

Abstract
Deposits of amyloid β-protein (Aβ) in neuritic plaques and cerebral vessels are a pathological hallmark of Alzheimer’s disease. Fibrillar Aβ deposits are closely associated with inflammatory responses such as activated microglia in brain with this disease. Increasing lines of evidence support the hypothesis that activated microglia, innate immune cells in the CNS, play a pivotal role in the progression of the disease: either clearing Aβ deposits by phagocytic activity or releasing cytotoxic substances and pro-inflammatory cytokines. Toll-like receptors (TLRs) are a family of pattern-recognition receptors in the innate immune system. Exogenous and endogenous TLR ligands activate microglia. To investigate the role of TLR4 in the amyloidogenesis in vivo, we determined the amounts of cerebral Aβ in Alzheimer’s disease mouse models with different genotypes of TLR4 using three distinct methods. We show that mouse models (Mo/Hu APPswe PS1dE9 mice) homozygous for a destructive mutation of TLR4 (TlrLps-d/TlrLps-d) had increases in diffuse and fibrillar Aβ deposits by immunocytochemistry, fibrillar Aβ deposits by thioflavine-S staining and buffer-soluble and insoluble Aβ by ELISA in the cerebrum, as compared with TLR4 wild-type mouse models. Although the differences in these parameters were less significant, mouse models heterozygous for the mutation (TlrLps-d/θ) showed co-dominant phenotypes. Consistent with these observations in vivo, cultured microglia derived from TlrLps-d/TlrLps-d mice failed to show an increase in Aβ uptake after stimulation with a TLR4 ligand but not with a TLR9 ligand in vitro. Furthermore, activation of microglia (BV-2 cell) with a TLR2, TLR4 or TLR9 ligand, markedly boosted ingestion of Aβ in vitro. These results suggest that TLR signalling pathway(s) may be involved in clearance of Aβ-deposits in the brain and that TLRs can be a therapeutic target for Alzheimer’s disease.
Keywords: Alzheimer’s disease, amyloid, toll-like receptors, microglia, transgenic mouse
Introduction
Alzheimer’s disease is a neurodegenerative disorder and the most common cause of dementia in the elderly. Patients show a gradual onset and progression of memory loss and other cognitive deficits. Cardinal pathological changes include neurofibrillary tangles, mainly composed of paired helical filaments consisting of hyper-phosphorylated tau and deposits of aggregated amyloid β-protein (Aβ) in neuritic plaques and cerebral vessels (cerebrovascular amyloid angiopathy). A severe loss of large neurons in the nucleus basalis of Meynert, hippocampus and neocortex is also characteristic of this disease. Aβ is produced from amyloid β-protein precursor (APP) by proteolytic processing. Aβ is heterogeneous at its C-terminus, resulting in peptides of 39–42 amino acids. Diffuse plaques mainly contain Aβ42 composed of 42 amino acids and are recognized by immunocytochemistry but not by Congo red/thioflavine staining. On the other hand, neuritic plaques contain fibrillar Aβ consisting of both Aβ40 and Aβ42 and are recognized by Congo red birefringence and thioflavine-S fluorescence (Iwatsubo et al., 1994; Savage et al., 1995). Aβ42 is less soluble and forms oligomers and fibrils faster than Aβ40 in vitro (Jarrett and Lansbury, 1993). Thus, diffuse plaques are thought to be precursors of neuritic plaques. Soluble Aβ oligomers are much more toxic than Aβ fibrils in vitro and in vivo (Lambert et al., 1998; Dahlgren et al., 2002; Klein, 2002; Walsh et al., 2002; Wang et al., 2002). Genetic analyses of DNA from patients with familial forms of Alzheimer’s dementia and molecular biological studies of APP metabolism led to the hypothesis that accumulation of Aβ, particularly Aβ42, in the brain is an early essential event for the pathogenesis of this illness (Hardy and Selkoe, 2002; Tanzi and Bertram, 2005).
The innate immune system is the vital first line of defence against a wide range of pathogens and tissue injuries, which trigger inflammation through activation of quiescent microglia and macrophages. Microglia are resident phagocytes of the innate immune system and are considered as macrophages in the CNS. Aβ deposits, particularly in fibrillar form, are closely associated with inflammatory responses in the Alzheimer’s disease brain, as evidenced by a local up-regulation of acute phase proteins, pro-inflammatory cytokines, complement components and proteases (Akiyama et al., 2000). Additionally, a number of epidemiological studies indicate that long-term use of nonsteroidal anti-inflammatory drugs reduces the risk of developing this type of dementia and delays its onset. (McGeer et al., 1990; Breitner et al., 1994; Breitner et al., 1995; Stewart et al., 1997; Veld et al., 2000; Szekely et al., 2004). These observations led to the hypothesis that the deposition of Aβ in the brain activates microglia, initiating a pro-inflammatory cascade that results in the release of potentially cytotoxic molecules, cytokines and other related compounds, causing neurodegeneration (the inflammatory hypothesis for Alzheimer’s disease) (Akiyama et al., 2000). Based upon more recent observations, this hypothesis has been challenged (Schenk and Yednock, 2002). Schenk et al. (1999) demonstrated that immunization of Alzheimer’s disease mouse models with synthetic Aβ prevented or reduced Aβ deposits and indicated that Aβ deposits were cleared through the Fc receptor-mediated phagocytosis by microglia (Schenk et al., 1999; Bard et al., 2000). In concord with this view, clearance of Aβ deposits in patients after vaccination with synthetic Aβ was associated with activated microglia that ingested Aβ (Nicoll et al., 2003; Ferrer et al., 2004). Furthermore, in vitro, microglia have been shown to be capable of phagocytosis of Aβ deposits (Ard et al., 1996; Webster et al., 2000; Koenigsknecht and Landreth, 2004) and slowly degrade phagocytosed Aβ (Paresce et al., 1996, 1997; Brazil et al., 2000). Consequently, some investigators have proposed that microglia could play a neuroprotective role as well as pathogenetic role in the aetiology of the disease (Town et al., 2001; Schenk and Yednock, 2002; Morgan et al., 2005; Walker and Lue, 2005).
Toll-like receptors (TLRs) are a class of pattern-recognition receptors in the innate immune system (Gordon, 2002). The role of TLRs is to activate phagocytes and tissue dendritic cells to respond to insults including pathogens and damaged host cells by secreting chemokines and cytokines, and to express the co-stimulatory molecules needed for protective immune responses, efficient clearance of damaged tissues and activating adaptive immunity. There are at least 11 known members of TLRs (TLR1 through 11). Each TLR now has exogenous ligands derived from a variety of microorganisms, such as bacteria, mycoplasma, yeasts, viruses and parasites, as well as endogenous ligands derived from damaged host cells, which includes lipids, proteins and nucleic acids (Ishii et al., 2005). Because microglia are activated by a number of TLR ligands (Bsibsi et al., 2002; Olson and Miller, 2004), activation of TLRs on microglia may modulate disease progression. In this study, in order to investigate the role of TLR4 in the amyloidogenesis in vivo, we determined the amounts of cerebral Aβ in Alzheimer’s disease mouse models with a destructive mutation of the TLR4 gene using three distinct methods: diffuse and fibrillar Aβ deposits by immunohistochemistry, fibrillar Aβ deposits by thioflavine-S staining and buffer-soluble and insoluble Aβ by enzyme-linked immunosorbent assay (ELISA). We used Mo/Hu APPswe PS1dE9 mice (Jankowsky et al., 2004) that develop numerous Aβ deposits in the brain. Mo/Hu APPswe PS1dE9 mice have been maintained by mating with B6C3F1 mice that were produced from a cross between C57BL/6J and C3H/HeJ mice (Jackson Laboratory, Bar Harbor, ME). C3H/HeJ mice are highly susceptible to Gram-negative infection and yet are resistant to bacterial lipopolysaccharide (LPS, endotoxin) due to a destructive mutation of the TLR4 gene (TlrLps-d), which causes lack of TLR4 activation with LPS (Poltorak et al., 1998). This mutation is thought to be co-dominant. Therefore, the phenotypic manifestation of the mutation makes Mo/Hu APPswe PS1dE9 mice suitable for studying the role of TLR4 in the amyloidogenesis in vivo. Additionally, we investigated Aβ uptake by primary microglial cultures derived from TLR4 wild-type and TlrLps-d/TlrLps-d mice. Using three different detection methods for Aβ uptake, we have confirmed a marked increase in Aβ uptake by BV-2 microglial cells upon activation of TLRs in vitro also.
