

Abstract
Lysosomal acid lipase (LAL) is an essential enzyme that hydrolyzes triglycerides (TGs) and cholesteryl esters (CEs) in lysosomes. Genetic LAL mutations lead to Wolman disease (WD) and cholesteryl ester storage disease (CESD). An LAL-null (lal−/−) mouse model resembles human WD/CESD with storage of CEs and TGs in multiple organs. Human LAL (hLAL) was expressed in Nicotiana benthamiana using the GENEWARE® expression system (G-hLAL). Purified G-hLAL showed mannose receptor-dependent uptake into macrophage cell lines (J774E). Intraperitoneal injection of G-hLAL produced peak activities in plasma at 60 min and in the liver and spleen at 240 min. The t1/2 values were: ∼90 min (plasma), ∼14 h (liver), and ∼32 h (spleen), with return to baseline by ∼150 h in liver and ∼200 h in spleen. Ten injections of G-hLAL (every 3 days) into lal−/− mice produced normalization of hepatic color, decreases in hepatic cholesterol and TG contents, and diminished foamy macrophages in liver, spleen, and intestinal villi. All injected lal−/− mice developed anti-hLAL protein antibodies, but suffered no adverse events. These studies demonstrate the feasibility of using plant-expressed, recombinant hLAL for the enzyme therapy of human WD/CESD with general implications for other lysosomal storage diseases.
Keywords: cholesteryl esters, triglyceride, plant-produced human enzyme, macrophage, pharmacokinetics, pharmacodynamics, enzyme therapy
Lysosomal acid lipase (LAL) hydrolyzes cholesteryl esters (CEs) and triglycerides (TGs) that are delivered to the lysosomes by receptor-mediated endocytosis (1). Mutations in the human LAL (hLAL) gene cause two distinct phenotypes, Wolman disease (WD) and cholesteryl ester storage disease (CESD) (2). WD is an infantile-onset disorder with an incidence of <1/300,000 live births (3). Affected WD infants display massive accumulations of CEs and TGs in macrophages throughout the viscera. CE and TG accumulations in liver and lung can lead to liver cirrhosis and pulmonary fibrosis (4, 5). Excess CEs in the zona reticularis of the adrenal gland lead to adrenal calcification and insufficiency (2, 6). WD patients develop cachexia from malabsorption related to the accumulation of engorged macrophages in the villi of the small intestine (2, 4). The mean life span for patients with WD is ∼6 months. In comparison, CESD is a later-onset disorder, and has greater prevalence than WD. The incidence is unknown, but may be two to three times that of WD. Hepatomegaly can be the only clinical manifestation of CESD and results primarily from the accumulation of macrophages (Kupffer cells) engorged with CEs (7). Some patients develop cirrhosis, and/or cholestasis, as well as premature atherosclerosis due to the impaired homeostasis of cholesterol, CE, and TG (7).
Bone marrow transplantation for the treatment of WD has had only minor therapeutic impact, owing to the need for matched donors and the cachexia of affected individuals (5). Unrelated umbilical cord blood transplantation to treat WD may be more promising (6). For CESD, therapeutic efforts have focused on diminishing the accumulation of CEs within the liver and spleen using HMG-CoA-reductase inhibitors. Some effects have been observed on lipoprotein metabolism, but no consistent phenotypic or outcome effects are apparent with these agents (8). A combination therapy of lovastatin and ezetimibe, an inhibitor of the Niemann-Pick type C1-like gene product that regulates sterol absorption in the small intestine (9–11), led to enhanced effects on lowering plasma LDL-cholesterol in one CESD patient (12).
Enzyme and gene strategies have been tested for therapeutic effects in the LAL-null (lal−/−) mouse model (13, 14). Peritoneal administration of hLAL, expressed in Pichia pastoris (phLAL), into lal−/− mice resulted in significant reductions of hepatomegaly and lipid storage in the liver, spleen, and small intestine. Chinese hamster ovary (CHO)-produced LAL (chLAL) showed similar results (15). However, with phLAL, the hepatic reduction of lipid storage was primarily in Kupffer cells, whereas the lipid storage in the hepatic parenchymal cells was unchanged (14). With the chLAL, hepatocyte and Kupffer cell storage was improved. The initial explanation for this was that the yeast production system for phLAL produced glycoproteins having α-mannosyl-terminated N-linked oligosaccharides (16). However, further testing with phLAL in macrophage mannose receptor (MMR) and LAL double-deficient mice (lal−/−;MMR−/−) showed therapeutic effects in both Kupffer cells and hepatocytes, suggesting a MMR-independent uptake system in vivo (15). chLAL has posttranslational modifications with complex glycosylations. Repeated intraperitoneal injections of chLAL into lal−/− mice showed lipid reductions in liver macrophages and hepatocytes (15). In comparison, a single intravenous injection of an adenovirus containing the hLAL cDNA into lal−/− mice showed significant corrections of lipid storage in the liver (Kupffer cells and hepatocytes), and peripheral uptake of hLAL in spleen and small intestine (13).
Each of the above approaches to therapy requires significant investment in infrastructure for clinical-grade enzyme or recombinant virus production. The potential for more-facile production of therapeutic proteins in plants has not been subjected to rigorous evaluation in preclinical models of human diseases. Here, purified hLAL expressed in Nicotiana benthamiana using the GENEWARE® expression system (G-hLAL) was shown to reduce hepatosplenomegaly and lipid storage in several target organs of lal−/− mice. These treatment outcomes and lack of toxicity are similar to those observed with hLAL from other recombinant sources.
MATERIALS AND METHODS
Animals
Mice were provided care in accordance with institutional guidelines under Institutional Animal Care and Use Committee (IACUC) approval at the Children's Hospital Research Foundation. The lal−/− mice were backcrossed for 10 generations into the friend virus B-type susceptibility/NIH background (FVB/N). Mice were hosted in microisolators, under 12 h/12 h dark/light cycles. Water and food (regular chow diets) were available ad libitum. The mice were genotyped by PCR-based screening of tail DNA (17).
