

Abstract
The mechanisms of adenovirus serotype 2 inactivation with either UV light (with a narrow emission spectrum centered at 254 nm) or monochloramine were investigated by assessing the potential inhibition of two key steps of the adenovirus life cycle, namely, E1A protein synthesis and viral genomic replication. E1A early protein synthesis was assayed by using immunoblotting, while the replication of viral DNA was analyzed by using slot blotting. Disinfection experiments were performed in phosphate buffer solutions at pH 8 and room temperature (UV) or 20°C (monochloramine). Experimental results revealed that normalized E1A levels at 12 h postinfection (p.i.) were statistically the same as the corresponding decrease in survival ratio for both UV and monochloramine disinfection. Normalized DNA levels at 24 h p.i. were also found to be statistically the same as the corresponding decrease in survival ratio for monochloramine disinfection. In contrast, for UV disinfection, genomic DNA levels were much lower than E1A or survival ratios, possibly as a result of a delay in DNA replication for UV-treated virions compared to that for controls. Future efforts will determine the pre-E1A synthesis step in the adenovirus life cycle affected by exposure to UV and monochloramine, with the goal of identifying the viral molecular target of these two disinfectants.
Human adenovirus is a nonenveloped, icosahedron-shaped virion, ranging in size from 70 to 100 nm. The virion consists of a protein capsid that protects a double-stranded DNA genome of 28 to 42 kbp and DNA-associated proteins (35). Adenovirus is a waterborne opportunistic pathogen that has been detected in tap and treated drinking water (10, 18, 21), surface water (18, 31), coastal seawater (10, 12, 18, 31), treated and untreated wastewater (10, 17, 18, 31, 41, 49), and swimming pool water (18, 48). Of the 51 adenovirus serotypes, serotypes 40 and 41 can cause enteric diseases and others can produce respiratory problems (18). Waterborne outbreaks of human adenovirus infection, especially affecting immunocompromised individuals and young children, have been reported for both enteric and respiratory serotypes (7, 18).
Free chlorine, the most common disinfectant currently used by U.S. drinking water utilities, is very effective for inactivating adenovirus (2). However, many utilities will be switching to alternative disinfection schemes to comply with the recently promulgated Long Term 2 Enhanced Surface Water Treatment Rule (LT2ESWTR) (45) and Stage 2 Disinfectants and Disinfection By-products Rule (Stage 2 DBPR) (44). An alternative treatment approach under consideration is to use UV light (UV) for primary disinfection at the treatment plant, followed by the use of combined chlorine species (predominantly monochloramine) in the distribution system. This disinfection alternative is being considered because UV can effectively inactivate Cryptosporidium parvum oocysts, the primary waterborne pathogen addressed in LT2ESWTR, which is highly resistant to free chlorine inactivation. Additionally, compared to free chlorine, monochloramine forms lower levels of trihalomethanes and haloacetic acids regulated under the Stage 2 DBPR.
This alternative method of disinfection has its drawbacks; switching from free chlorine to UV treatment may result in water containing active virus particles since viruses are more resistant to UV inactivation than are protozoan (oo)cysts. Adenovirus, in particular, is more resistant to UV than any other viral, bacterial, and protozoan pathogen of current concern in drinking water (13, 17, 24, 26, 29, 38, 41, 43, 50). Furthermore, adenovirus is also resistant to inactivation by monochloramine (1, 39), suggesting that a UV-monochloramine treatment scheme would be ineffective. Thus, waterborne adenovirus, which was not a concern historically because of its susceptibility to free chlorine inactivation (2), is now emerging as a health risk issue. As an example of this growing concern, adenovirus is currently included in the Drinking Water Contaminant Candidate List 2 for potential future regulatory development (46). Such regulatory decisions would require a comprehensive characterization of the inactivation kinetics of adenovirus with both UV and monochloramine under the range of conditions of relevance to drinking water disinfection. Furthermore, elucidation of the corresponding inactivation mechanisms is required to better understand the relatively weak effect of UV and monochloramine on adenovirus particles and to guide the development of effective disinfection methods to neutralize adenovirus in drinking water.