Material and methods
Animals
A pathogen-free transgenic line of Alzheimer’s disease mouse model, Mo/Hu APPswe PS1dE9 mice (Jankowsky et al., 2004), was obtained from Jackson Laboratory (Bar Harbor, ME) and maintained by crossing transgenic males with B6C3F1 females that were purchased from Jackson Lab, also. The genotyping for the APPswe transgene was performed by the PCR-based method provided by the Jackson Lab. Mo/Hu APPswe PS1dE9 mice used were 14–16 months old. For each experimental group, 4–9 mice were used.
B6C3F1 females were mated with B6C3F1 males to produce B6C3F2 mice that were genotyped for the TLR4 mutation (TlrLps-d) as described below. B6C3F2 mice homozygous for the TLR4 mutation were crossed with each other to produce TlrLps-d/TlrLps-d pups for isolation of microglial cells. TLR4 wild-type pups were produced by mating B6C3F2 mice free of the TLR4 mutation.
All animal protocols used for this study were prospectively reviewed and approved by the Institutional Animal Care and Use Committees of the University of Alabama at Birmingham and the University of Illinois College of Medicine at Peoria.
TLR4 genotyping
Mouse tail DNA was extracted using the Extract-N-Amp tissue PCR kit (Sigma-Aldrich, St Louis, MO) according to the manufacturer’s protocol. The PCR was carried out in 25 μl vol using 1 μl of tail DNA extract and 200 nM each of two primers for the TLR4 gene through 35 cycles consisting of 30 s at 94°C, 30 s at 55°C and 1 min at 72°C. The last elongation step was done at 72°C for 5 min. The two primer sequences, TLR4sen193 = 5′-GAG ATG AAT ACC TCC TTA GTG TTG G-3′ and TLR4ant607 = 5′-ATT CAA AGA TAC ACC AAC GGC TCT GA-3, were used to amplify a part of exon 3 of the TLR4 gene, on which the mutation in C3H/HeJ was localized (Poltorak et al., 1998). After purification by phenol–chloroform extraction, the PCR products were digested with Nla III restriction enzyme and subjected to electrophoresis through a 1.5% agarose gel. Mousetail DNAs extracted from a C3H/HeJ and C57BL/6J mouse were used as a positive and negative control for the TLR4 mutation, respectively.
Quantification of Aβ deposits by morphometry
Mice were sacrificed by lethal injection of sodium pentobarbital and the brains were quickly removed. After removing the cerebellums, the right hemispheres were processed for morphometric analysis as described previously (Kim et al., 2004). In brief, four micron-tissue sections were immunostained with 6E10 antibody using Vectastain ABC reagent for detection of Aβ deposits. Ten micron brain sections were subjected to thioflavine-S staining for detection of Aβ fibrils. The amyloid burden was quantified by histomorphometry consisting of an Olympus BX61 automated microscope capable of both brightfield and fluorescence, an Olympus Fluoview system and the Image Pro Plus v4 image analysis software (Media Cybernetics, Silver Spring, MD) capable of colour segmentation and automation via programmable macros. The entire hippocampus and neocortex in each slide were scanned. Approximately 40 fields (1 mm2 each, using a × 10 objective and a × 1 eyepiece lens) from 5 to 7 coronal brain sections, each separated by >240 μm interval, from each mouse were analyzed with each staining method. Amyloid burden was expressed as a percentage of total area covered by Aβ immunoreactivity or thioflavine-S fluorescence. Data were expressed as mean ± standard error. Intergroup differences were assessed by two-tailed Student’s t-test. P < 0.05 was considered statistically significant.
Quantification of brain Aβ by ELISA and brain APP and PS1 by western blot analysis
The left hemispheres were dounce-homogenized in carbonate buffer (100 mM Na2CO3, 50 mM NaCl, pH 11.5) containing protease inhibitors [10 μg/ml aprotinin and 1 mM 4-(2-aminoethyl)benzenesulphonyl fluoride hydrochloride (AEBSF)] and centrifuged at 16 000 g for 30 min at 4°C. After diluting the supernatants by phosphate-buffered saline (PBS), protein concentrations were determined by the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA), and levels of buffer-soluble Aβ were determined by the Aβ42 and Aβ40 ELISA kits (Biosource International, Camarillo, CA) according to the manufacturer’s protocol. The pellets were further dounce-homogenized in guanidine hydrochloride (final concentration, 5 M) and then rock-shaken for 3–4 h at room temperature. The solubilized pellets were then diluted with PBS at various concentrations for determination of Aβ concentration by the Aβ42 and Aβ40 ELISA kits.
Expression levels of APP and presenilin 1 (PS1) transgenes in brain were determined by western blot analysis followed by densitometric scanning. Aliquots of carbonate homogenates of the left hemispheres described above were further homogenized with the same volume of 2 × Laemmli buffer (1× = 62.5 mM Tris–HCl, pH 6.8, 2% SDS, 10% glycerol, 5% 2-mercaptoethanol and 0.001% bromophenol blue) with protease inhibitors (1× final concentration of Complete Mini, Roche, Mannheim, Germany) and subjected to western blot analysis as described below.
Primary microglial culture
Primary TLR4 wild-type and TlrLps-d/TlrLps-d microgia were prepared from postnatal 1–3 day mice and cultured as described by Woodroofe and Cuzner (1995). Briefly, cerebral cortex from 8 to 10 neonatal mice were minced after removing meninges and dissociated in Earle’s Balanced Salt Solution containing trypsin (0.125%), DNase I (10 μg/ml) and collagenase (100 U/ml). After passing through 70 μm nylon mesh, cells in Dulbecco’s Minimum Essential Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin were plated into a 75 cm2 flask at the density of 8.5 × 105 cells/ml, 10 ml/flask and incubated at 37°C. After 8 days, post-incubation, flasks were shaken for 20 h at 180 r.p.m. at 37°C. Microglia released into the supernatant were harvested and plated into a fresh flask at the density of 1 × 105 cells/ml. After 1 h, any non-attached cells were removed and attached microglia were harvested using cell scrapers (Fisher Scientific, Pittsburg, PA). Isolated microglia were plated into four chamber Tab-Tek culture slides (Nunc, Naperville, IL). The purity of the culture (>90%) was confirmed by expression of a microglial marker, using anti-mouse CD11b antibody (clone 5C6) (Serotec, Raleigh, NC).
BV-2 cell culture and TLR ligand treatment for ELISA and western blot analysis
A mouse cell line of microglia, BV-2 (gift from Dr Michael McKinney), was maintained in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin in 5% CO2 atmosphere at 37°C. One day before treatment with specific TLR ligands, BV-2 cells were inoculated at a concentration of 1.1 × 105 cells/1.5 ml/well into Falcon Multiwell 24-well plates (Becton Dickison, Franklin Lakes, NJ). The BV-2 cultures were treated with specific TLR ligands for 24 h. The concentrations used for TLR ligands were LPS from Escherichia coli 055:B55 (Sigma-Aldrich) at 1.2, 3.7, 11, 33 and 100 ng/ml, CpG oligodeoxynucleotides (ODN 1826; Invivogen, San Diego, CA) at 0.02, 0.06, 0.18 and 0.54 μM and peptidoglycan (PGN) from Staphylococcus aureus (Fluka) at 0.5, 1.5, 4.5 and 40 μg/ml. The culture was treated with 0.51 μM of control oligodeoxynucleotides (control ODN; Invitrogen), also. After washing the culture with PBS twice, 300 μl of OPTI-MEM I (Invitrogen) that contained 1 μM of oligomerized Aβ42 were added to the culture. After incubation for 24 h, the media and cells were harvested and centrifuged at 1500 r.p.m. in a bench top centrifuge for 10 min. The media were used to determine the concentrations of Aβ42, which was done by using the Aβ42 ELISA kits (Biosource International Camarillo, CA) according to the manufacturer’s protocol.
The cell pellets were further washed with cold PBS twice and then suspended in 30 μl of 2× Laemmli buffer with protease inhibitors. The cell lysates were used for western blotting. Three independent cultures were prepared for each TLR ligand and control treatment.
To determine the effects of chemical inhibitors on Aβ uptake, BV-2 cells were incubated with LPS (100 ng/ml) for 24 h. The culture was washed with PBS twice and placed in OPTI-MEM I. Pertussis toxin (PTX; 100 or 500 ng/ml; List Biological Laboratories, Campbell, CA), anti-mouse CD14 antibody (20 μg/ml; Clone 4CI/CD14; BD Biosciences, San Jose, CA) or fucoidan (50 μg/ml; Sigma) was added to the culture 30 min prior to the addition of oligomerized Aβ (1 μM). After 24 h incubation, the cells were harvested and processed as described above for western blotting. Three independent experiments were performed.
