples. The exposure time was 100 s for each measurement. The details of data reduction and analysis for SAXS were carried out as described previously (Nakasako et al., 2004 ▶).
3. Results and discussion
3.1. Crystallization and X-ray diffraction experiments
Small crystals appeared within two weeks of setup using a precipitant solution containing 1.5 M ammonium sulfate, 12%(v/v) glycerol and 0.1 M Tris–HCl pH 8.7. The crystallization conditions were further optimized by varying the concentrations of ammonium sulfate and pH in the precipitant solution. Finally, crystals suitable for X-ray diffraction experiments (Fig. 2 ▶ a) were obtained using a precipitant solution containing 1.7 M ammonium sulfate, 15%(w/v) glycerol and 0.1 M Tris–HCl pH 8.0. Harvested crystals were dialyzed against a cryo-buffer containing 2.2 M ammonium sulfate, 22%(w/v) glycerol, 0.1 M Tris–HCl pH 8.0 for more than 6 h and were flash-cooled with cold nitrogen gas at 100 K.
Figure 2.

(a) A photograph of an AMDase crystal. The scale bar is 0.2 mm in length. (b) Native PAGE of AMDase solutions obtained by dissolving crystals soaked in crystallization buffer containing 3 mM heavy-atom reagents for 2 h. Lanes are native crystal (1), crystals soaked in the cryo-buffer containing 3 mM K2PtCl4 (2), K2PtCl6 (3), K2Pt(CN)6 (4), HgCl2 (5), mersalyl acid (6), mercury acetate (7), HgO (8), Hg(CN)2 (9) and p-chloromercuribenzoate (10). The PAGE pattern of the AMDase solution dissolved from native crystals is the same as that of the enzyme solution after purification.
A diffraction pattern from an AMDase crystal is shown in Fig. 3 ▶. The statistics of the diffraction data are summarized in Table 1 ▶. Systematic absences of reflections in the diffraction data indicated that the crystal belonged to space group P212121. According to the acceptable range of partial specific volumes (V M) for crystals of soluble proteins (Matthews, 1968 ▶), the possible number of AMDase molecules occupying the crystallographic asymmetric unit was four (V M = 3.1 Å3 Da−1), five (2.5 Å3 Da−1) or six (2.1 Å3 Da−1).
Figure 3.

An X-ray diffraction pattern with 0.6° oscillation obtained at our laboratory. The arrow indicates a Bragg spacing of 3.0 Å.
Table 1. Statistics of the collected diffraction data.
Values in parentheses are for the highest shell.
| Oscillation range (°) | 0.6 |
| Exposure time (min) | 30 |
| No. of plates | 150 |
| Unit-cell parameters | |
| a (Å) | 83.13 |
| b (Å) | 99.62 |
| c (Å) | 139.64 |
| Resolution (Å) | 50.00–3.00 (3.11–3.00) |
| No. of reflections | 85421 |
| No. of unique reflections | 23727 |
| Completeness† (%) | 99.6 (98.9) |
| 〈I/σ(I)〉 | 19.3 (4.2) |
| Rmerge‡ for I > 1σ(I) | 0.062 (0.273) |
Percentage of unique reflections measured against the theoretical prediction.
R
merge = 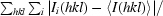
 , where I
i(hkl) is the intensity of the ith observation of reflection hkl.
, where I
i(hkl) is the intensity of the ith observation of reflection hkl.
Because the primary sequence of AMDase showed a low homology of ∼30% with those of GluR from Aq. pyrophilus (Hwang et al., 1999 ▶) and AspR from P. horikoshii (Liu et al., 2002 ▶), we first tried to solve the crystal structure by the molecular-replacement (MR) method. A self-rotation function suggested that four molecules would occupy the crystallographic asymmetric unit. However, all MR trials were unsuccessful and were abandoned. In addition, it seems likely that there will be difficulties in applying the multiple-wavelength anomalous dispersion method with selenomethionine-substituted AMDase because 16 or more AMDase molecules, each containing six methionine residues, in a unit cell will result in a large number of cross-vectors between Se atoms. As a result, we have begun a search for heavy-atom derivatives in order to apply the multiple isomorphous replacement method.