Purification of G-hLAL
G-hLAL was purified from Nicotiana benthamiana plants by Large Scale Biology Corporation (Vacaville, CA). The GENEWARE® technology is a tobacco mosaic virus (TMV)-based transient vector system (18). The G-hLAL was secreted from tobacco leaf cells into the apoplast. The G-hLAL present in the interstitial space was recovered by vacuum infiltration and centrifugation (2,800 g, 20 min). Sodium chloride was added to a final concentration of 700 mM, and the interstitial fluid was adjusted to pH 5.0–5.2 to facilitate removal of photosynthetic plant impurities using centrifugation (10,000 g, 10 min) and filtration through a 0.65/0.45 μm capsule filter. The clarified interstitial fluid was loaded onto butyl-Sepharose FF resin equilibrated with buffer A (25 mM sodium acetate, 700 mM NaCl, pH 5.2), washed to ultraviolet (UV) baseline with buffer A, and eluted with a sodium taurocholate gradient. The eluate was dialyzed against buffer B (25 mM sodium acetate, pH 5.2) and then loaded onto SP Sepharose high performance equilibrated with 25 mM sodium acetate, 1 mM sodium taurocholate, pH 5.2. The SP Sepharose HP column was then washed to UV baseline with equilibration buffer and eluted with a salt gradient. This SP pool was dialyzed against PBS, pH 7.4, concentrated and filtered with a 0.2 μm membrane. The product was stored in liquid or lyophilized states. Purified G-hLAL was dispensed as liquid directly into borosilicate vials, or sucrose was added to a 3.5% final concentration, and then dispensed prior to lyophilization and stored at 2–8°C. G-hLAL purity was monitored by SDS-PAGE using 10–20% Tris-glycine gels (InVitrogen; Carlsbad, CA). N-terminal sequence analysis of the purified G-hLAL protein showed signal peptidase cleavage identical to that of the native human protein.
Peptide analysis and mass spectrometry
For peptide analyses, aliquots of typically digested G-hLAL (5 mg/ml) were mixed 1:1 with sample matrix [recrystallized α-cyano-4-hydroxycinnamic acid (5 mg/ml) in acetonitrile (ACN)-ethanol-deionized water-trifluoroacetic acid (TFA) (30:30:40:0.1; v/v/v/v)]. A 1 μl aliquot was applied to the matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF MS) plate for mass analysis. Sample spots were analyzed in reflectron positive-ion mode with 20 kV accelerating voltage, 72% grid voltage, 200 ns delay time, and guide wire voltage varying from 0.02% to 0.05% using a Voyager-DE STR Biospectrometry™ Workstation (Applied Biosystems; Foster City, CA). MALDI-TOF spectra were obtained over a mass range of 500–8,000 Da. External calibration was performed using the protonated molecular ions of adrenocorticotropic hormone (fragment 1-17, molecular weight = 2,093.0867) and bradykinin (fragment 2-9, MW = 904.4681). All spectral data were processed using Applied Biosystems Data Explorer 4.0.0.0. Protein identity was verified using General Protein/Mass Analysis for Windows, version 5.0 software (Lighthouse Data; Denmark).
Enzymatic deglycosylation and fluorescent labeling
G-hLAL samples were dialyzed against deionized water. An internal glycan standard, M5 (α1-3, α1-6 mannopentaose), was added, and the samples were dried. Each sample (100 μg) was resuspended in pronase buffer (10 mM Tris, pH 7.8, 10 mM CaCl2) with pronase (0.01 μg/μl). Samples were incubated for 18 h at 37°C, followed by heat inactivation. Pronase-digested G-hLAL samples were mixed with 0.2 mU of glycosidase A (CalBiochem; San Diego, CA) in 0.2% acetic acid and incubated for 18 h at 37°C. Released glycans were separated from peptides/proteins on C4 Spin Columns (Silica C-4 Macro-spin column; Harvard Apparatus, Inc., Hollistone, MA). The collected glycans were heated together with 2-aminobenzoic acid and sodium cyanoborohydride in DMSO-acetic acid for 2 h at 65°C, and then cooled.
Glycan HPLC separation
The glycan preparation was reconstituted in 50% ACN, 0.1% aqueous TFA, and subjected to normal-phase HPLC (1090M; Hewlett-Packard, Palo Alto, CA) using an LC-NH2 Supercosil™ column. The glycans were resolved in 0.1% formic acid with a gradient of deionized water and ACN. Eluted peaks were detected with a fluorescent detector (Hewlett-Packard 1046A) at excitation and emission wavelengths of 225 and 424 nm, respectively. Fractions were collected by an automatic fraction collector (Eyela DC-40; Tokyo Rikakikai Co. Ltd., Tokyo, Japan). HPLC peaks were integrated and analyzed quantitatively using an internal standard calibrated by glycan standards of 2-AB asialo-, galactosylated biantennary, core-substituted with fucose.
Glycan mass spectrometry
Dried glycan samples were reconstituted in water to approximately 1 pmole/μl and spotted on the MALDI-TOF plate using a matrix composed of 2,5-dihydroxybenzoic acid (2 mg/ml) in acetonitrile-water (30:70; v/v). Sample spots were analyzed in the reflectron negative-ion mode with 20 kV accelerating voltage, 72% grid voltage, 150 ns delay time, and guide wire voltage of 0.02%.
Enzyme uptake into J774E and J774A.1 macrophages
J774E and J774A.1 cells were maintained in MEM with 6-thioguanine (60 μM) or in DMEM medium, respectively, supplemented with 10% fetal calf serum, penicillin, and streptomycin (37°C; 5% CO2). For uptake studies, cells were seeded at 2 × 105 per well 1 day before adding G-hLAL or imiglucerase (Cerezyme™; Genzyme Corp., Cambridge, MA). The indicated amount of G-hLAL, with or without imiglucerase, was added to the cells for 24 h. The cells were washed twice with PBS, collected by cell scraper, and centrifuged (13,000 g, 1 min) at room temperature. The cellular proteins were extracted and analyzed by Western blot as described (14).
Study design
Three preclinical studies were designed to evaluate pharmacokinetics, pharmacodynamics, and therapeutic effects. A pilot study of three doses (0, 12, 24, or 48 U correspond to 0, 33, 66, or 133 μg) of G-hLAL (lot # LSBC040623L) was conducted using intraperitoneal injection into lal−/− mice. Mice were harvested 4 h post injection. Liver and spleen were collected in Boiun's fixative solution for immunohistochemical staining and kept frozen until assayed for LAL activity. Based on the preliminary studies, the initial pharmacokinetic study used 24 U per 25 g body weight with a total of 25 lal−/− mice. Blood samples were collected from the retro-orbital plexus at 0, 5, 10, 15, 30, 45, 75, 90, and 105 min and from inferior vena cava at 1, 2, 3, and 4 h post injection. Three time points were collected from each mouse and five mice were used in each time point. Mice were euthanized at postinjection times of 1, 2, 3, and 4 h. Liver, spleen, kidney, small intestine, lung, and brain were collected and fixed for histological analyses. Livers and spleens were collected for enzymatic assay.