Preparation of oligomeric Aβ
Synthetic Aβ42 was purchased from US Peptide. Oligomeric Aβ was prepared as described by Dahlgren et al. (2002). The peptide was dissolved in 1 mM hexafluoroisopropanol (Sigma) and then dried under vacuum in a Speed Vac (Savant, Holbrook, NY). The residual peptide was re-suspended in dimethyl sulphoxide (Sigma) to a concentration of 5 mM. By adding phenol red free Ham’s F-12 medium (Mediatech, Herndon, VA) to the re-suspended peptide, the concentration was made to 100 μM and the peptide was kept at 4°C for 24 h. Before adding to cell cultures, the concentrations of Aβ42 were adjusted by adding OPTI-MEM I. To verify the degree of Aβ oligomerization, oligomerized Aβ42 was analysed by SDS–PAGE followed by western blotting for each cell culture experiment.
Western blotting and densitometric analysis
Western blot analysis was used to quantify brain APP and PS1 in Mo/Hu APPswe PS1dE9 mice and intracellular Aβ after stimulating BV-2 cells with TLR ligands. Protein concentrations of brain homogenates and BV-2 cell lysates were determined by Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Thirty micrograms of total protein from each sample were applied to 4–15% Tris–HCl linear gradient (brain samples) and 16.5% Tris–Tricine (BV-2 cells) SDS–PAGE and electrotransferred to polyvinylidine difluoride membranes (Immobilon-P, Millipore, Bedford, MA). Oligomerized Aβ of 30 and/or 90 ng were applied to SDS–PAGE for verification, also. The membranes were blocked with PBS containing 5% non-fat dried milk (w/v), 0.02% sodium azide and 0.02% Tween-20, incubated at 4°C for 16 h with 6E10 antibody (Signet Laboratories, Dedham, MA) for detection of Aβ and APP or with H-70 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) for detection of PS1, and visualized by western blot chemiluminescence reagent plus (Perkin Elmer, Boston, MA) according to the manufacturer’s protocol. The membranes were reprobed with monoclonal antibody against actin (brain homogenates) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (cell lysates) (both were from Chemicon International, Temecula, CA). The relative concentrations of APP, PS1 and Aβ were determined by densitometric scanning of the membranes using HP Scanjet 4470c and an HP Precisionscan Pro 3.1 (Hewlett-Packard Development Company, Houston, TX) and normalized by the GAPDH or actin signals.
For chemical inhibitor experiments, the signals from the entire lane for each sample were quantified and normalized by those from GAPDH. The normalized signals of the samples without Aβ (0% uptake) were subtracted from the others and, then, the subtracted values were divided by those of the samples treated only with LPS and Aβ (100% uptake). Data were expressed as mean ± standard error. Intergroup differences were assessed by analysis of variance (ANOVA) and two-tailed Student’s t-test. P < 0.05 was considered statistically significant.
Immunofluorescent labelling of intracellular Aβ
Intracellular Aβ was quantified as described by Chu et al. (1998) with a minor modification. BV-2 and primary microglia were plated at concentrations of 5 × 104 cells/1.5 ml/chamber into 2-chamber slides and 1 × 104 cells/0.75 ml/chamber into 4-chamber slides, respectively and 16 h after plating, BV-2 cells were treated with each TLR ligand (100 ng/ml of LPS, 13.3 μg/ml of PGN or 0.51 μM of CpG-ODN) or control ODN (0.51 μM) for 24 h, while primary microglia were treated with 100 ng/ml of LPS or 0.51 μM of CpG-ODN, for 24 h. After washing twice with PBS, cells were cultured in 400 μl of OPTI-MEM I containing 0.25 or 1 μM of Aβ42 for 24 h for primary cultures or BV-2 cells, respectively. The cells were washed three times with cold PBS, fixed with cold methanol at −30°C for 15 min and, then, washed three times with cold PBS. After blocking with 5% goat serum in PBS at room temperature for 2 h, cells were incubated with 6E10 antibody in PBS containing 5% goat serum at 4°C for 16 h. After washing four times with cold PBS, cells were treated with goat anti-mouse IgG antibody coupled with Alexa Fluor 488 in PBS containing 1.5% goat serum at room temperature for 2 h. After washing four times with cold PBS, cover glasses were mounted on slides with n-PG (0.2% n-propyl gallate, 90% glycerol and 10% PBS). The slides were kept at 4°C in the dark until analysis under a histomorphometry consisting of an OLYMPUS IX71 microscope equipped for fluorescence microscopy and an OLYMPUS DP70 digital camera (OLYMPUS). Three independent experiments were performed. For BV-2 cells, >2400 cells for each TLR ligand were analyzed for each independent experiment. A custom Pascal macro sub-routine was written to calculate both % of cells stained with 6E10 (fluorescent cells) and % of cell area showing fluorescence. For primary cultures, >150 cells in 4–8 random fields were manually counted under the fluorescence microscope for each independent experiment.
Results
TLR4 genotyping
Because C3H/HeJ mice carry a point mutation (TlrLps-d) in the TLR4 gene (Poltorak et al., 1998), the TLR4 genotype of Mo/Hu APPswe PS1dE9 mice that have been maintained by mating with B6C3F1 mice, can be homozygous (TlrLps-d/TlrLps-d), heterozygous (TlrLps-d/θ) or of wild-type for the mutation. We selected two oligonucleotides complementary to sequences flanking the mutation as primers (TLR4sen193 and TLR4ant607), which span 415 bp in the mouse TLR4 gene. PCR amplification of mouse genomic DNA with the primers produced a single DNA fragment with 415 bp in length (Fig. 1). The mutation (TlrLps-d) in C3H/HeJ mice is caused by a C to A transversion resulting in substitution of histidine for proline (Poltorak et al., 1998). The mutation creates an Nla III site (CATG) in the 415 bp DNA fragment. To verify the genotyping method, we used DNAs extracted from three different strains of mice with known TLR4 genotypes: C3H/HeJ (TlrLps-d/TlrLps-d), B6C3F1 (TlrLps-d/θ) and C57BL/6J (wild-type). Digestion of PCR products from C3H/HeJ mice with Nla III produced two DNA fragments with 304 and 111 bp in length and PCR products from C57BL/6J mice were not cut with Nla III, as expected (Fig. 1). Three DNA fragments with 415, 304 and 111 bp in length were observed in Nla III-digested PCR products from B6C3F1 (Fig. 1). We have used this method to determine the TLR4 genotype of transgenic progeny produced by a crossing between Mo/Hu APPswe PS1dE9 and B6C3F1 mice.
Fig. 1.
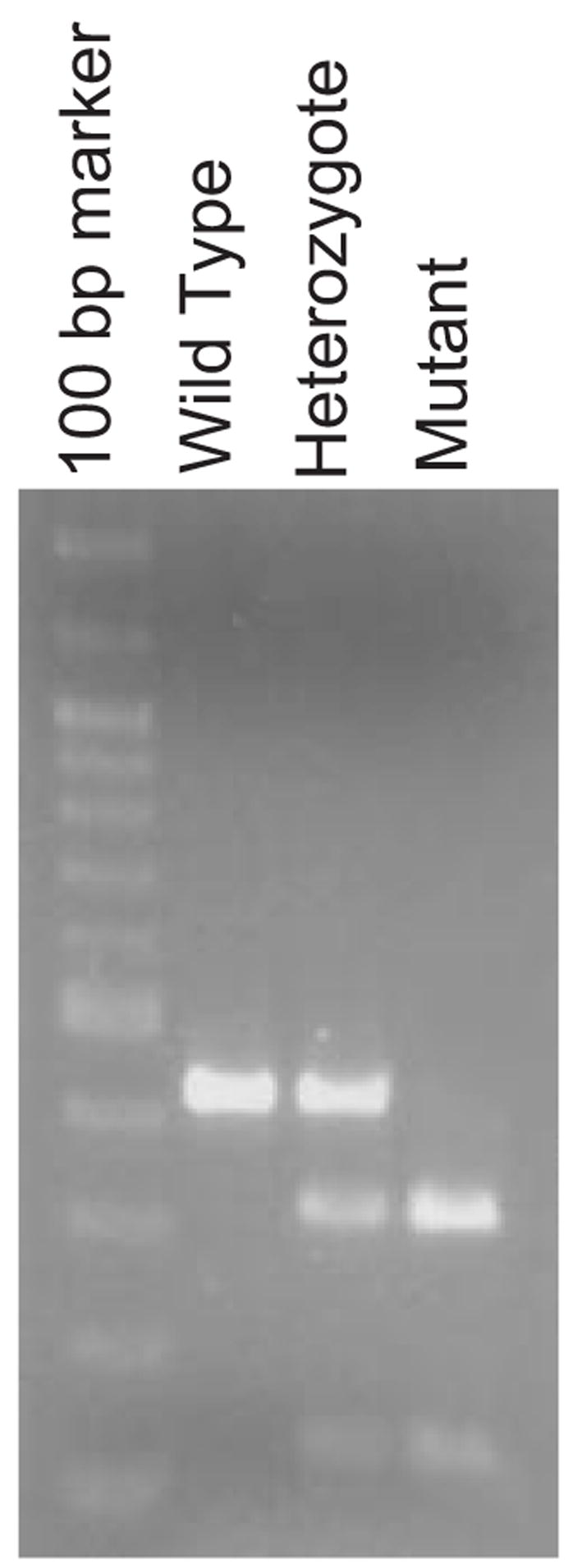
TLR4 genotyping. A 415 bp DNA fragment was amplified by PCR using mouse tail DNA and two TLR4-specific primers, TLR4sen193 and TLR4ant607. The mutation in the TLR4 gene in C3H/HeJ mice creates an Nla III site in the 415 bp DNA fragment. Nla III digestion of the amplified DNA fragment from a mouse with the mutation produces a 304 and 111 bp DNA fragments. The restriction enzyme polymorphism is shown in the picture for each TLR4 genotype.
