

Dihydrodipicolinate synthase (DHDPS), an enzyme of the lysine-biosynthetic pathway, is a promising target for antibiotic development against pathogenic bacteria. Here, the expression, purification, crystallization and preliminary diffraction analysis to 1.45 Å resolution of DHDPS from methicillin-resistant S. aureus is reported.
Keywords: antibiotics, antibiotic resistance, dihydrodipicolinate synthase, drug discovery, lysine biosynthesis, Staphylococcus aureus
Abstract
In recent years, dihydrodipicolinate synthase (DHDPS; EC 4.2.1.52) has received considerable attention from both mechanistic and structural viewpoints. DHDPS is part of the diaminopimelate pathway leading to lysine, coupling (S)-aspartate-β-semialdehyde with pyruvate via a Schiff base to a conserved active-site lysine. In this paper, the cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of DHDPS from methicillin-resistant Staphylococcus aureus, an important bacterial pathogen, are reported. The enzyme was crystallized in a number of forms, predominantly from PEG precipitants, with the best crystal diffracting to beyond 1.45 Å resolution. The space group was P1 and the unit-cell parameters were a = 65.4, b = 67.6, c = 78.0 Å, α = 90.1, β = 68.9, γ = 72.3°. The crystal volume per protein weight (V M) was 2.34 Å3 Da−1, with an estimated solvent content of 47% for four monomers per asymmetric unit. The structure of the enzyme will help to guide the design of novel therapeutics against the methicillin-resistant S. aureus pathogen.
1. Introduction
The branch-point reaction in the biosynthetic pathway leading to meso-diaminopimelate and (S)-lysine in plants and bacteria is catalysed by dihydrodipicolinate synthase (DHDPS; EC 4.2.1.52). This reaction couples pyruvate and (S)-aspartate-β-semialdehyde in an aldol-like condensation to form (4S)-4-hydroxy-2,3,4,5-tetrahydro-(2S)-dipicolinic acid. Since (S)-lysine biosynthesis does not occur in animals, specific inhibitors of this pathway are less likely to exhibit mammalian host toxicity. Although the enzymes of this pathway, such as DHDPS, are attractive targets for rational antibiotic design (Hutton et al., 2003 ▶, 2007 ▶; Mitsakos et al., 2008 ▶), no potent inhibitors have yet been identified.
The catalytic mechanism of DHDPS has been extensively studied (Blickling et al., 1997 ▶; Dobson, Gerrard et al., 2004 ▶; Dobson, Griffin et al., 2004 ▶; Dobson, Valegård et al., 2004 ▶; Dobson, Devenish et al., 2005 ▶), with many enzymes shown to be allosterically feedback-inhibited by (S)-lysine. Varying levels of feedback inhibition have been observed, ranging from strong inhibition in plant DHDPS (IC50 = 0.01–0.05 mM; Kumpaisal et al., 1989 ▶; Wallsgrove & Mazelis, 1981 ▶; Matthews & Widholm, 1978 ▶; Ghislain et al., 1990 ▶; Frisch et al., 1991 ▶; Dereppe et al., 1992 ▶) to relatively weak inhibition in bacterial DHDPS (IC50 = 0.25–1 mM; Yugari & Gilvarg, 1965 ▶; Kefala et al., 2008 ▶). Furthermore, there are examples of DHDPS enzymes, typically from Gram-positive bacteria, in which no significant lysine-mediated feedback inhibition of DHDPS is observed (Stahly, 1969 ▶; Barnes et al., 1969 ▶). Despite extensive study of the regulation of DHDPS in plants and bacteria, the mechanism of inhibition remains poorly understood (Yugari & Gilvarg, 1965 ▶; Stahly, 1969 ▶; Kumpaisal et al., 1989 ▶; Laber et al., 1992 ▶; Blickling et al., 1997 ▶, 1998 ▶; Dobson, Griffin et al., 2004 ▶).
The structures of DHDPS from a variety of organisms have been solved (Mirwaldt et al., 1995 ▶; Blickling et al., 1998 ▶; Dobson, Griffin et al., 2005 ▶; Blagova et al., 2006 ▶; Pearce et al., 2006 ▶; Kefala et al., 2008 ▶). In most cases, DHDPS is a tetrameric protein and exists as a dimer of tight dimers, with two distinct arrangements observed to date. There are four active sites per tetramer and the active site of each monomer, which is located within a pocket of an (α/β)8-barrel, contains a residue from the other subunit of the tight dimer (i.e. in Escherichia coli DHDPS Tyr107 from chain B contributes to the active site of chain A and vice versa). Two allosteric binding sites are located at the tight-dimer interface, approximately 20 Å away from the active sites (Dobson, Griffin et al., 2004 ▶).