For pharmacodynamics, 24 U (85 μg) of G-hLAL (lot # LSBC040811L) per 25 g mouse was used. A total of 55 lal−/− mice at 2 to 4 months of age received G-hLAL via intraperitoneal injection. Mice were euthanized post injection at 4, 7, and 16 h, and 2, 3, 4, 5, 7, 10, and 14 days. Plasma, liver, and spleen were harvested to determine LAL activity. Samples of liver and spleen were also fixed for histological analyses.
To evaluate therapeutic effects, twenty lal−/− mice at age 2.5 months were divided into four groups. Three tested groups received 24 U, 48 U, or 72 U (85, 170, or 255 μg) of G-hLAL (lot# LSBC040811L). The control group received PBS. Each mouse received an injection every third day for a total of 10 injections. Mice were sacrificed at 3.5 months of age. Liver, spleen, adrenal glands, and small intestine were collected in Boiun's fixative for paraffin blocks and in 4% paraformaldehyde for frozen blocks.
Histological analyses and immunohistochemical staining
Representative paraffin-embedded sections stained with hematoxylin-eosin (H and E) were analyzed by light microscopy. Immunohistochemical analyses of paraffin-embedded tissue sections were performed using a VECTASTAIN ABC kit (Vector Laboratories Inc., Burlingame, CA) (14). Frozen sections were prepared from liver, spleen, intestine, adrenal gland, and lymph nodes by standard cryostat procedures. Sections (10 μm) were stained with Oil Red O (0.5% in propylene glycol) in a 60°C oven for 10 min and then placed in 85% propylene glycol (1 min). The slides were counterstained with hematoxylin (19).
Tissue protein extraction and LAL enzyme activity
Aliquots (∼100 mg) of liver and spleen were homogenized, sonicated, and extracted in LAL tissue extraction buffer (∼750 μl) (10 mM NaPO4, pH 6.8, 1 mM EDTA, 0.02% NaAzide, 10 mM DTT, 0.5% NP-40). Protein concentrations were determined by bicinchoninic acid assay (Pierce; Rockford, IL) using BSA as the standard. Tissue extract aliquots (∼100 μl each) were stored at −20°C. G-hLAL activities were determined using 4-methylumbelliferyl-oleate (4-MUO) as substrate. All assays were conducted with two dilutions and in duplicate. Assays were linear within the time frame of these assays, and less than 10% of substrate was cleaved. One unit is 1 μmol of 4-MUO cleaved per min under standard assay conditions.
Tissue lipid extraction and cholesterol and triglyceride concentrations
Total lipids were extracted from liver, spleen, and small intestine by the method of Folch, Lees, and Sloane Stanley (20). Triglyceride and cholesterol contents were determined (21, 22).
Western blot analyses
Mouse sera, from PBS- or G-hLAL-injected mice, at dose 0, 5, and 10 were collected and assessed for anti-G-hLAL antibodies by Western blot analyses using G-hLAL (50 ng) as antigen. The titers of the anti-G-hLAL serum antibodies were estimated by Western blot analysis using 2-fold dilutions (up to 1:25,600).
Statistics analysis
Data were analyzed by nonparametric t-test using PRISM software (GraphPad; San Diego, CA). All data are presented as mean ± SEM. Statistical significance was set at P < 0.05.
RESULTS
Production and purification of G-hLAL
The wild-type, hLAL cDNA, encoding the mature hLAL and its 21 amino acid-secretory peptide was cloned into the TMV expression vector, pLSBC735, to produce the final expression vector, pLSBC G-hLAL.16 (18); all data were derived from studies using G-hLAL expressed from this construct. The major bands of the butyl Sepharose FF- and SP Sepharose HP-purified G-hLAL were resolved on SDS-PAGE (Fig. 1). These bands were recognized by anti-hLAL antibody on immunoblots (data not shown). These broad bands in the purified G-hLAL were due to heterogeneity of N-glycosylation (Table 1). The identity of G-hLAL was further verified by molecular weight of either full-length or tryptic peptides using MALDI-TOF MS and N-terminal sequencing (Table 2). Three G-hLAL protein lots had N-terminal sequence identical to that of the mature human protein. These lots had high specific activities (281–364.5 U/mg protein) and no residual TMV infectivity and passed Federal Department of Agriculture specifications for lack of bioburden and endotoxin, and TMV infectivity (Table 2).
Fig. 1.
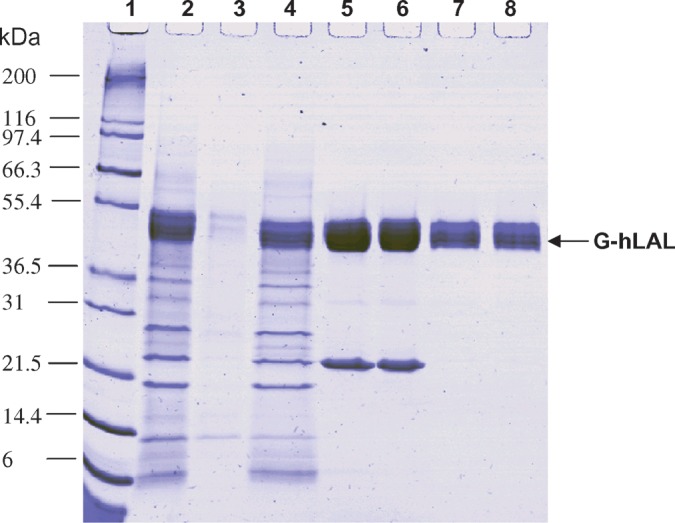
SDS-PAGE analysis of G-hLAL (human lysosomal acid lipase expressed in Nicotiana benthamiana using the GENEWARE® expression system) purification. Protein samples taken at each step of the purification procedure were separated by SDS-PAGE using a 10–20% Tris-glycine gel and stained with Coomassie Brilliant Blue stain. Lane 1, protein molecular mass marker, with corresponding molecular mass shown at left; lane 2, crude plant interstitial fluid extract; lane 3, proteins removed by centrifugation; lane 4, material loaded on a butyl Sepharose column; lane 5, pooled fractions eluted from butyl Sepharose; lane 6, material loaded onto an SP Sepharose column; lane 7, pooled fractions eluted from SP Sepharose; lane 8, dialyzed product before 0.2 μm filtration and vialing. The migration position of G-hLAL is indicated by the arrow.
TABLE 1.