One approach for assessing the inactivation mechanism of UV or monochloramine is to determine the step of the adenovirus life cycle that is inhibited by the disinfectant. This approach is possible since the molecular steps of the adenovirus life cycle have been well characterized (3, 6, 16, 36). An event occurring after virus attachment and subsequent entry into the host cell is the synthesis of viral early proteins, such as the E1A product. Then, after viral genomic DNA replication, late genes are synthesized to create core components of the virion. The genetic material is packaged into particles, creating mature virions, which are released from the host cell via lysis.
The objective of this study is to elucidate the mechanisms of adenovirus serotype 2 inactivation with either UV light or monochloramine by assessing the potential inhibition of two key steps of the adenovirus life cycle, the synthesis of E1A proteins and the replication of viral DNA. Results shown here demonstrate that each disinfectant has a unique effect on viral replication.
MATERIALS AND METHODS
Viruses and cells.
Human adenovirus serotype 2 strain adenoid 6 (VR-846; ATCC, Manassas, VA), was used as a model virus for this study (38). The virus stock was propagated in human lung carcinoma A-549 cells (CCL-185; ATCC, Manassas, VA) before use in experiments according to the following protocol, with the exception of experiment UV-1 using the virus stock directly upon shipment. The A-549 cells were maintained at 37°C inside an incubator with humidified air containing 5% CO2 and were fed with nutrient mixture F-12 Ham Kaighn's modification (Ham's F-12K) medium, containing 10% fetal bovine serum (FBS), 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 0.25 μg/ml of amphotericin B. The virus stock was inoculated onto subconfluent A-549 cellular monolayers at a target multiplicity of infection (MOI) of 160 to 500 PFU/cell. The medium used for suspending viruses was a modified version of the Ham's F-12K medium, containing 2% FBS, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 0.25 μg/ml of amphotericin B. Following inoculation, the cell monolayers were rocked for 2 to 3 s at 15-min intervals for 90 min, after which the virus inoculum was removed. Infected cellular monolayers were then incubated at 37°C with humidified air containing 5% CO2 and were fed with Ham's F-12K medium, containing 10% FBS, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 0.25 μg/ml of amphotericin B. The infected cells were harvested after 3 to 4 days postinfection (p.i.), when the cells showed cytopathic effect. Cells were dislodged using a cell scraper. The suspension in the flasks was freeze-thawed three times. Cell debris and released virus were collected and centrifuged at 230 × g at room temperature for 10 min. The cell debris pellet was discarded, and the supernatant was filtered through a polyvinylidene fluoride membrane with a nominal pore size of 0.45 μm by using a vacuum-driven Stericup filter unit (Millipore, Billerica, MA) to remove fine debris and virus aggregates that had not been removed in the centrifugation step. The filtrate was aliquoted in 10 mM phosphate-buffered saline (PBS; pH 8) and was stored at −80°C until used. The final titer was in the range of 5 × 108 to 1 × 109 PFU/ml.
UV treatment of adenovirus.
Two replicate experiments (UV-1 and UV-2) were performed to characterize the kinetics of UV inactivation for adenovirus serotype 2 to determine the optimal dosage of UV-C treatment for future studies. For these experiments, purified virions were tested in 0.01 M PBS at pH 8.0 ± 0.2 and ambient temperature (22 ± 2°C). Additionally, a collimated beam system (Calgon Carbon Corporation, Pittsburgh, PA) installed with a low-pressure Hg lamp emitting a narrow emission spectrum centered at 254 nm was used as the UV-C source. The light intensity used was I = 0.043 ± 0.003 mW/cm2, and adenovirus samples were collected at various times corresponding to light fluences (i.e., IT or product of light intensity and contact time) in the range of 10 to 160 mJ/cm2.