Although the active site is highly conserved and the tight-dimer interface, or at least at the loop bearing Tyr107, is well conserved amongst DHDPS enzymes, the weak-dimer interface shows little conservation. Since dimeric mutants of E. coli DHDPS have drastically altered activity and substrate specificity (Griffin et al., 2008 ▶), the weak-dimer interface represents a target for the design of pathogen-specific drugs. Thus, we are engaged in a study of DHDPS enzymes from a variety of pathogenic bacteria in order to characterize the structure of the enzyme for the design of molecules that disrupt the quaternary structure of DHDPS.
Here, we present preliminary crystallographic studies of DHDPS from methicillin-resistant S. aureus (MRSA-DHDPS) as a first step towards the development of novel therapeutics targeting S. aureus.
2. Materials and methods
2.1. Cloning, expression and purification of MRSA-DHDPS
The dapA structural gene encoding DHDPS and the flanking nucleotide sequence was amplified by PCR (primers OSA1, GTATTGGAACAAGTTATGCG, and OSA2, TCTGCTAATCTAGCAAGCGC) from genomic DNA derived from methicillin-resistant S. aureus subsp. aureus MRSA252. The amplified product was cloned into pCR-Blunt II-TOPO (Invitrogen) to produce the vector pBB01. Following verification of the nucleotide sequence, the primers OSA3 (TGACACATTTATTTGAGGGTG) and OSA4 (TCACTCATTTTCACCCGC) facilitated PCR amplification and cloning of the dapA open reading frame from pBB01 into the pET11a expression vector to produce pBB02.
E. coli BL21 (DE3) cells transformed with pBB02 were cultured at 310 K in Luria–Bertani broth containing ampicillin (50 µg ml−1) to an OD600 of 0.6. Expression of DHDPS was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 1 mM before incubation at 310 K for 3 h. Cells were harvested by centrifugation at 10 000g for 15 min. The cell pellet was resuspended in buffer A (20 mM potassium phosphate pH 6.0) and stored at 193 K prior to use.
Cell pellets were thawed on ice and lysed by sonication with an MSE Soniprep 150 sonicator at 14 µm amplitude, following a 5 min cycle of 3 s bursts with a 10 s rest between bursts. Cellular debris was cleared by centrifugation (10 000g, 10 min, 277 K) and the supernatant was collected. The pellet was resuspended in buffer A for a repeated round of sonication and centrifugation, with the supernatant from each centrifugation pooled and retained as the crude cell lysate. The crude cell lysate was applied onto a Q-Sepharose Fast Flow anion-exchange column (50 ml) pre-equilibrated with ten bed volumes of buffer A at 277 K and washed until a stable baseline was reached. The enzyme was eluted over five column volumes with a 0–1 M NaCl gradient in buffer A. Active fractions were conservatively selected for purity and pooled prior to a one-in-four dilution with buffer B (20 mM Tris–HCl pH 8.0). The protein was concentrated to 10 mg ml−1 with a Vivaspin20 10 kDa molecular-weight cutoff concentrator prior to use or storage at 193 K. Slowly thawed protein was further purified by size-exclusion liquid chromatography using a 10/300 Sephacryl S-200 column (GE Healthcare) prior to use. Protein-purification steps were assessed by SDS–PAGE and monitored for enzymatic activity using the qualitative ο-aminobenzaldehyde assay (Dobson, Devenish et al., 2005 ▶).
2.2. Crystallization of MRSA-DHDPS
Initial protein-crystallization experiments were performed at the CSIRO node of the Bio21 Collaborative Crystallization Centre (C3; http://www.csiro.au/c3/) using the PACT Suite and the JCSG+ Suite crystal screens (Qiagen) at 281 and 293 K (Newman et al., 2005 ▶). Conditions from these initial screens that yielded a variety of small crystal forms were factorially randomized in a subsequent screen at 293 K. These screens were set up using the sitting-drop vapour-diffusion method, with droplets consisting of 100 nl protein solution and 100 nl reservoir solution. The crystal shown in Fig. 1 ▶ was obtained at 293 K from a 400 nl drop formed of 200 nl protein solution (10.6 mg ml−1 in 20 mM Tris–HCl pH 8.0) and 200 nl precipitant [16.7%(w/v) polyethylene glycol 6000, 135 mM sodium fluoride, 296 mM lithium chloride, 100 mM sodium acetate pH 4.9; condition A]. A single crystal (indicated with an arrow in Fig. 1 ▶) was gently separated from attached crystal plates using a crystal dissection kit. The crystal was briefly soaked in reservoir liquor containing glycerol [20%(v/v)] before being flash-frozen in liquid nitrogen.
Figure 1.

Crystal of recombinant MRSA-DHDPS used in the diffraction experiment.