Glycan forms identified from enzymatically deglycosylated G-hLAL
| Identity | Molecular Weight | Relative Percent | Mannose Terminal |
|---|---|---|---|
| M3XGlcNAc2 | 1,042.4 | 3.30 | + |
| M3XGlcNAc3 | 1,245.5 | 2.70 | + |
| M3FXGlcNAc2 | 1,188.4 | 29.20 | + |
| M5GlcNAc2 and M3GlcNAc4a | 1,234.4 and 1,316.5 | 4.80 | +/− |
| M3FXGlcNAc3 | 1,391.5 | 17.90 | + |
| M6GlcNAc2 | 1,396.5 | 4.20 | + |
| M3FXGlcNAc4 | 1,594.6 | 23.90 | — |
| M7GlcNAc2 and M4XGlcNAc4a | 1,558.5 and 1,610.6 | 6.00 | +/− |
| M3GlcNAc6 | 1,722.6 | 3.90 | — |
| Unidentifiedb | 1.30 | ||
| Unidentified | 1.30 | ||
| Unidentified | 1.30 | ||
| Unidentified | 0.40 |
G-hLAL, human lysosomal acid lipase expressed in Nicotiana benthamiana using the GENEWARE® expression system; M, mannose; X, xylose; F, fucose; GlcNAc, N-acetyl glucosamine. Numbers refer to the quantity of residues present for each sugar structure. No specific number represents one residue in a given structure.
Two glycan structures were coeluted in two distinct overlapping peaks. In each of these cases, the combined relative percent of the integrated peaks is reported.
The particular structures for four glycan peaks were not identified from available data.
TABLE 2.
Analytical characterization of three lots of G-hLAL
| Protein Property | LSBC040929L | LSBC040811L | LSBC040623L |
|---|---|---|---|
| Puritya | 99.00% | 99.60% | 98.40% |
| Identity: Average molecular mass (Da)b | 50,383 | 50,402 | 50,772 |
| Identity: Principal molecular mass (Da)b | 49,032, 49,635, 50,700, 52,167 | 49,303, 50,660, 51,244 | 49,003, 50,808, 52,506 |
| Identity: Matched tryptic peptidesb | 10 | 10 | 10 |
| Identity: N-terminal sequencec | Passed | Passed | Passed |
| Protein concentration (mg/ml) | 1.33 | 0.69 | 1.64 |
| Protein aggregation (monomer %:multimer %)d | 97.8:2.2 | 85:15 | 97:3 |
| Specific activity (U/mg protein) | 364.5 | 281 | 360 |
| pH | 7.0 | 7.3 | 7.3 |
| Bioburden | Passedg | Passed | Passed |
| Endotoxin (EU/mg)e | <1 | <1 | <1 |
| Residual TMVf | Passedh | Passed | Passed |
EU, endotoxin unit; TMV, tobacco mosaic virus.
Purity was determined by SDS-PAGE and densitometry scanning analysis.
The identity of the protein was determined by molecular mass of the full-length protein and the molecular matching of 10 tryptic peptides using matrix-assisted laser desorption-ionization time-of-flight mass spectrometry.
The identity of the protein was determined by N-terminal amino acid sequencing of the first 10 residues (sequenced by Protein Structure Core Facility, University of Michigan).
Protein aggregation (monomer vs. multimer) was determined by size exclusion chromotography.
Endotoxin was determined by Limulus Amebocyte Lysate assay as described (28).
Infectivity of G-hLAL was determined by local lesion bioassay as described (28).
The criteria for “passed” is zero colony-forming units per mg protein.
The criteria for “passed” is zero infectious TMV particles per mg protein.
Cellular uptake of G-hLAL, ex vivo and in vivo
Cellular targeting of recombinant-produced glycoproteins is largely dependent on the posttranslational modification derived from the host system (15). As shown in Table 1, ∼62.7% of glycan forms from G-hLAL were α-mannose-terminated, i.e., the cellular uptake of G-hLAL was likely to be MMR dependent. This was evaluated ex vivo using the murine macrophage cell lines J774E (MMR positive) or J774A.1 (MMR negative). Dose-dependent uptake of G-hLAL occurred in J774E cells, but not in J774A.1 cells (Fig. 2A, B). Coincubation of increasing amounts of G-hLAL with a fixed dose of imiglucerase, mannose-terminal glucocerebrosidase, with J774E cells showed competition for cellular incorporation (Fig. 2C, D). These data indicated uptake of G-hLAL into J774E cells through the MMR.
Fig. 2.
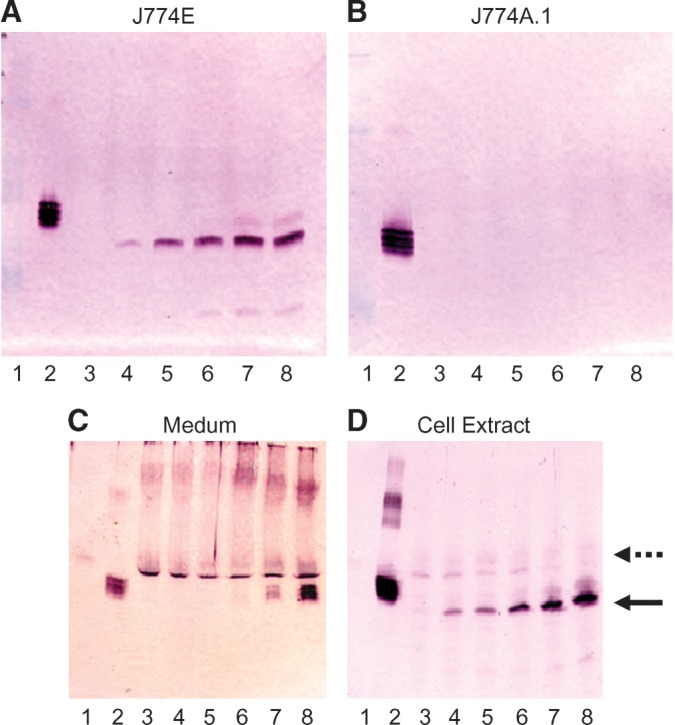
Mannose receptor-dependent macrophage uptake of G-hLAL. Cell extracts (5 μg protein) of J774E (A) and J774A.1 (B) were loaded onto 12.5% SDS-PAGE for Western blot analyses using rabbit anti-hLAL antibody. Lane 1, molecular weight markers; lane 2, G-hLAL (50 ng). Lanes 3–8: Cells were incubated with variable amounts of G-hLAL from 0 μg (lane 3), 0.6 μg (lane 4), 1.2 μg (lane 5), 2.4 μg (lane 6), 6.0 μg (lane 7), and 12 μg (lane 8). A dose-dependent uptake into J774E but not J774A.1 cells was evident. A constant amount of imiglucerase (12 μg) and a varied amount of G-hLAL (from 0 to 12 μg, lanes 3–8) were coincubated with J774E cells for 24 h. Cell medium (C) and cell extracts (D) were analyzed by Western blot analyses using a rabbit anti-hLAL antibody. Lane 1, molecular weight markers; lane 2, G-hLAL (50 ng). Lanes 3–8: Cells were incubated with a constant amount of imiglucerase (12 μg) and variable amounts of G-hLAL from 0 μg (lane 3), to 0.6 μg (lane 4), 1.2 μg (lane 5), 2.4 μg (lane 6), 6.0 μg (lane 7), and 12 μg (lane 8). Notice that the intracellular uptake of imiglucerase (indicated as dashed arrow) decreases as G-hLAL increase (indicated as solid arrow in D).