Prior to surveying heavy-atom derivatives by X-ray diffraction experiments, crystals soaked in Hg reagents were found to be potential derivatives with the help of the native polyacrylamide gel electrophoresis (PAGE) method (Boggon & Shapiro, 2000 ▶; Fig. 2 ▶ b). In the experiment, AMDase crystals were soaked in crystallization buffers containing heavy-atom reagents at 3 mM for 2 h. The crystals were then dissolved by adding 10 mM Tris–HCl pH 8.0 buffer. Crystals treated with Hg reagents of small molecular size displayed abnormal patterns relative to native crystals and those treated with platinum reagents. The Hg reagent probably attached to the SH groups of solvent-accessible cysteine residues and the additional charge of Hg2+ resulted in retardation of mobility.
Whilst we were preparing this crystallization communication, the crystal structure of AMDase from Bordetella bronchiseptica was published (Küttner et al., 2008 ▶; PDB code 2vlb). The enzyme was crystallized under different conditions from those described above. Unfortunately, the active-site cysteine residue of the enzyme was covalently modified by β-ME in the sample buffer. The authors reported that incubation of the enzyme solution in the presence of β-ME for 4–5 weeks was necessary for crystallization. The differences in the crystallizations may arise from their His-tagged AMDase sample. Thus, at the present time, the structure of a modification-free active site is still unknown. Thus, our sample preparation and crystallization conditions may have the benefit of revealing the active-site structure, as the period of crystallization was far shorter than that reported by Küttner and coworkers.
3.2. Functional unit of AMDase
The Guinier plot (Guinier & Fournet, 1955 ▶) of the wild-type enzyme at a concentration of 1 mg ml−1 displayed an inflection in the very small-angle region, indicating slight aggregation (Fig. 4 ▶). In contrast, the plot of the mutated enzyme at the same concentration showed monodisperse characteristics (Fig. 3 ▶). The radius-of-gyration (R g) value of the mutated enzyme calculated by Guinier analysis (see legend of Fig. 4 ▶) was 18.8 ± 0.5 Å. The profile of the wild-type enzyme was closely similar to that of the mutated enzyme in the region S > 0.005 Å−1. Ignoring the inflection in the very small-angle region, the R g value of the wild-type enzyme was 18.4 ± 0.6 Å. Considering the molecular weight of AMDase and the molecular dimensions of GluR and AspR, the R g value strongly suggested that both wild-type and mutated enzymes were monomeric in solution. In addition, the forward-scattering intensities gave an apparent molecular weight of ∼25 kDa for both the wild-type and mutated enzymes using bovine serum albumin (molecular weight 65 kDa) as a reference. From these results, it was concluded that the oligomeric assembly of AMDase in the crystal was induced during the crystallization process and that the molecules in the assembly are related by noncrystallographic symmetry.
Figure 4.

Guinier plots (Guinier & Fournet, 1955 ▶; the logarithm of the scattering intensity plotted against the square of the scattering vector) for SAXS of wild-type AMDase (open squares) and Gly74Cys/Cys188Ser mutant enzyme (filled squares). In the Guinier approximation (Guinier & Fournet, 1955 ▶), the small-angle scattering intensity I(S), as a function of the scattering vector S, is expressed as ln[I(S)] = ln[I(0)] − 4π2/3R g 2 S 2 and S = 2sinθ/λ, where 2θ is the scattering angle, λ is the wavelength of the X-rays and R g is the radius of gyration. The grey line was calculated by the least-squares method for the region 10 × 10−6 Å−2 < S 2 < 150 × 10−6 Å−2 of the SAXS of the mutant enzyme.
Acknowledgments
This work was supported by grants-in-aid from MEXT, Japan to MN (Nos. 15076210 and 19204042). The authors thank Dr K. Ito of RIKEN Harima Institute and Mr K. Sakamoto of Keio University for their help in the scattering experiments at SPring-8.
References
- Boggon, T. J. & Shapiro, L. (2000). Structure, 8, R143–R149. [DOI] [PubMed]
- Fujisawa, T., Inoue, K., Oka, T., Iwamoto, H., Uruga, T., Kumasaka, T., Inoko, Y., Yagi, N., Yamamoto, M. & Ueki, T. (2000). J. Appl. Cryst.33, 797–800.