2.3. Data collection and processing
Intensity data were collected at 100 K at the Australian Synchrotron using the 3-BM1 beamline (McPhillips et al., 2002 ▶; Cohen et al., 2002 ▶; Evans & Pettifer, 2001 ▶) producing X-rays at a wavelength of 0.9536 Å. An ADSC Q210r image-plate detector positioned 140 mm from the crystal was used to collect 360° of data with an exposure time of 5 s and 0.5° steps. Diffraction data sets were processed and scaled using the MOSFLM package (Leslie, 1992 ▶) and SCALA (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
Initial screening for crystallization conditions of MRSA-DHDPS was performed at the CSIRO node of the Bio21 Collaborative Crystallization Centre (C3) using the JCSG+ and PACT crystallization screens (Qiagen). Several conditions of the PACT suite produced small crystal plates after 1 d at 293 K, which were further optimized to produce large crystal plates (∼300 × 70 × 30 µm; Fig. 1 ▶) in condition A after 3 d of growth.
An X-ray diffraction data set was collected to a resolution of 1.45 Å from a crystal of MRSA-DHDPS grown in condition A using 20%(v/v) glycerol as a cryoprotectant. The crystal displayed diffraction beyond this resolution (to ∼1.35 Å in the corners of the CCD detector); however, these data could not be collected with reasonable completeness. The data-collection information and statistics are listed in Table 1 ▶. The Matthews coefficient (V M; Matthews, 1968 ▶) was calculated to be 2.34 Å3 Da−1 assuming the presence of four MRSA-DHDPS monomers in the asymmetric unit, with a corresponding solvent content of 47%.
Table 1. X-ray data-collection statistics.
Statistical values for the highest resolution shells are given in parentheses. The Matthews coefficient and solvent content are based on four monomers of molecular weight 32 480 Da in the asymmetric unit.
| Wavelength (Å) | 0.9536 |
| No. of images | 720 |
| Oscillation (°) | 0.5 |
| Space group | P1 |
| Unit-cell parameters (Å, °) | a = 65.4, b = 67.6, c = 78.0, α = 90.1, β = 68.9, γ = 72.3 |
| Resolution (Å) | 38.49–1.45 (1.53–1.45) |
| Observed reflections | 750976 |
| Unique reflections | 190869 |
| Completeness (%) | 91.4 (71.9) |
| Rmerge† | 0.057 (0.147) |
| Rp.i.m.‡ | 0.034 (0.087) |
| Mean I/σ(I) | 18.8 (8.4) |
| Redundancy | 3.9 (3.9) |
| Wilson B value (Å2) | 9.8 |
| Molecules in ASU | 4 |
| VM (Å3 Da−1) | 2.34 |
| Solvent content (%) | 47 |
R
merge = 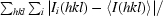
 .
.
R
p.i.m. = 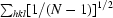
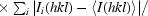 .
.
Verified bacterial DHDPS enzymes to date have been shown to crystallize as homotetramers. The tetramer is also present in solution and represents the most active form of the enzyme. Whilst still subject to further refinement, the crystal structure of MRSA-DHDPS has been solved by molecular replacement and, although the tight-dimer structure is preserved, the archetypal subunit orientation in the crystal structure of other DHDPS enzymes is not observed for the MRSA enzyme. The refinement of this structure will therefore provide important information regarding the structural evolution of DHDPS and the design of antibiotics targeting lysine biosynthesis in S. aureus.
Acknowledgments
We would like to thank Tom Caradoc-Davis, Trevor Huyton and Michael Gorman for helpful/entertaining discussions and assistance at the Australian Synchrotron (Victoria, Australia). We also thank Margaret Jane Whipp at the Medical Diagnostics Unit (Melbourne, Australia) for providing genomic DNA from MRSA (strain 252) and the Defence Threat Reduction Agency (DTRA Project ID AB07CBT004) and Australian Research Council (DP 0770888) for funding. MWP is supported by an Australian Research Council Federation Fellowship and an NHMRC Honorary Fellowship.