Pilot studies of four dose levels (0, 12, 24, and 48 U) were conducted with lal−/− mice using intraperitoneal injection (three mice per dose) based on our previous studies with phLAL (15). Dose-dependent LAL activity was detected in liver and spleen at 4 h. The LAL activity increased 2.2-fold in liver and 5.5-fold in spleen (12 U dose), 6.2-fold in liver and 12.3-fold in spleen (24 U dose), and 11.8-fold in the liver and 26-fold in spleen (48 U dose) (Fig. 3A). Cellular targeting (liver and spleen) of the G-hLAL in vivo was evaluated by immunohistochemistry with rabbit anti-hLAL. Uptake of G-hLAL was mainly in Kupffer cells and in splenic macrophages (arrows in panels C and D of Fig. 3B). The total recovery of LAL activity was 43.7% in liver and 0.23% in spleen.
Fig. 3.


Dose-dependent uptake of G-hLAL and cellular targeting of G-hLAL in the liver and spleen of LAL-null (lal−/−) mice. A: Mice (n = 3 for each dose) were harvested 4 h post intraperitoneal injection with 0, 12, 24, or 48 U of G-hLAL. Liver and spleen tissues (∼100 mg) were homogenized, sonicated, and extracted in LAL tissue extraction buffer. LAL enzyme activities in tissue extracts were determined and normalized for tissue extract protein concentrations. Error bars indicate ± SEM. B: Paraffin-embedded sections of liver and spleen from PBS-injected lal−/− mice (A and B, respectively) or G-hLAL-injected lal−/− mice (C and D, respectively) were processed by immunohistochemical staining with anti-hLAL antibody. Positive signals were evident in the Kupffer cells and in splenic macrophages of representative sections from lal−/− mice (arrows). Original magnification: 400× for liver and 200× for spleen.
Plasma clearance
LAL activities in plasma were assessed from 0 to 16 h following a single intraperitoneal dose (24 U/25 g body weight) of G-hLAL (Fig. 4A). Maximal plasma activity was achieved by ∼1 h, and a biphasic plasma clearance was observed. The disappearance half-lives of G-hLAL were ∼90 min and 5.5 h for phases 1 and 2, respectively. This pattern was similar to that observed with slow intravenous infusions of lysosomal enzymes in humans (23).
Fig. 4.



Time course of G-hLAL activities in plasma, liver, and spleen. A: Plasma LAL activity in blood samples collected from the retro-orbital plexus after single-dose (24 U per 25 g body weight) intraperitoneal injection of lal−/− mice. LAL activity in tissue extract of liver (B) or spleen (C), normalized for tissue extract protein concentrations, was determined for lal−/− mice that received a single dose (24 U per 25 g body weight) intraperitoneal injection. B,C: LAL activities in the liver and spleen during a 14 day time course. Inserts for B, C: details of LAL activity during the first 24 h in liver and 70 h in spleen post injection (as marked by hash bars on the X axis). Each data point represents the mean ± SE of five mice assayed in duplicate with two dilutions.
Tissue distribution and enzyme stability in vivo
After a single dose of G-hLAL (24 U/25 g body weight), tissues were harvested from 1 h to 14 days. The LAL activities were maximal at 4 h post injection in liver and spleen (inserts Fig. 4B, C). The half-lifes of G-hLAL were ∼14 h and ∼32 h in liver and spleen, respectively. The LAL activities return to background levels by ∼6 and ∼8 days in liver and spleen (Fig. 4B, C), respectively.
Therapeutic effects of G-hLAL
After 10 intraperitoneal administrations of G-hLAL to lal−/− mice, the yellow hepatic color, a manifestation of CE and TG storage, decreased in a dose-dependent manner (Fig. 5A). Similar color changes were observed in spleen and kidney (Fig. 5B). In lal−/− mice, the enlargement of a lipid-laden mesenteric lymph node is typical. In mice receiving G-hLAL, the size, but not the color, of this lymph node was significantly reduced (Fig. 5B, last row). This effect was not observed in lal−/− mice treated with hLAL from Pichia pastoris or CHO sources (14, 15). Differential effects on liver mass were observed between male and female mice receiving G-hLAL. In males, the liver was reduced from 15.6 to 9.3% of body weight (WT = 4.5–5%) and in females from 18.9 to 12.3%. This corresponds to reductions of the excess liver mass by ∼40.6% in males and 34.8% in females. Histological analyses of liver, spleen, and small intestine showed a dose-dependent reduction of lipid-laden macrophages (Fig. 6). Oil Red O staining of frozen sections showed dose-dependent clearance of lipid-laden macrophages (Fig. 7). However, hepatocytes were not cleared (Fig. 7, upper panels). To further confirm that lipid-storing macrophages were being cleared, staining with rat anti-mouse macrophage-specific antibody clearly showed a dose-dependent disappearance of positive cells. Two macrophage populations (storage cells) were present in the lal−/− mouse liver: large, clustered cells with lipid storage, and less-engorged, individual Kupffer cells (Fig. 8, arrow and arrowhead). G-hLAL treatment reduced the size and number of macrophages in the large clusters, but the number of individual Kupffer cells was unchanged (Fig. 8). Total macrophage numbers were greatly reduced in spleen and small intestine in G-hLAL-treated lal−/− mice (Fig. 8). Although the size of the mesenteric lymph node was reduced in treated mice (Fig. 5B), the H and E sections showed no differences in the remaining pathology between control and treated mice (data not shown). Also, the adrenal glands showed no effect of treatment (data not shown). Our previous findings with phLAL in lal−/− mice were similar. Significant reductions of cholesterol and TG levels were observed with all three doses in liver, spleen, lymph node, and small intestine (Fig. 9).
Fig. 5.