Three additional UV disinfection experiments (UV-M-1, UV-M-2, and UV-M-3) were performed to assess the effect of UV treatment on the virus life cycle, using the same conditions described above, except that two fluences of <30 mJ/cm2 (samples S1 and S2) were utilized, with the goal of having adenovirus survivals, as measured by the plaque assay described under “Virus viability assay,” greater than approximately 10%. The limited exposure to UV was selected so that signals obtained would be above the detection limit of the molecular techniques described under “Immunoblotting assay” and “DNA analysis using a modified Southern blot assay (slot blotting).”
For all of the experiments where adenovirus was treated with UV-C, virus stock was added to 15 ml of PBS in a 60-mm-diameter tissue culture dish to achieve an initial virus titer N0 = (3.8 ± 3.3) × 106 PFU/ml (N0 values for specific experiments are listed in Table 1). The reactor was then placed under the light beam of the collimated system, mixed continuously by magnetic stirring, and removed from the light beam once the target IT value was reached. UV fluences were determined by radiometry (4). Treated virions were then used to infect A-549 cellular monolayers as described below.
TABLE 1.
Experimental conditions for inactivating adenovirus type 2 virions with UV-C irradiationa
| Expt | N0 (PFU/ml) | tf (min) |
|---|---|---|
| UV-1 | 5.82 × 105 | 60.0 |
| UV-2 | 2.14 × 106 | 48.0 |
| UV-M-1 | 7.12 × 106 | 12.4 |
| UV-M-2 | 5.53 × 106 | 11.8 |
N0, initial virus titer; tf, maximum time for which viruses were exposed to UV-C. Other conditions equal for all experiments: UV light intensity I, 0.043 ± 0.003 mW/cm2; pH 8.0 ± 0.2; PBS concentration, 0.01 M; ambient temperature, 22 ± 2°C.
Monochloramine disinfection of adenovirus.
The inactivation kinetics of adenovirus serotype 2 with monochloramine was characterized in an earlier body of work (39). Consequently, only three additional experiments (MN-M-1, MN-M-2, and MN-M-3) were performed in which adenovirus was treated with monochloramine, for the purpose of investigating the inactivation mechanism of this chemical. These additional monochloramine experiments were performed at pH 8.0 ± 0.2 (0.01 M PBS) and 20°C. Monochloramine disinfection experiments were performed in a batch reactor. The reactor was immersed in a water bath fed with a recirculating CH/P temperature control system, model 2002 (Forma Scientific, Inc., Marietta, OH). Preformed monochloramine was prepared immediately before use by slow addition of 100 ml of a 5- to 68-mg-Cl2/liter sodium hypochlorite solution in 0.01 M PBS at pH 8.0 to 200 ml of a 25-mg-N/liter ammonium chloride solution in 0.01 M PBS at the target pH, with vigorous stirring. PBS was subsequently added to reach a final volume of 500 ml. The resulting solution was stirred for 15 min before its concentration was measured by the DPD colorimetric method (9). Thirty milliliters of the preformed monochloramine stock solution was subsequently added into a 250-ml Erlenmeyer flask. The solution was acclimated to the water bath temperature for 15 min. The monochloramine disinfection experiments were then started by adding various volumes of virus stock into the flask containing 30 ml of monochloramine solution. The initial monochloramine concentration was c0 = 5.8 to 7.5 mg/liter as Cl2, the ammonia nitrogen-to-chlorine molar ratio was x = 3.5 to 4.4, and the initial adenovirus concentration in the reactor was N0 = (4.6 ± 0.7) × 106 PFU/ml (conditions for specific experiments are listed in Table 2).
TABLE 2.