TLR4 mutation does not influence steady state levels of APP and PS1
Mo/Hu APPswe PS1dE9 mice carry mouse APP with the double mutations (K670N and M671L) and human PS1 with a deletion of exon 9 found in familial Alzheimer’s disease patients. Mo/Hu APPswe PS1dE9 mice, however, develop amyloid deposits composed of human Aβ in the brain because the Aβ sequence in the mouse APP transgene has been humanized (Jankowsky et al., 2004). To determine if the Lps-d mutation affects the expression levels of APP and PS1 in Mo/Hu APPswe PS1dE9 mice, brain homogenates extracted from the transgenic mice with three different genotypes of the mutation were quantified by western blotting followed by densitometric analysis. No significant differences were found in the steady state APP and PS1 levels in the brain among the three different genotypes of the TLR4 mutation (data not shown).
TLR4 mutation exacerbates Aβ load in Alzheimer’s disease mouse models
Mo/Hu APPswe PS1dE9 mice develop amyloid deposits as early as 4 months of age and amyloid deposits increase during ageing (data not shown). To determine if the Lps-d mutation influences Aβ pathology in Alzheimer’s disease mouse models, we evaluated Aβ load in Mo/Hu APPswe PS1dE9 mice at 14–16 months of age by three different methods: immunohistochemistry by anti-Aβ antibody (6E10), thioflavine-S staining and Aβ40- and Aβ42-specific sandwich ELISA.
Diffuse and fibrillar Aβ deposits were detected by inmmunohistochemistry using 6E10 antibody (Fig. 2). The β-amyloid loads were indicated by average percentages of areas showing Aβ immunoreactivity in the hippocampus and neocortex. In the neocortex, the Aβ load in TlrLps-d/TlrLps-d mice (4.11 ± 0.13%) was greater that that in TLR4 wild-type mice (2.73 ± 0.20%, n = 6, P = 0.0001; Fig. 2C). TlrLps-d/θ mice (3.28 ± 0.30%; n = 9) had more Aβ load than TLR4 wild-type mice (P = 0.04). On average, the Aβ load in TlrLps-d/TlrLps-d mice was more than that in TlrLps-d/θ mice in the neocortex but the difference was not significant. In the hippocampus, the Aβ load in TlrLps-d/TlrLps-d mice (2.69 ± 0.20%, n = 6) was greater than that in TLR4 wild-type mice (1.74 ± 0.25%, n = 6, P = 0.01; Fig. 2D). The Aβ load on average in TlrLps-d/θ mice (2.43 ± 0.24%) was more than that in TLR4 wild-type mice but the difference was not significant because of relatively high variations (P = 0.076).
Fig. 2.

Detection of diffuse and fibrillar Aβ deposits by anti-Aβ antibody in Mo/Hu APPswe PS1dE9 mice with different TLR4 genotypes. Aβ deposits in the brain are visualized by immunohistochemistry using 6E10 antibody in TLR4 wild-type mice (A) and TlrLps-d/TlrLps-d mice (B). Average percentages of areas showing Aβ immunoreactivity measured by morphometry in the neocortex (C) and hippocampus (D) are shown. In the neocortex, the Aβ load in TLR4 wild-type mice is less than that in TlrLps-d/θ and TlrLps-d/TlrLps-d mice. In the hippocampus, the Aβ load in TlrLps-d/TlrLps-d mice is greater than that in TLR4 wild-type mice. Scale bars 250 μm.
Fibrillar Aβ deposits were visualized by thioflavine-S fluorescence and the results are shown in Fig. 3. The Aβ load in TlrLps-d/TlrLps-d mice (2.58 ± 0.42% for the neocortex; 1.95 ± 0.27% for the hippocampus; n = 6) was greater than that in TlrLps-d/θ mice (1.47 ± 0.06%, P = 0.006 for the neocortex; 1.09 ± 0.06%; P = 0.002 for the hippocampus; n = 9) and in TLR4 wild-type mice (1.56 ± 0.14%, P = 0.04 for the neocortex; 1.21 ± 0.15%; P = 0.03 for the hippocampus; n = 6; Fig. 3C and D). There was no difference in Aβ load between TlrLps-d/θ and wild-type mice in the neocortex and hippocampus.
Fig. 3.

Detection of fibrillar Aβ deposits by thioflavine-S fluorescence in Mo/Hu APPswe PS1dE9 mice with different TLR4 genotypes. Fibrillar Aβ deposits show thioflavine-S fluorescence in the brain of TLR4 wild-type mice (A) and TlrLps-d/TlrLps-d mice (B). Average percentages of areas showing thioflavine-S fluorescence measured by morphometry in the neocortex (C) and hippocampus (D) are shown. The Aβ load in TlrLps-d/TlrLps-d mice is greater than that in TlrLps-d/θ mice and TLR4 wild-type mice for the neocortex and hippocampus. Scale bars 250 μm.
The amount of buffer-soluble and insoluble Aβ in the cerebrum was determined by the Aβ40- and Aβ42-specific ELISA (Fig. 4). The cerebral buffer-soluble Aβ40 content in TlrLps-d/TlrLps-d mice (4.85 ± 0.92 ng/mg protein, n = 4) was higher than that in TLR4 wild-type mice (2.05 ± 0.51 ng/mg protein, n = 5, P = 0.02; Fig. 4A). On average, TlrLps-d/θ mice (3.91 ± 0.86 ng/mg protein, n = 8) had more buffer-soluble Aβ40 than TLR4 wild-type mice and less buffer-soluble Aβ40 than TlrLps-d/TlrLps-d mice but these differences were not significant. The cerebral buffer-soluble Aβ42 content in TlrLps-d/TlrLps-d mice (10.02 ± 1.71 ng/mg protein, n = 4) was higher than that in TlrLps-d/θ mice (5.56 ± 1.09 ng/mg protein, n = 8, P = 0.04; Fig. 4B). The cerebral buffer-soluble Aβ42 content in TlrLps-d/TlrLps-d mice tended to be more than that in TLR4 wild-type mice (4.99 ± 1.46 ng/mg protein, n = 5) but did not reach the statistical significance because of greater variations in the wild-type mice (P = 0.058). The amount of total buffer-soluble Aβ (Aβ40 + Aβ42) in TlrLps-d/TlrLps-d mice (14.87 ± 2.48 ng/mg protein, n = 4) was higher than that in TLR4 wild-type mice (7.05 ± 1.79 ng/mg protein, n = 5, P = 0.04; Fig. 4C). The cerebral insoluble Aβ40 content in TlrLps-d/TlrLps-d mice (13.47 ± 2.61 μg/mg protein, n = 4) was higher than that in TlrLps-d/θ mice (7.03 ± 1.38 μg/mg protein, n = 8, P = 0.03; Fig. 4D). TlrLps-d/TlrLps-d mice had more insoluble Aβ40 than TLR4 wild-type mice (6.71 ± 2.02 μg/mg protein, n = 5) on average but the difference was not significant due to a greater variation in the wild-type mice (P = 0.07). The cerebral insoluble Aβ42 content in TlrLps-d/TlrLps-d mice (14.26 ± 1.36 μg/mg protein, n = 4) was higher than that in TLR4 wild-type mice (8.75 ± 0.90 μg/mg protein, n = 5, P = 0.009; Fig. 4E). TlrLps-d/θ mice (11.94 ± 1.36 μg/mg protein, n = 8) were not different from the other groups in the cerebral insoluble Aβ42 content. The amount of total insoluble Aβ (Aβ40 + Aβ42) in TlrLps-d/TlrLps-d mice (27.73 ± 3.94 μg/mg protein, n = 4) was higher than that in TLR4 wild-type mice (15.46 ± 2.87 μg/mg protein, n = 5, P = 0.03; Fig. 4F).