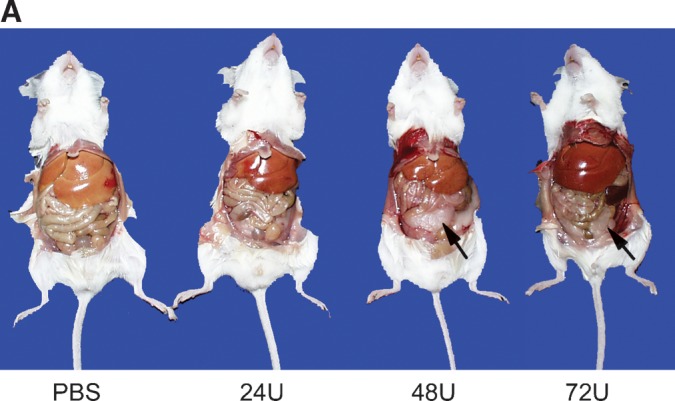

Correction of gross appearance and reduction of hepatomegaly by G-hLAL in lal−/− mice. A: Ventral views showing the yellow fatty liver in a typical PBS control lal−/− mouse. In G-hLAL-treated lal−/− mice, there is a dose-dependent correction of liver to normal hepatic color. Notice a recovery of white adipose tissue in 48 U and 72 U G-hLAL-injected lal−/− mice (arrow). B: Gross view of liver (top row), spleen (second row), kidney (third row), and mesenteric lymph node (bottom row) from PBS-treated lal−/− mice and G-hLAL-treated mice (24, 48, and 72 U). Notice a color correction in spleen and kidney in G-hLAL-treated lal−/− mice, and a size reduction of mesenteric lymph node in G-hLAL-treated lal−/− mice.
Fig. 6.

Hematoxylin-eosin (H and E) staining of liver, spleen, and small intestine of wild-type, and PBS- or G-hLAL-treated lal−/− mice. Sections were from age-matched lal+/+ (A, D, G), lal−/− untreated mice (B, E, H), or G-hLAL-treated with 72 U (C, F, I). The lipid-laden Kupffer cells (B), spleen (E), and small intestine (H) macrophages were clearly evident in untreated lal−/− mice (arrows). Complete correction of lipid storage in macrophages (arrows in C, I) was observed in macrophages of these tissues of G-hLAL-treated mice. Hepatocytes was not corrected (arrowhead). Top row, liver (Kupffer cells); middle row, spleen; bottom row, small intestine.
Fig. 7.

Oil Red O staining of liver, spleen, and small intestine sections of PBS- or G-hLAL-treated lal−/− mice. Sections were from livers (A–D), spleen (E–H), and small intestine (I–L) from PBS (A, E, I), G-hLAL 24 U (B, F, J), 48 U (C, G, K), and 72 U (D, H, L) -treated lal−/− mice. The neutral lipids, cholesterol esters, and triglycerides, were stained red with Oil Red O. A dose-dependent reduction of lipid-laden Kupffer cells (arrows in B, C, and D) and intestinal macrophages (arrows in J, K, L) of G-hLAL-treated lal−/− mice was evident. There is a complete correction of lipid storage in the spleens (arrows) of G-hLAL treated lal−/− mice. Oil Red O-positive lipid storage in hepatocytes was not corrected (arrowhead).
Fig. 8.

Dose-dependent reduction of macrophages in tissues of G-hLAL-treated lal−/− mice. Immunohistochemical staining of macrophages in the liver (A–D), spleen (E–H), and small intestine sections (I–L) from age-matched lal−/− mice untreated (A, E, I), or G-hLAL-treated with 24 U (B, F, J), 48 U (C, G, K), and 72 U (D, H, L). The macrophages were dark-brown color by immunohistochemical staining with rabbit anti-mouse macrophage membrane antibody. A dose-dependent reduction of lipid-laden Kupffer cells, splenic macrophages, and intestinal macrophages of G-hLAL-treated lal−/− mice (arrows) was evident. Regular Kupffer cells, which are not lipid laden, are indicated by arrowheads.
Fig. 9.

Dose-dependent reduction of tissue cholesterol and triglyceride in G-hLAL-injected lal−/− mice. Total cholesterol (left) and total TG (right) in liver, spleen, and mesenteric lymph node, and lipid concentration in intestine were compared between PBS-injected and G-hLAL-injected lal−/− mice (n = 5). The P values were from unpaired t-tests between the PBS- and G-hLAL-treated groups. Error bars indicate ± SEM.
Immune responses
Wild-type or the lal−/− mice receiving a single intraperitoneal dose of G-hLAL did not develop antibodies against G-hLAL. All multiply injected lal−/− mice developed anti-hLAL IgG antibodies at 1:100 dilution of mouse serum. A few of these mice showed antibody titers of 1:1600. The PBS-injected control group did not exhibit anti-hLAL antibodies. Western analyses, using plasma as the antibody source, showed that these antibodies reacted against deglycosylated G-hLAL. This result does not categorically exclude the possibility of antibodies recognizing plant glycan structures, but shows that the major antibodies were directed against the protein backbone. No gross untoward effects on the treated mice were observed after any injection, and no mice died of injection-related events. To address whether the antibody that developed in the mice could affect cellular uptake of sequential injected G-hLAL, mouse sera (positive for anti-G-hLAL antibody) were preincubated with different amounts of G-hLAL (0.0, 0.5, 1.0, 2.0, 4.0, and 8.0 μg) at room temperature for 30 min. The mixtures were then added to the media of J774A.1 and J774E cells that are Fcγ receptor positive. J774E cells are also MMR positive. Preincubation of mouse serum with G-hLAL did not affect the dose-dependent uptake in J774E cells, and enhanced the uptake of G-hLAL into the MMR receptor-negative, but Fcγ receptor-positive, J774A.1 cells. The uptake of G-hLAL into J774A.1 cells was G-hLAL dose independent (data not shown). These results suggest that anti G-hLAL antibody may facilitate ex vivo uptake of G-hLAL into Fcγ receptor-positive cells.
DISCUSSION
Significant therapeutic effects on the clinical, histologic, and biochemical manifestations of lal−/− were achieved after several injections of hLAL produced in Nicotiana plants. The present studies also showed the safety and efficacy of plant-based recombinant protein for enzyme therapy in the preclinical lal−/− mouse model. The plant-based recombinant protein production system, termed GENEWARE®, has significant advantages as a high-level expression system (18). This system lacks: 1) the less-desirable properties associated with the use of food/feed crops; 2) the risks of horizontal transmission; and 3) the slow expression cycles (18). Additionally, this system: 1) lacks contamination by mammalian pathogens; 2) has ease and speed for genetic manipulation; 3) retains the eukaryotic protein modification (proteolytic and glycosylation) machinery; 4) has potential for economical production; and 5) achieves lower endotoxin levels than microbial systems.