Experimental conditions for inactivating adenovirus type 2 virions with monochloraminea
| Expt | N0 (PFU/ml) | c0 (mg/liter as Cl2) | x (dimensionless) | tf (min) |
|---|---|---|---|---|
| MN-M-1 | 4.39 × 106 | 7.07 | 3.50 | 44.2 |
| MN-M-2 | 5.23 × 106 | 7.46 | 3.53 | 42.2 |
| MN-M-3 | 3.92 × 106 | 5.82 | 4.43 | 51.2 |
Experiments MN-M-1, MN-M-2, and MN-M-3 were performed to assess the molecular mechanism of monochloramine. N0, initial virus titer; c0, initial disinfectant concentration; x, ammonia nitrogen-to-chlorine (NH3-N/Cl2) molar ratio; tf, maximum time for which viruses were exposed to monochloramine. Other conditions equal for all experiments: pH 8.0 ± 0.2; PBS concentration, 0.01 M; temperature, 20 ± 1°C.
Sample volumes of 1 ml were withdrawn from the reactor at predetermined times and transferred to 1.5-ml microcentrifuge tubes containing 0.1 ml of 0.35% (wt/vol) sodium thiosulfate to quench the residual monochloramine. The quenched samples were kept on ice until all samples were taken. Virus viability was assessed as described subsequently, immediately after taking the last sample at time tf listed in Table 2. Each experiment was performed at two relatively low monochloramine CT (product of monochloramine concentration and contact time) values (samples S1 and S2) that resulted in less than 1 log of inactivation (i.e., survival greater than 10%) for the same reason provided above for the corresponding UV disinfection experiments.
The first sample of each experiment, taken within approximately 10 to 20 s, was used as the initial concentration of the virus suspension, N0 (Table 2), to plot inactivation curves. Control reactors without disinfectant addition, tested at each pH and temperature combination, confirmed that no measurable virus inactivation took place within such a short initial exposure, and they also revealed that in the absence of monochloramine viruses remained viable for the duration of all disinfection experiments under the conditions investigated.
Virus viability assay.
Adenovirus viability was determined by plaque assays (47) using A-549 cells. Three days prior to the plaque assay, A-549 cells were placed in T-25 tissue culture flasks at a concentration of (3.5 ± 0.5) × 106 cells per flask. On the day of the plaque assay, 0.4 ml of virus samples at appropriate dilutions was inoculated onto the cellular monolayers. After a 90-min absorption period, 10 ml of an overlay mixture was added to each flask. The overlay mixture was prepared by mixing 100 ml of 3% autoclaved agar solution, kept at 57°C, with 100 ml of nutrient mixture. The nutrient mixture was prepared by mixing 83 ml of 2× minimum essential medium, 3 ml FBS, 1 ml 1 M MgCl2, 2 ml 0.1% neutral red, and 5 ml sterile distilled deionized water. The nutrient mixture was incubated at 37°C and dosed with 6 ml of 7.5% sodium bicarbonate solution immediately before mixing with the agar solution. The overlaid flasks were incubated at 37°C under humidified air containing 5% CO2. PFU were enumerated daily 3 to 10 days later. Virus titers were calculated by assessing the number of plaques present in flasks containing 20 to 300 PFU/plate at 10 days p.i. No significant increase in PFU was observed after day 10, and the A-549 cell monolayer started to degrade after day 10, resulting in disappearance of the plaques (data not shown).
Cell infection for E1A protein synthesis and DNA replication assessment.