Fig. 4.

Quantification of buffer-soluble and insoluble Aβ in the cerebrum by the Aβ40- and Aβ42-specific ELISA. The cerebral buffer-soluble Aβ40 content in TlrLps-d/TlrLps-d mice is higher than that in TLR4 wild-type mice (A). The cerebral buffer-soluble Aβ42 content in TlrLps-d/TlrLps-d mice is higher than that in TlrLps-d/θ mice (B). The amount of total buffer-soluble Aβ (Aβ40 + Aβ42) is higher than that in TLR4 wild-type mice (C). The cerebral insoluble Aβ40 content in TlrLps-d/TlrLps-d mice is higher than that in TlrLps-d/θ mice (D). The cerebral insoluble Aβ42 content in TlrLps-d/TlrLps-d mice is higher than that in TLR4 wild-type mice (E). The amount of total insoluble Aβ (Aβ40 + Aβ42) in TlrLps-d/TlrLps-d mice is higher than that in TLR4 wild-type mice (F).
Aβ42 uptake by TlrLps-d/TlrLps-d microglial is stimulated by a TLR9 ligand but not by a TLR4 ligand
To investigate roles of TLR4 in Aβ uptake by microglia in vitro, primary microglial cultures were prepared from TLR4 wild-type and TlrLps-d/TlrLps-d neonatal mice. Ingested Aβ by primary microglia was detected by fluorescent immunocytochemistry using anti-Aβ antibody and quantified by histomorphometry (Fig. 5). Upon treatment with LPS (100 ng/ml), the percentages of Aβ-immunoreactive cells in the TLR4 wild-type cultures increased from 21.58 ± 3.29 to 44.52 ± 0.97% (P = 0.003; Fig. 5A and C) while those in the TlrLps-d/TlrLps-d cultures were unchanged before (16.85 ± 3.31% ) and after treatment with LPS (22.67 ± 1.53%, P > 0.05; Fig. 5D and F, respectively). When the cultures were treated with CpG-ODN (0.51 μM), Aβ-immunoreactive cells increased (42.82 ± 1.56%, P = 0.007 for wild-type cells and 38.57 ± 2.67%, P = 0.004 for TlrLps-d/TlrLps-d cells; Fig. 5B and E, respectively). Accordingly, Aβ-uptake by LPS-stimulated microglia is affected by the TLR4 mutation.
Fig. 5.

Detection of ingested Aβ42 in primary TLR4 wild-type and TlrLps-d/TlrLps-d microglia by fluorescent immunocytochemistry. Primary TLR4 wild-type (A–C) and TlrLps-d/TlrLps-d (D–F) microglia were treated with LPS (100 ng/ml) (A and D), CpG-ODN (0.51 μM) (B and E) or PBS (C and F) and exposed to oligomerized Aβ (0.25 μM) for 24 h. Ingested Aβ42 in the cells shows fluorescence by immunocytochemistry using 6E10 antibody and anti-mouse IgG antibody coupled with Alexa Fluor 488. Arrows indicate ingested Aβ. Scale bars 40 μm.
TLR ligands stimulate Aβ42 uptake by a microglial cell line
To determine if stimulation of TLRs with their specific ligands influences Aβ42 clearance in vitro, we investigated Aβ42 uptake by a microglial cell line, BV-2 cells, using three different methods: ELISA (Fig. 6), western blotting (Fig. 7) and immunohistomorphometry (Fig. 8). We tested three different TLR ligands: PGN for TLR2, LPS for TLR4 and CpG-ODN for TLR9.
Fig. 6.
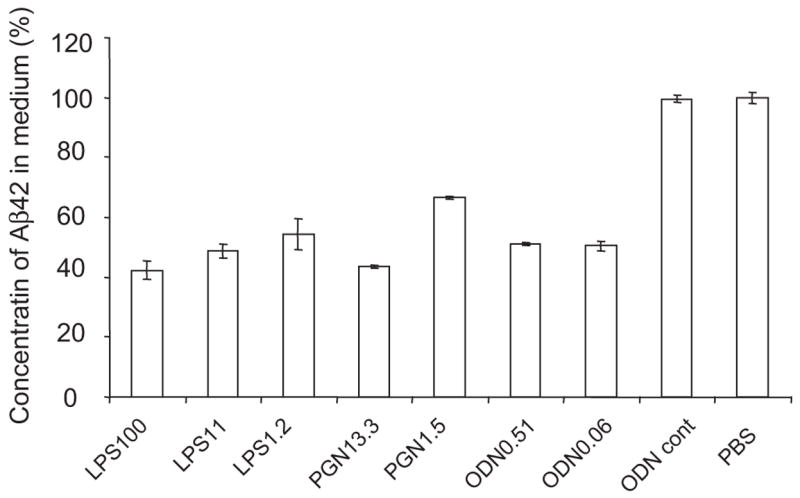
The clearance of Aβ42 from culture media by activation of TLRs on microglia (BV-2 cells). BV-2 cells were treated with LPS at the concentrations of 1.2, 11 and 100 ng/ml, CpG oligodeoxynucleotides (CpG-ODN) at 0.06 and 0.51 μM or peptidoglycan (PGN) at 1.5 and 13.3 μg/ml. BV-2 cells treated with the TLR ligands were incubated with oligomerized Aβ42 for 24 h. As controls, BV-2 cells treated with Control ODN or PBS were used. The concentrations of residual Aβ42 in the culture media were determined by Aβ42-specific sandwich ELISA. The ratios of the Aβ42 concentrations in TLR ligand-treaded media to that in PBS-treated medium are shown. After stimulation with every TLR ligand tested, residual Aβ42 in the medium was reduced by ~50% at the TLR ligand concentrations tested, compared with that in the media from BV-2 cells treated with PBS or control ODN (P < 0.01 for every TLR ligand treatment).
Fig. 7.
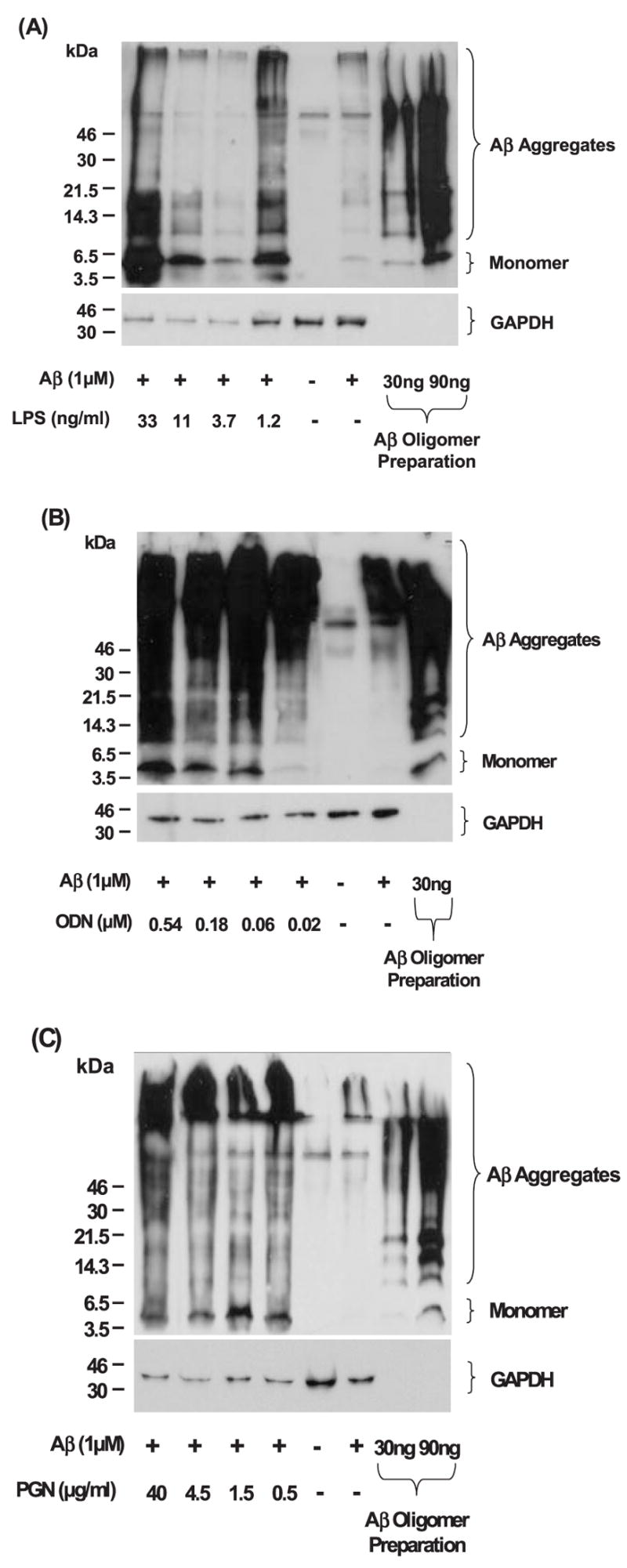
Detection of Aβ42 ingested by BV-2 cells after stimulation with TLR ligands. After activation of microglia with TLR ligands, ingested Aβ42 by BV-2 cells are detected by western blots using 6E10 antibody. To monitor the degree of Aβ oligomerization for each experiment, oligomerized Aβ42 was similarly analyzed (the extremely right lanes). Intracellular monomer and aggregates of Aβ are readily detectable in the cell lysates from BV-2 cells treated with TLR ligands at the concentrations of 1.2, 3.7, 11 and 33 ng/ml for LPS (A), 0.02, 0.06, 0.18 and 0.54 μM for CpG-ODN (B) and 0.5, 1.5, 4.5 and 40 μg/ml for PGN (C), while Aβ is barely visible in the cell lysate from BV-2 cells without treatment with TLR ligands. The membranes were reprobed with GAPDH-specific antibody for normalization.