References
- Barnes, I. J., Bondi, A. & Moat, A. G. (1969). J. Bacteriol.99, 169–174. [DOI] [PMC free article] [PubMed]
- Blagova, E., Levdikov, V., Milioti, N., Fogg, M. J., Kalliomaa, A. K., Brannigan, J. A., Wilson, K. S. & Wilkinson, A. J. (2006). Proteins, 62, 297–301. [DOI] [PubMed]
- Blickling, S., Beisel, H., Bozic, D., Knablein, J., Laber, B. & Huber, R. (1998). J. Mol. Biol.274, 608–621. [DOI] [PubMed]
- Blickling, S., Renner, C., Laber, B., Pohlenz, H.-D., Holak, T. A. & Huber, R. (1997). Biochemistry, 36, 24–33. [DOI] [PubMed]
- Cohen, A. E., Ellis, P. J., Miller, M. D., Deacon, A. M. & Phizackerley, R. P. (2002). J. Appl. Cryst.35, 720–726. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dereppe, C., Bold, G., Ghisalba, O., Ebert, E. & Schar, H. P. (1992). Plant Physiol.98, 813–821. [DOI] [PMC free article] [PubMed]
- Dobson, R. C. J., Devenish, S. R. A., Turner, L. A., Clifford, V. R., Pearce, F. G., Jameson, G. B. & Gerrard, J. A. (2005). Biochemistry, 44, 13007–13013. [DOI] [PubMed]
- Dobson, R. C. J., Gerrard, J. A. & Pearce, F. G. (2004). Biochem. J.377, 757–762. [DOI] [PMC free article] [PubMed]
- Dobson, R. C. J., Griffin, M. D. W., Jameson, G. B. & Gerrard, J. A. (2005). Acta Cryst. D61, 1116–1124. [DOI] [PubMed]
- Dobson, R. C. J., Griffin, M., Roberts, S. J. & Gerrard, J. A. (2004). Biochimie, 86, 311–315. [DOI] [PubMed]
- Dobson, R. C. J., Valegård, K. & Gerrard, J. A. (2004). J. Mol. Biol.338, 329–339. [DOI] [PubMed]
- Evans, G. & Pettifer, R. F. (2001). J. Appl. Cryst.34, 82–86.
- Frisch, D. A., Gengenbach, B. G., Tommey, A. M., Sellner, J. M., Somers, D. A. & Myers, D. E. (1991). Plant Physiol.96, 444–452. [DOI] [PMC free article] [PubMed]
- Ghislain, M., Frankard, V. & Jacobs, M. (1990). Planta, 180, 480–486. [DOI] [PubMed]
- Griffin, M. D. W., Dobson, R. C. J., Pearce, F. G., Antonio, L., Whitten, A. E., Liew, C. K., Mackay, J. P., Trewhella, J., Jameson, G. B., Perugini, M. A. & Gerrard, J. A. (2008). In the press. [DOI] [PubMed]
- Hutton, C. A., Perugini, M. A. & Gerrard, J. A. (2007). Mol. Biosyst.3, 458–465. [DOI] [PubMed]
- Hutton, C. A., Southwood, T. J. & Turner, J. J. (2003). Mini Rev. Med. Chem.3, 115–127. [DOI] [PubMed]
- Kefala, G., Evans, G. L., Griffin, M. D. W., Devenish, S. R. A., Pearce, F. G., Perugini, M. A., Gerrard, J. A., Weiss, M. S. & Dobson, R. C. J. (2008). Biochem. J.411, 351–360. [DOI] [PubMed]
- Kumpaisal, R., Hashimoto, T. & Yamada, Y. (1989). Agric. Biol. Chem.53, 355–359.
- Laber, B., Gomis-Rüth, F.-X., Româo, M. J. & Huber, R. (1992). Biochem. J.288, 691–695. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- McPhillips, T. M., McPhillips, S. E., Chiu, H.-J., Cohen, A. E., Deacon, A. M., Ellis, P. J., Garman, E., Gonzalez, A., Sauter, N. K., Phizackerley, R. P., Soltis, S. M. & Kuhn, P. (2002). J. Synchrotron Rad.9, 401–406. [DOI] [PubMed]
- Matthews, B. F. & Widholm, J. M. (1978). Planta, 141, 315–321. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Mirwaldt, C., Korndorfer, I. & Huber, R. (1995). J. Mol. Biol.246, 227–239. [DOI] [PubMed]
- Mitsakos, V., Dobson, R. C. J., Pearce, F. G., Devenish, S. R., Evans, G., Burgess, B. R., Perugini, M. A., Gerrard, J. A. & Hutton, C. A. (2008). Bioorg. Med. Chem. Lett.18, 842–844. [DOI] [PubMed]
- Newman, J., Egan, D., Walter, T. S., Meged, R., Berry, I., Ben Jelloul, M., Sussman, J. L., Stuart, D. I. & Perrakis, A. (2005). Acta Cryst. D61, 1426–1431. [DOI] [PubMed]
- Pearce, F. G., Perugini, M. A., McKerchar, H. J. & Gerrard, J. A. (2006). Biochem. J.400, 359–366. [DOI] [PMC free article] [PubMed]
- Stahly, D. P. (1969). Biochim. Biophys. Acta, 191, 439–451. [DOI] [PubMed]
- Wallsgrove, R. M. & Mazelis, M. (1981). Phytochemistry, 20, 2651–2655.
- Yugari, Y. & Gilvarg, C. (1965). J. Biol. Chem.240, 4710–4716. [PubMed]