Similar to the sampling procedure for viability assessment, sample volumes of 3 ml were withdrawn from the reactor at predetermined times corresponding to two different levels of inactivation and transferred to 15-ml centrifuge tubes, which also contained 0.3 ml of 0.35% (wt/vol) sodium thiosulfate to quench the residual disinfectant. The UV and quenched monochloramine samples were kept on ice until the last sample was taken at time tf listed in Tables 1 and 2 and then used to infect A-549 cell monolayers. Each cell monolayer was prepared 1 day prior to virus infection by plating approximately 5 × 105 A-549 cells into the wells of a 60-mm tissue culture dish and incubated overnight at 37°C inside a 5% CO2 incubator. Cells were infected with 0.5 ml of either untreated/control (C), UV-treated, or monochloramine-treated (S1 and S2) virions at an MOI of 5 PFU/cell, based on the pretreatment titer. The volume added for disinfected samples (S1 and S2) was the same as that for the control (C), such that the MOI calculated from plaque formation was decreased by a factor equal to the survival ratio. After a 90-min absorption phase, the virus-containing suspension was aspirated and 2 ml of “complete” medium (Ham's F-12K medium [Fisher Scientific, Waltham, MA] containing 10% FBS, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 0.25 μg/ml of amphotericin B) was added to cellular monolayers. Virus inoculum suspended in the 0.01 M, pH 8 PBS possessed the same ability to infect cells as that of viruses suspended in the complete medium (data not shown). The infected monolayer was incubated at 37°C and harvested at either 12 or 24 h p.i., and samples were prepared for subsequent immunoblotting or slot blotting procedures (see below).
Immunoblotting assay.
Cells previously infected with either untreated virions (C) or treated virions (S1 and S2) were harvested at 12 h p.i. To this end, the infected cellular monolayer was rinsed three times with complete medium, and cells were removed from the tissue culture dish by scraping. Cells were collected by centrifugation at 230 × g centrifugation at 4°C for 10 min. Supernatants were removed, and the cellular pellets were resuspended in 100 μl of cytoplasmic extraction buffer (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA [pH 8.0], 0.1 mM EGTA, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 0.05% NP-40, 2 μg/ml aprotinin, 0.5 μg/ml leupeptin, 20 mM β-glycerophosphate, 10 mM NaF, 1 mM Na3VO4) containing 1% HALT protease inhibitor (Pierce Biotechnology, Rockford, IL). After a 10-min incubation period on ice, samples were centrifuged at 10,000 × g for 10 min. Supernatants were removed to new tubes and stored at −20°C. Total protein (cellular and viral) concentrations for each sample were measured by a bicinchoninic acid assay (40) using a BCA Protein Assay kit (Pierce Biotechnology, Rockford, IL). For all experiments, equal amounts of cytoplasmic extracts were further analyzed by immunoblotting (see below). Cytoplasmic extracts were mixed with 5× ImmunoPure Non-reducing Lane Marker Sample Buffer (Pierce Biotechnology, Rockford, IL) and 5% 2-mercaptoethanol. Samples were boiled for 5 min and then incubated on ice. Next, samples were loaded into individual wells of a sodium dodecyl sulfate-12% polyacrylamide gel, and the proteins were separated by using electrophoresis. The proteins were then electrophoretically transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA), and the membrane was incubated in Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBS-T) and 5% nonfat dry milk. After a 30-min incubation period at room temperature, membranes were incubated with mouse monoclonal anti-E1A immunoglobulin G (M73; Santa Cruz Biotechnology, Santa Cruz, CA), diluted 1:500. After incubation either overnight at 4°C or for 1 h at room temperature, membranes were washed in TBS-T containing 0.5% nonfat dry milk three times to remove excess antibody. Membranes were incubated with horseradish peroxidase-conjugated immunoglobulin G specific for mouse antibodies (Fisher Scientific, Waltham, MA; 1:12,000 dilution) for 1 h at room temperature. Interactions between the anti-E1A antibody and E1A proteins, if present, were then detected via a chemiluminescent reaction using the Supersignal West Femto system (Pierce Biotechnology, Rockford, IL), and the signal was visualized on X-ray film (Kodak, Rochester, NY). To quantify the chemiluminescence signals, X-ray films were digitized and then analyzed by densitometry, using the Scion Image software (Scion Corporation, Frederick, MD). The signals were quantified using the Analyzing One-Dimensional Electrophoretic Gels program of Scion Image, with the application of a macro called GelPlot2. This method was used to compare the ratio of band density only within each film image, where the film exposure time was identical. A value for E1A-specific signal was computed by dividing the value of signal obtained from lanes containing virus-infected cell lysates by the value of signal from lanes containing lysates of untreated viruses. Lysates from uninfected cells were harvested as mentioned above and were used as negative controls. Blots were reprobed to detect cellular actin protein levels by using rabbit polyclonal antiactin antibodies, diluted at 1:1,000 (Sigma-Aldrich, St. Louis, MO). After washing, membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit antibodies (Fisher Scientific, Waltham, MA; 1:10,000 dilution). Blots were incubated with Supersignal West Pico reagents (Pierce Biotechnology, Rockford, IL), and chemiluminescence was detected with X-ray film (Kodak, Rochester, NY).