Fig. 8.
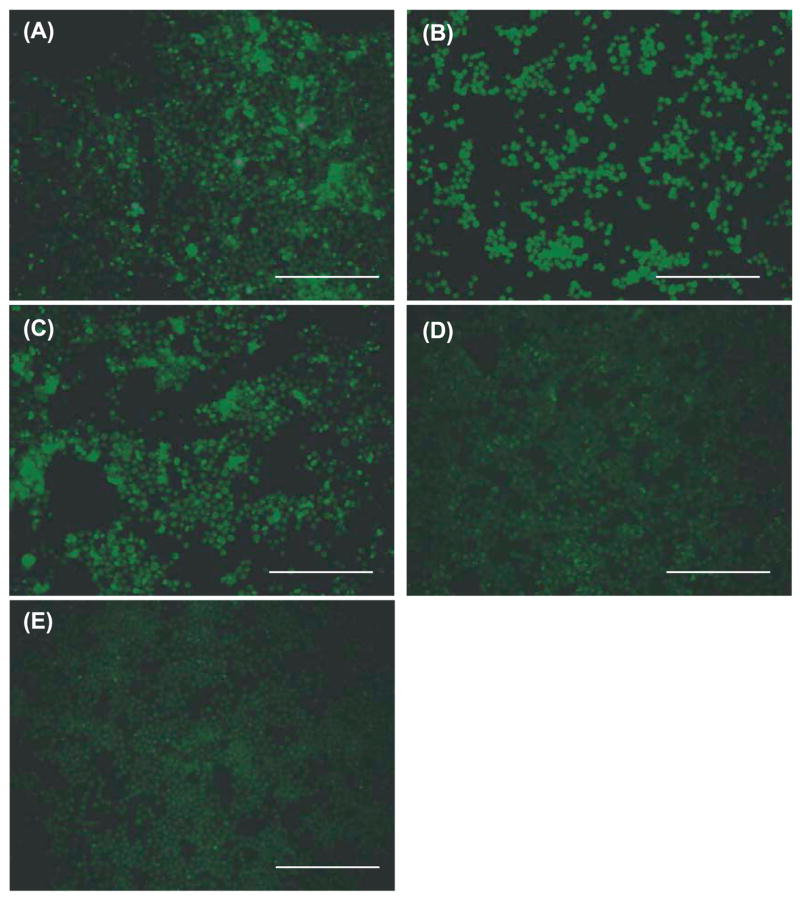
Detection of ingested Aβ42 in BV-2 cells by fluorescent immunocytochemistry. After treatment with LPS (100 ng/ml) (A), PGN (13.3 μg/ml) (B) or CpG-ODN (0.51 μM) (C), ingested Aβ42 in BV-2 cells shows fluorescence by immunocytochemistry using 6E10 antibody and anti-mouse IgG antibody coupled with Alexa Fluor 488. BV-2 cells treated with control ODN (D) or PBS (E) show little to no fluorescence. Scale bars 250 μm.
BV-2 cells were treated with several different concentrations of TLR ligands and incubated with oligomerized Aβ42 for 24 h, which contained Aβ monomer as well as oligomers with a wide range of molecular weight (Fig. 7). The concentrations of residual Aβ42 in the culture media were determined by Aβ42-specific sandwich ELISA. After stimulation with every TLR ligand tested, residual Aβ42 in the medium was reduced by ~50%, compared with that in the medium from BV-2 cells treated with control ODN or PBS (P < 0.01 for every treatment; Fig. 6). To detect Aβ42 ingested by BV-2 cells after stimulation with TLR ligands, cells were lysed with 4% SDS and subjected to western blot analysis using anti-Aβ antibody. Intracellular oligomers and high molecular weight aggregates of Aβ were readily detectable in the cell lysate from BV-2 cells treated with every TLR ligand tested, while Aβ was barely detected in the cell lysate from BV-2 cells untreated with TLR ligands (Fig. 7). Monomeric Aβ was also found in the cell lysates from BV-2 cells treated with TLR ligands (Fig. 7). Aβ was visualized within BV-2 cells by fluorescent immunocytochemistry using anti-Aβ antibody and quantified by histomorphometry (Fig. 8). After treatment with LPS (100 ng/ml), PGN (13.3 μg/ml) or CpG-ODN (0.51 μM), 18.96 ± 7.62, 16.83 ± 5.46 and 10.21 ± 0.74% of BV-2 cells showed Aβ-immunoreactivity, respectively, while very limited Aβ-immunoreactivity (<0.5%) was found in BV-2 cells that had been treated with control ODN or PBS (P < 0.001 for all TLR ligands tested) (Fig. 8). Thus, TLR ligands conspicuously stimulated Aβ-uptake by BV-2 cells.
Effects of chemical inhibitors on Aβ uptake by microglial cells
CD14 (Fassbender et al., 2004; Liu et al., 2005), scavenger receptors (Paresce et al., 1996; Bamberger et al., 2003) and mFPR2 G-protein coupled receptor (Chen et al., 2006) have been postulated to be receptors for aggregated Aβ. Therefore, we have investigated possible roles of these receptors in Aβ uptake by LPS-stimulated BV-2 cells using chemical inhibitors. After activation of BV-2 cells with LPS, anti-CD14 antibody, fucoidan (scavenger receptor inhibitor) and PTX (G-protein receptor deactivator) were added to the culture followed by incubation with oligomerized Aβ that contained monomeric and highly aggregated Aβ (Fig. 9A). Aβ uptake was quantified by densitometric analysis of the western blots (Fig. 9A). The amounts of Aβ uptake by LPS-activated BV-2 cells after treatment with chemical inhibitors were compared with those by LPS-activated BV-2 cells without inhibitors. The relative amounts of Aβ uptake decreased to 32.8 ± 0.46% (P = 0.03) for fucoidan treatment, to 14.1 ± 5.35% (P = 0.02) by anti-CD14 treatment and to 27.7 ± 2.57 (P = 0.03) and 2.49 ± 1.48% (P = 0.01) for 100 and 500 ng/ml PTX treatment, respectively (Fig. 9B). These results indicate that Aβ uptake by LPS-activated BV-2 cells is mediated in part by these receptors.
Fig. 9.