- Gallo, K. A. & Knowles, J. R. (1993). Biochemistry, 32, 3981–3990. [DOI] [PubMed]
- Gallo, K. A., Tanner, M. E. & Knowles, J. R. (1993). Biochemistry, 32, 3991–3997. [DOI] [PubMed]
- Gerlt, J. A., Babbitt, P. C. & Rayment, I. (2005). Arch. Biochem. Biophys.433, 59–70. [DOI] [PubMed]
- Glavas, S. & Tanner, M. E. (1999). Biochemistry, 38, 4106–4113. [DOI] [PubMed]
- Glavas, S. & Tanner, M. E. (2001). Biochemistry, 40, 6199–6204. [DOI] [PubMed]
- Guinier, A. & Fournet, G. (1955). Small-Angle Scattering of X-rays. New York: John Wiley.
- Hatakeyama, K., Asai, Y., Uchida, Y., Kobayashi, M., Terasawa, M. & Yukawa, H. (1997). Biochem. Biophys. Res. Commun.239, 74–79. [DOI] [PubMed]
- Hwang, K. Y., Co, C.-S., Kim, S. S., Sung, H.-C., Yu, Y. G. & Cho, Y. (1999). Nature Struct. Biol.6, 422–426. [DOI] [PubMed]
- Ijima, Y., Matoichi, K., Terao, Y., Doi, N., Yanagawa, H. & Ohta, H. (2005). Chem. Commun.239, 877–879. [DOI] [PubMed]
- Küttner, E. B., Keim, A., Kircher, M., Rosmus, S. & Sträter, N. (2008). J. Mol. Biol.377, 386–394. [DOI] [PubMed]
- Liu, L., Iwata, K., Kita, A., Kawarabayashi, Y., Yohda, M. & Miki, K. (2002). J. Mol. Biol.319, 479–489. [DOI] [PubMed]
- Matoishi, K., Ueda, M., Miyamoto, K. & Ohta, H. (2004). J. Mol. Catal. B Enzym.27, 161–168.
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Miyamoto, K. & Ohta, H. (1990). J. Am. Chem. Soc.112, 4077–4078.
- Miyamoto, K. & Ohta, H. (1991). Biocatalysis, 5, 49–60.
- Miyamoto, K. & Ohta, H. (1992a). Appl. Microbiol. Biotechnol.38, 234–238. [DOI] [PubMed]
- Miyamoto, K. & Ohta, H. (1992b). Eur. J. Biochem.210, 475–481. [DOI] [PubMed]
- Miyamoto, K., Tsutsumi, T., Terao, Y. & Ohta, H. (2007). Chem. Lett.36, 656–657.
- Miyazaki, M., Kakidani, H., Hanzawa, S. & Ohta, H. (1997). Bull. Chem. Soc. Jpn, 70, 2765–2769.
- Nakasako, M., Iwata, T., Matsuoka, D. & Tokutomi, S. (2004). Biochemistry, 43, 14881–14890. [DOI] [PubMed]
- Ohta, H. (1997). Bull. Chem. Soc. Jpn, 70, 2895–2911.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Tanner, M. E. (2002). Acc. Chem. Res.35, 237–246. [DOI] [PubMed]
- Terao, Y., Ijima, Y., Miyamoto, K. & Ohta, H. (2007). J. Mol. Catal. B Enzym.45, 15–20.
- Terao, Y., Miyamoto, K. & Ohta, H. (2006a). Chem. Commun.34, 3600–3602. [DOI] [PubMed]
- Terao, Y., Miyamoto, K. & Ohta, H. (2006b). Appl. Microbiol. Biotechnol.73, 647–653. [DOI] [PubMed]
- Watabe, K., Ishikawa, T., Mukohara, Y. & Nakamura, H. (1992). J. Bacteriol.174, 3461–3466. [DOI] [PMC free article] [PubMed]
- Yohda, M., Okada, H. & Kumagai, H. (1991). Biochim. Biophys. Acta, 1089, 234–240. [DOI] [PubMed]