In vitro cellular targeting showed MMR-dependent uptake of G-hLAL in J774E cells. This is consistent with the N-linked oligosaccharide modification of G-hLAL that has mostly mannose termini (Table 1), similar to those of hLAL produced in Pichia pastoris (phLAL) (14). In vivo G-hLAL was delivered to Kupffer cells of the liver, and to macrophages of the spleen and small intestine that are significantly affected by this enzyme deficiency. This is expected because macrophages in these organs express high levels of MMR, whereas hepatocytes express high levels of mannose-6-phosphate receptor, but not MMR. Pharmacokinetic and pharmacodynamic analyses of G-hLAL in lal−/− mice showed slow appearance in plasma (peaked at 60 min) and in liver and spleen (4 h), and slow clearance rates in plasma (t1/2 = 1.5 h and 5.5 h) and in the liver and spleen (t1/2 = 14 h and 32 h, respectively). To our knowledge, this study is the first report of systematic analysis of enzyme replacement via intraperitoneal injection and the first with human proteins made in plants. Most reported enzyme therapy studies in mouse models have used intravenous bolus injections that are dissimilar to the clinical usage of the drugs; e.g., bolus injection of imiglucerase showed peak activities in the liver and spleen at 20 min and half-lives of 40 and 60 min in the liver and spleen, respectively (19). The longer half-lives of G-hLAL in the liver (14 h) and spleen (32 h) could be attributed to: a) differences in administration routes, because intraperitoneal administration is slower than intravenous bolus injection for body absorption. We reasoned that intraperitoneal injections may resemble the slow intravenous infusion route of recombinant enzyme therapy in human patients; and b) in vivo stability of G-hLAL activity compared with that of imiglucerase. The stability of injected enzyme activity in vivo would clearly be of benefit for treatment.
Dose-dependent therapeutic effects of G-hLAL were evident between 24 and 72 U per dose; i.e., between 85 and 256 μg per 25 g mouse/dose. An effective dose for reducing gross hepatomegaly was 24 U. Higher doses (48 U and 72 U) were significantly more effective than the lower dose in reducing the amount of neutral lipid stored in the macrophages of the liver, spleen, and small intestine of lal−/− mice. A more general metabolic effect of enzyme therapy was evidenced by the redeposition of subcutaneous and mesenteric white adipose tissue that was absent in PBS-treated lal−/− mice (data not shown and Fig. 5A). Interestingly, although the administration of G-hLAL did not alter the histology of lipid-stored macrophages in mesenteric lymph nodes in comparison to PBS-injected control mice, their size and total lipid amount were clearly reduced. This specific effect has not been observed in lal−/− mice receiving administration of hLAL produced in Pichia pastoris (phLAL) or CHO cells (chLAL) (14, 15).
Plant-based expression systems N-glycosylated recombinant proteins (24), e.g., human α-galactosidase expressed in GENEWARE system. Each of the N-glycosylation sites known to be occupied in mammalian-produced enzymes were occupied in plant-produced α-galactosidase A (25). Another example is human glucocerebrosidase, expressed in carrot cells, with terminal mannose residues on its complex glycans, and with biological activity similar to that of imiglucerase produced in CHO cells (26). Like mammals, plants produce proteins with complex N-linked glycans that have a core of two GlcNAc residues and three mannose residues. In plant glycoproteins, this core often has a β[1,2]-linked xylose residue (core xylose) and an α[1,3]-linked fucose (core α[1,3]fucose) (Fig. 10) (24), while nonplant glycoproteins have an α[1,6]-linked fucose attached to an N-linked glycan core. The immunogenicity of plant N-glycans with core xylose and core fucose has been evaluated in rats and mice. The data showed that rats and selected mouse strains (e.g., in C57BL/6, but not in BALB/c mice) developed weak antibodies specific for core xylose and core α[1,3]fucose epitopes (27). Additionally, 50% of sera from nonallergic human blood donors had antibody titers specific to core xylose, and 25% had antibody specific for core α[1,3]fucose (27). Whether this could lead to adverse effects with administrated plant-derived enzymes is not known. Here, the lal−/− mouse showed an immune response to G-hLAL protein, but no evidence was found for antibody to the plant oligosaccharides. Also, the antibody to G-hLAL did not affect the efficacy of treatment.
Fig. 10.

N-glycan structures identified from G-hLAL. Three major N-glycan structures identified from G-hLAL are shown: M3FXGlcNAc2 (A), M3FXGlcNAc3 (B), and M3FXGlcNAc4 (C).
In summary, evidence is provided that plant-produced recombinant protein G-hLAL is active in vitro and in vivo; the intraperitoneally administered active enzyme has a relatively long half-life in plasma, liver, and spleen that could be of therapeutic benefit in vivo. Finally, plant-derived G-hLAL has therapeutic effects without toxicity in the range of doses tested in the lal−/− mouse.
Acknowledgments
The authors would like to thank Lisa McMillin for her help with the histology, and Nathan F. Wenzel, Elizabeth DuPont, Huimin Li, and Lori Stanton for their technical assistance.
Published, JLR Papers in Press, April 15, 2008.
Footnotes
This work was partially supported by National Institutes of Health Grant DK-36729 (G.A.G.) and a grant from the Large Scale Biology Corp (G.A.G).
References
- 1.Goldstein J. L., S. E. Dana, J. R. Faust, A. L. Beaudet, and M. S. Brown. 1975. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J. Biol. Chem. 250 8487–8495. [PubMed] [Google Scholar]
- 2.Assmann, G., and U. Seedorf. (2001). Acid lipase deficiency: Wolman disease and cholesteryl ester storage disease. In Metabolic and Molecular Bases of Inherited Diseases. C. R. Scriver, A. L. Beaudet, D. Valle, and W. S. Sly, editors. McGraw-Hill, New York. 3551–3572.