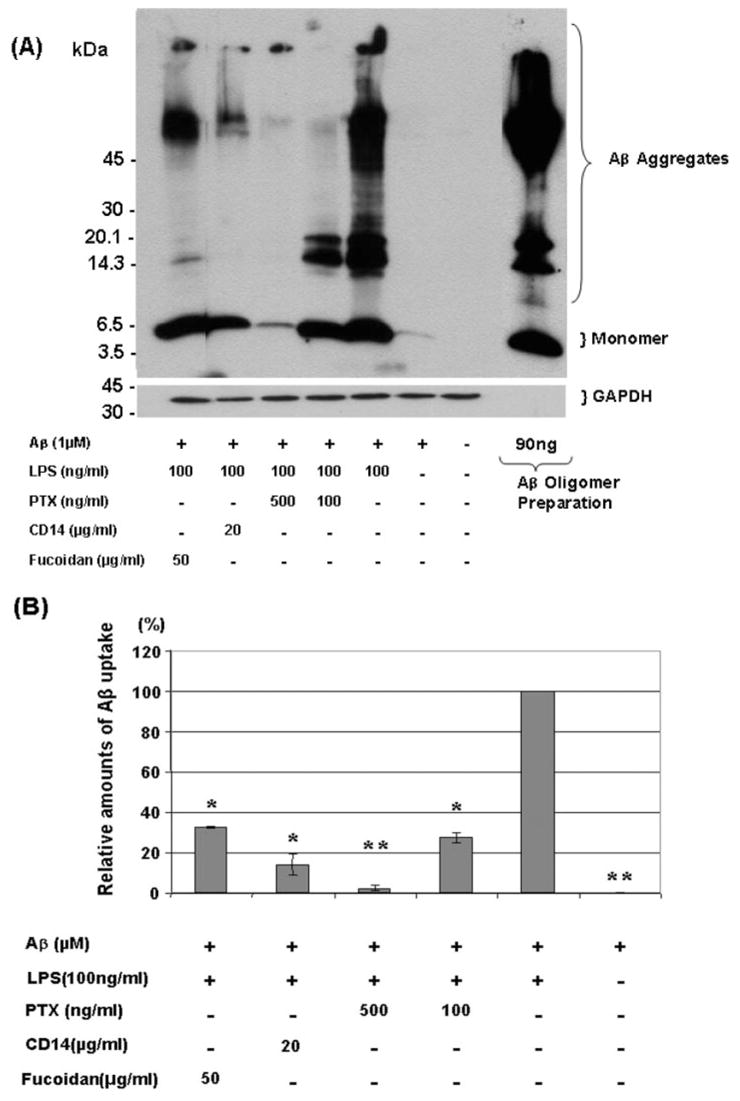
Effects of inhibitors on Aβ42 uptake by LPS-activated BV-2 cells. After stimulation with LPS (100 ng/ml), BV-2 cells were treated with pertussis toxin (PTX) (100 or 500 ng/ml), anti-mouse CD14 antibody (20 μg/ml) or fucoidan (50 μg/ml) and incubated with oligomerized Aβ for 24 h. Ingested Aβ42 was detected by western blots using 6E10 antibody (A). Oligomerized Aβ42 was similarly analysed for verification (A, the extreme right lane). The membranes were reprobed with GAPDH-specific antibody for normalization. The amounts of Aβ uptake by LPS-activated BV-2 cells after treatment with chemical inhibitors were compared with those by LPS-activated BV-2 cells without inhibitors by densitometric analysis of the western blots (B). *P < 0.05 and **P < 0.01 compared with LPS-activated BV-2 cells without inhibitors, n = 3, independent samples, t-test.
Discussion
In this study, we demonstrate that Mo/Hu APPswe PS1dE9 mice with TlrLps-d/TlrLps-d genotype showed an increase in cerebral Aβ load, compared to Mo/Hu APPswe PS1dE9 mice with TLR4 wild-type alleles using three different methods. Uptake of Aβ by cultured microglia from TlrLps-d/TlrLps-d mice was less than that from TLR4 wild-type mice when stimulated by LPS but not by ODN. Activation of any of the three TLRs (TLR2, TLR4 or TLR9) with their specific ligands markedly boosted uptake of Aβ by microglial cell line (BV-2 cells). Uptake of Aβ by activated BV-2 cells was reduced by treatment with anti-CD14 antibody, PTX or fucoidan. These results suggest that TLR signalling pathway(s) may be involved in clearance of cerebral Aβ deposits and that activation of TLRs on microglia may be beneficial to patients.
C3H/HeJ mice have a defective response to bacterial LPS (endotoxin), rendering them resistant to septic shock and highly susceptible to Gram-negative infection due to a destructive mutation (TlrLps-d) of the TLR4 gene. This mutation has been shown to be co-dominant in terms of resistance to endotoxin yet highly susceptible to Gram-negative infection (Poltorak et al., 1998). In this study, the amounts of cerebral Aβ load on average in Mo/Hu APPswe PS1dE9 mice heterozygous for the mutation (TlrLps-d/θ) were greater than those in TLR4 wild-type mice and less than those in TlrLps-d/TlrLps-d mice in most of the different parameters although the values were not always significantly different from other genotypic groups, suggesting that TlrLps-d is co-dominant for Aβ load, also. These results suggest that Aβ load in the brain is modulated in part by TLR4 signalling pathways in mouse models. Because activation of these TLRs (TLR2, TLR4 and TLR9) on microglia distinctly increased Aβ uptake by microglia, protein kinase signalling pathways common to these TLRs may be involved in the clearance of Aβ deposits. Thus, it is interesting to ascertain if cerebral Aβ load is influenced by other TLRs in mouse models.
Microglial activation is complex and can produce diverse phenotypes depending upon their environment, activating ligands and genetic backgrounds (Town et al., 2001; Schenk and Yednock, 2002; Miller et al., 2005; Morgan et al., 2005; Walker and Lue, 2005). Lotz et al. (2005) reported that, in vitro, co-administration of Aβ40 with a TLR4 (LPS) or TLR2 (synthetic lipopeptide Pam3cys-Ser-Lys4) ligand into microglial cultures led to an additive release of nitric oxide and tumour necrosis factor alpha (TNF-α) from microglia. Sheng et al. (2003) reported that daily intraventricular injection of LPS for 14 days induced premature fibrillar Aβ deposits in young mouse models. Hence, activation of TLR signalling can exacerbate Alzheimer’s disease by induction of oxidative stress, inflammation and Aβ deposition. On the other hand, Chen et al. (2005) and Iribarren et al. (2005) reported that activation of microglia through a TLR2 (Pam3cys-Ala-Gly) or TLR9 (CpG-ODN) ligand, respectively, promoted cell uptake of Aβ in vitro by up-regulation of mFPR2 that served as a receptor for Aβ. They suggested a neuroprotective role of microglia through activation of TLR2 and TLR9. Several groups performed hippocampal or intraperitoneal injection of bacterial LPS (a TLR4 ligand) for activation of microglia in old mouse models and reported a reduction in diffuse Aβ plaques but not in fibrillar Aβ plaques, suggesting that activation of TLR4 on microglia may be therapeutic to old mouse models (DiCarlo et al., 2001; Quinn et al., 2003; Herber et al., 2004; Malm et al., 2005). These positive effects of microglial activation are consistent with our experimental results. Our results also suggest that Aβ uptake by microglia upon activation of TLR signalling may be mediated in part by scavenger receptors, mFPR2 and CD14, which have been claimed as receptors for aggregated Aβ (Paresce et al., 1996; Chen et al., 2005; Iribarren et al., 2005; Liu et al., 2005). TLR signalling induces a battery of genes involved in phagocytosis and inflammation (Doyle et al., 2004; Akira et al., 2006; McKimmie et al., 2006). Particularly, a G protein-coupled receptor, mFPR2, may play a predominant role in Aβ uptake by LPS-stimulated BV-2 cells because >95% of Aβ uptake was inhibited by PTX (500 ng/ml). Our results are concordant with the report by Cui et al. (2002a), where up-regulation of mFPR2 in a microglial cell line (N9) and primary microglia was demonstrated by LPS treatment. Up-regulation of mFPR2 by TLR2 and TLR9 ligands involves activation of mitogen activated protein kinase (MAPK) p38 and NF-κB (Iribarren et al., 2005; Chen et al., 2006). TNFα activates mFPR2 in both N9 cells and primary microglia through MAPK p38 and NF-κB dependent signalling pathways (Cui et al., 2002). Thus, up-regulation of mFPR2 in microglia by a TLR4 ligand is likely to be mediated via MAPK p38 and NF-κB activation. It may be possible to reduce Aβ load and cell injuries in Alzheimer’s disease brain by modulating the innate immune responses. Further elucidation of molecular mechanisms that govern the innate immune responses to Aβ deposits may make such beneficial modulation possible.
Acknowledgments
We thank Drs David Borchelt and Joanna Jankowsky for providing the Mo/Hu APPswe PS1dE9 mice and Karen Minter for her help with preparing this manuscript. This study was supported in part by the National Institutes of Health grants to K.F. (NS43947) and to L.L. (AG025949) and the Alzheimer’s Association grant to K.F. (ZEN-03-5834).
Abbreviations
- Aβ
amyloid β-protein
- APP
amyloid β-protein precursor
- LPS
lipopolysaccharide
- PS1
presenilin 1
- PTX
pertussis toxin
- TLR
toll-like receptor
References
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD. Scavenging of Alzheimer’s amyloid beta-protein by microglia in culture. J Neurosci Res. 1996;43:190–202. doi: 10.1002/(SICI)1097-4547(19960115)43:2<190::AID-JNR7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–74. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Brazil MI, Chung H, Maxfield FR. Effects of incorporation of immunoglobulin G and complement component C1q on uptake and degradation of Alzheimer’s disease amyloid fibrils by microglia. J Biol Chem. 2000;275:16941–7. doi: 10.1074/jbc.M000937200. [DOI] [PubMed] [Google Scholar]
- Breitner JC, Gau BA, Welsh KA, Plassman BL, McDonald WM, Helms MJ, et al. Inverse association of anti-inflammatory treatments and Alzheimer’s disease: initial results of a co-twin control study. Neurology. 1994;44:227–32. doi: 10.1212/wnl.44.2.227. [DOI] [PubMed] [Google Scholar]
- Breitner JC, Welsh KA, Helms MJ, Gaskell PC, Gau BA, Roses AD, et al. Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging. 1995;16:523–30. doi: 10.1016/0197-4580(95)00049-k. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–21. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Chen K, Iribarren P, Hu J, et al. Activation of toll-like receptor 2 on microglia promotes cell uptake of Alzheimer’s disease-associated amyloid beta peptide. J Biol Chem. 2006;281:3651–9. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- Chu T, Tran T, Yang F, Beech W, Cole GM, Frautschy SA. Effect of chloroquine and leupeptin on intracellular accumulation of amyloid-beta (A beta) 1–42 peptide in a murine N9 microglial cell line. FEBS Lett. 1998;436:439–44. doi: 10.1016/s0014-5793(98)01161-2. [DOI] [PubMed] [Google Scholar]
- Cui YH, Le Y, Gong W, Proost P, Van DJ, Murphy WJ, et al. Bacterial lipopolysaccharide selectively up-regulates the function of the chemotactic peptide receptor formyl peptide receptor 2 in murine microglial cells. J Immunol. 2002a;168:434–42. doi: 10.4049/jimmunol.168.1.434. [DOI] [PubMed] [Google Scholar]
- Cui YH, Le Y, Zhang X, Gong W, Abe K, Sun R, et al. Up-regulation of FPR2, a chemotactic receptor for amyloid beta 1–42 (A beta 42), in murine microglial cells by TNF alpha. Neurobiol Dis. 2002b;10:366–77. doi: 10.1006/nbdi.2002.0517. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–53. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–12. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- Doyle SE, O’Connell RM, Miranda GA, Vaidya SA, Chow EK, Liu PT, et al. Toll-like receptors induce a phagocytic gene program through p38. J Exp Med. 2004;199:81–90. doi: 10.1084/jem.20031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender K, Walter S, Kuhl S, Landmann R, Ishii K, Bertsch T, et al. The LPS receptor (CD14) links innate immunity with Alzheimer s disease. FASEB J. 2004;18:2035. doi: 10.1096/fj.03-0364fje. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Boada RM, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell. 2002;111:927–30. doi: 10.1016/s0092-8674(02)01201-1. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D, et al. Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190:245–53. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Iribarren P, Chen K, Hu J, Gong W, Cho EH, Lockett S, et al. CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid beta 1–42 peptide by up-regulating the expression of the G-protein- coupled receptor mFPR2. FASEB J. 2005;19:2032–4. doi: 10.1096/fj.05-4578fje. [DOI] [PubMed] [Google Scholar]
- Ishii KJ, Coban C, Akira1 S. Manifold mechanisms of toll-like receptor-ligand recognition. J Clin Immunol. 2005;25:511–21. doi: 10.1007/s10875-005-7829-1. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–70. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT., Jr Seeding ‘one-dimensional crystallization’ of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–8. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- Kim HD, Kong FK, Cao Y, Lewis TL, Kim H, Tang DC, et al. Immunization of Alzheimer model mice with adenovirus vectors encoding amyloid beta-protein and GM-CSF reduces amyloid load in the brain. Neurosci Lett. 2004;370:218–23. doi: 10.1016/j.neulet.2004.08.059. [DOI] [PubMed] [Google Scholar]
- Klein WL. Abeta toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41:345–52. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci. 2004;24:9838–46. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta 1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain. 2005;128:1778–9. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- Lotz M, Ebert S, Esselmann H, Iliev AI, Prinz M, Wiazewicz N, et al. Amyloid beta peptide 1–40 enhances the action of toll-like receptor-2 and -4 agonists but antagonizes toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem. 2005;94:289–98. doi: 10.1111/j.1471-4159.2005.03188.x. [DOI] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–42. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer E, Rogers J, Sibley J. Anti-inflammatory drugs and Alzheimer disease. Lancet. 1990;335:1037. doi: 10.1016/0140-6736(90)91101-f. [DOI] [PubMed] [Google Scholar]
- McKimmie CS, Roy D, Forster T, Fazakerley JK. Innate immune response gene expression profiles of N9 microglia are pathogen-type specific. J Neuroimmunol. 2006;175:128–41. doi: 10.1016/j.jneuroim.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–53. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–24. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–65. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer’s disease amyloid beta-protein by microglial cells. J Biol Chem. 1997;272:29390–7. doi: 10.1074/jbc.272.46.29390. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in TLR 4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Quinn J, Montine T, Morrow J, Woodward WR, Kulhanek D, Eckenstein F. Inflammation and cerebral amyloidosis are disconnected in an animal model of Alzheimer’s disease. J Neuroimmunol. 2003;137:32–41. doi: 10.1016/s0165-5728(03)00037-7. [DOI] [PubMed] [Google Scholar]
- Savage MJ, Kawooya JK, Pinsker LR, Emmons TL, Mistretta SSR, Greenberg BD. Elevated A-beta levels in Alzheimer’s disease brain are associated with selective accumulation of A-beta 42 in parenchymal amyloid plaques and both A-beta40 and A-beta42 in cerebrovascular deposits. Int J Exp Clin Invest. 1995;2:234–40. [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Schenk DB, Yednock T. The role of microglia in Alzheimer’s disease: friend or foe? Neurobiol Aging. 2002;23:677–9. doi: 10.1016/s0197-4580(02)00034-9. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14:133–45. doi: 10.1016/s0969-9961(03)00069-x. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 1997;48:626–32. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JC, et al. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: a systematic review. Neuroepidemiology. 2004;23:159–69. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, et al. Role of CD40 ligand in amyloidosis in transgenic Alzheimer’s mice. Nat Neurosci. 2002;5:1288–93. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–55. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Town T, Tan J, Sansone N, Obregon D, Klein T, Mullan M. Characterization of murine immunoglobulin G antibodies against human amyloid-beta1-42. Neurosci Lett. 2001;307:101–4. doi: 10.1016/s0304-3940(01)01951-6. [DOI] [PubMed] [Google Scholar]
- Townsend KP, Town T, Mori T, Lue LF, Shytle D, Sanberg PR, et al. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur J Immunol. 2005;35:901–10. doi: 10.1002/eji.200425585. [DOI] [PubMed] [Google Scholar]
- Veld BA, Ruitenberg A, Launer LJ. Duration of nonsteroidal antiinflammatory drug use and risk of Alzheimer’s disease. The Rotterdam study Neurobiol Aging. 2000;21:S204. [Google Scholar]
- Walker DG, Lue LF. Investigations with cultured human microglia on pathogenic mechanisms of Alzheimer’s disease and other neurodegenerative diseases. J Neurosci Res. 2005;81:412–25. doi: 10.1002/jnr.20484. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang SS, Becerra-Arteaga A, Good TA. Development of a novel diffusion-based method to estimate the size of the aggregated Abeta species responsible for neurotoxicity. Biotechnol Bioeng. 2002;80:50–9. doi: 10.1002/bit.10347. [DOI] [PubMed] [Google Scholar]
- Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp Neurol. 2000;161:127–38. doi: 10.1006/exnr.1999.7260. [DOI] [PubMed] [Google Scholar]
- Woodroofe MN, Cuzner ML, Cohen J, Wilkin GP. Neural cell culture: a practical approach. IRL Press (at Oxford University Press); 1995. Microglia: the tissue macrophage of the CNS; pp. 107–19. [Google Scholar]