- 3.Meikle P. J., J. J. Hopwood, A. E. Clague, and W. F. Carey. 1999. Prevalence of lysosomal storage disorders. J. Am. Med. Assoc. 281 249–254. [DOI] [PubMed] [Google Scholar]
- 4.Boldrini R., R. Devito, R. Biselli, M. Filocamo, and C. Bosman. 2004. Wolman disease and cholesteryl ester storage disease diagnosed by histological and ultrastructural examination of intestinal and liver biopsy. Pathol. Res. Pract. 200 231–240. [DOI] [PubMed] [Google Scholar]
- 5.Krivit W., C. Peters, K. Dusenbery, Y. Ben-Yoseph, N. K. Ramsay, J. E. Wagner, and R. Anderson. 2000. Wolman disease successfully treated by bone marrow transplantation. Bone Marrow Transplant. 26 567–570. [DOI] [PubMed] [Google Scholar]
- 6.Stein J., B. Z. Garty, Y. Dror, E. Fenig, M. Zeigler, and I. Yaniv. 2006. Successful treatment of Wolman disease by unrelated umbilical cord blood transplantation. Eur. J. Pediatr. 166 663–666. [DOI] [PubMed] [Google Scholar]
- 7.Beaudet A. L., G. D. Ferry, B. L. Nichols, Jr., and H. S. Rosenberg. 1977. Cholesterol ester storage disease: clinical, biochemical, and pathological studies. J. Pediatr. 90 910–914. [DOI] [PubMed] [Google Scholar]
- 8.Ginsberg H. N., N. A. Le, M. P. Short, R. Ramakrishnan, and R. J. Desnick. 1987. Suppression of apolipoprotein B production during treatment of cholesteryl ester storage disease with lovastatin. Implications for regulation of apolipoprotein B synthesis. J. Clin. Invest. 80 1692–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altmann S. W., H. R. Davis, Jr., L. J. Zhu, X. Yao, L. M. Hoos, G. Tetzloff, S. P. Iyer, M. Maguire, A. Golovko, M. Zeng, et al. 2004. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 303 1201–1204. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Calvo M., J. Lisnock, H. G. Bull, B. E. Hawes, D. A. Burnett, M. P. Braun, J. H. Crona, H. R. Davis, Jr., D. C. Dean, P. A. Detmers, et al. 2005. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc. Natl. Acad. Sci. USA. 102 8132–8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smart E. J., R. A. De Rose, and S. A. Farber. 2004. Annexin 2-caveolin 1 complex is a target of ezetimibe and regulates intestinal cholesterol transport. Proc. Natl. Acad. Sci. USA. 101 3450–3455. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Tadiboyina V. T., D. M. Liu, B. A. Miskie, J. Wang, and R. A. Hegele. 2005. Treatment of dyslipidemia with lovastatin and ezetimibe in an adolescent with cholesterol ester storage disease. Lipids Health Dis. 4 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du H., M. Heur, D. P. Witte, D. Ameis, and G. A. Grabowski. 2002. Lysosomal acid lipase deficiency: correction of lipid storage by adenovirus-mediated gene transfer in mice. Hum. Gene Ther. 13 1361–1372. [DOI] [PubMed] [Google Scholar]
- 14.Du H., S. Schiavi, M. Levine, J. Mishra, M. Heur, and G. A. Grabowski. 2001. Enzyme therapy for lysosomal acid lipase deficiency in the mouse. Hum. Mol. Genet. 10 1639–1648. [DOI] [PubMed] [Google Scholar]
- 15.Du H., M. Levine, C. Ganesa, D. P. Witte, E. S. Cole, and G. A. Grabowski. 2005. The role of mannosylated enzyme and the mannose receptor in enzyme replacement therapy. Am. J. Hum. Genet. 77 1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trimble R. B., C. Lubowski, C. R. Hauer, III, R. Stack, L. McNaughton, T. R. Gemmill, and S. A. Kumar. 2004. Characterization of N- and O-linked glycosylation of recombinant human bile salt-stimulated lipase secreted by Pichia pastoris. Glycobiology. 14 265–274. [DOI] [PubMed] [Google Scholar]
- 17.Du H., M. Duanmu, D. Witte, and G. A. Grabowski. 1998. Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Hum. Mol. Genet. 7 1347–1354. [DOI] [PubMed] [Google Scholar]
- 18.Pogue G. P., J. A. Lindbo, S. J. Garger, and W. P. Fitzmaurice. 2002. Making an ally from an enemy: plant virology and the new agriculture. Annu. Rev. Phytopathol. 40 45–74. [DOI] [PubMed] [Google Scholar]
- 19.Lian X., C. Yan, L. Yang, Y. Xu, and H. Du. 2004. Lysosomal acid lipase deficiency causes respiratory inflammation and destruction in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 286 L801–L807. [DOI] [PubMed] [Google Scholar]
- 20.Folch J., M. Lees, and G. H. Sloane Stanley. 1957. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226 497–509. [PubMed] [Google Scholar]
- 21.Du H., M. Heur, M. Duanmu, G. A. Grabowski, D. Y. Hui, D. P. Witte, and J. Mishra. 2001. Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J. Lipid Res. 42 489–500. [PubMed] [Google Scholar]
- 22.Rudel L. L., and M. D. Morris. 1973. Determination of cholesterol using o-phthalaldehyde. J. Lipid Res. 14 364–366. [PubMed] [Google Scholar]
- 23.Barton N. W., R. O. Brady, J. M. Dambrosia, A. M. Di Bisceglie, S. H. Doppelt, S. C. Hill, H. J. Mankin, G. J. Murray, R. I. Parker, and C. E. Argoff. 1991. Replacement therapy for inherited enzyme deficiency–macrophage-targeted glucocerebrosidase for Gaucher's disease. N. Engl. J. Med. 324 1464–1470. [DOI] [PubMed] [Google Scholar]
- 24.Lerouge P., M. Cabanes-Macheteau, C. Rayon, A. C. Fischette-Laine, V. Gomord, and L. Faye. 1998. N-glycoprotein biosynthesis in plants: recent developments and future trends. Plant Mol. Biol. 38 31–48. [PubMed] [Google Scholar]
- 25.Gelderman M. P., K. L. Oliver, A. T. Yazdani, G. J. Murray, G. F. Miller, T. I. Cameron, S. J. Garger, T. H. Turpen, R. B. Holtz, and R. O. Brady. 2004. Preclinical studies with plant-produced alpha-galactosidase A in Fabry mice show potential for replacement therapy. Preclinica. 2 67–74. [Google Scholar]
- 26.Shaaltiel Y., D. Bartfeld, S. Hashmueli, G. Baum, E. Brill-Almon, G. Galili, O. Dym, S. A. Boldin-Adamsky, I. Silman, J. L. Sussman, et al. 2007. Production of glucocerbrosidase with terminal mannose glycans for enzyme replacement therapy of Gaucher's disease using a plant cell system. Plant Biotechnol. J. 5 579–590. [DOI] [PubMed] [Google Scholar]
- 27.Bardor M., C. Faveeuw, A. C. Fitchette, D. Gilbert, L. Galas, F. Trottein, L. Faye, and P. Lerouge. 2003. Immunoreactivity in mammals of two typical plant glyco-epitopes, core alpha(1,3)-fucose and core xylose. Glycobiology. 13 427–434. [DOI] [PubMed] [Google Scholar]
- 28.Palmer K. E., A. Benko, S. A. Doucette, T. I. Cameron, T. Foster, K. M. Hanley, A. A. McCormick, M. McCulloch, G. P. Pogue, M. L. Smith, et al. 2006. Protection of rabbits against cutaneous papillomavirus infection using recombinant tobacco mosaic virus containing L2 capsid epitopes. Vaccine. 24 5516–5525. [DOI] [PubMed] [Google Scholar]