DNA analysis using a modified Southern blot assay (slot blotting).
After absorption of virus into A-549 cellular monolayers, infected monolayers were harvested at 24 h p.i. At this time, monolayers were rinsed three times with complete medium. Next, cellular monolayers were detached from their plates by scraping, and detached cells were collected by centrifugation at 230 × g and 4°C for 10 min. The supernatant was aspirated, and the cellular pellets were resuspended in PBS for subsequent DNA extraction. Total DNA (cellular and viral) was extracted from harvested cells using the QIAamp blood DNA kit (Qiagen Inc., Valencia, CA), following the manufacturer's instructions, resulting in 200 μl of DNA samples in Tris-Cl-EDTA solution. The DNA samples were stored at −20°C. Total DNA concentrations for all samples were measured spectrophotometrically at a wavelength of 260 nm.
DNA samples were either used directly or diluted 1:5 in Tris-EDTA buffer (pH 8.0) for a final volume of 50 μl. Samples were boiled for 5 min and immediately cooled on ice. Next, each sample was loaded onto a Hybond-N positively charged nylon membrane (Amersham Biosciences, Piscataway, NJ) by using a Hoefer PR 648 slot blot manifold (Hoefer Inc., San Francisco, CA) connected to a vacuum device. Nylon membranes containing DNA samples were UV cross linked. The presence of adenovirus genomes was detected by incubating membranes with an alkaline phosphatase-labeled probe that specifically recognized a portion of the adenovirus genome. The primers utilized to amplify adenovirus type 2 DNA are ADFOR (5′-GGCCCTAGACAAATATTACGCGCTAG-3′) and ADREV (5′-GGATTGAAGCCAATATGATAATGAGGGGG-3′). These primers were designed such that the N terminus of the linear double-stranded DNA genome of adenovirus 2, specifically nucleotides 25 to 300, would be PCR amplified. A method using a similar approach and using a similar amplified portion of the adenovirus genome was used previously to assess viral genomic DNA levels by slot blotting (11). The 300-bp amplicon was purified by using the PCR product purification kit (Qiagen Inc., Valencia, CA) and subsequently labeled with alkaline phosphatase using the AlkPhos Direct Labeling and Detection System (Amersham Biosciences, Piscataway, NJ). The modified probe was suspended in hybridization buffer (prepared as directed for the CDP-Star chemiluminescent detection system; Amersham Biosciences, Piscataway, NJ) and incubated with the viral DNA-containing nylon membrane overnight at 65°C. After overnight incubation, the membrane was washed with primary and secondary wash buffers, per the manufacturer's instructions, to remove excess nonspecifically bound probes. The membrane was incubated with chemiluminescence reagents from the CDP-Star chemiluminescent detection system (Amersham Biosciences, Piscataway, NJ). The membrane was exposed to X-ray film, and chemiluminescence signals were developed. Lysate from uninfected cells was harvested as mentioned above and was used as a negative control. The signal density for this control sample, quantified with the Scion Image software following the same protocol described for E1A analysis under “Immunoblotting assay,” was below detection.
Statistical analysis.
Virus titers were determined by weighed regression analysis (20). Statistical differences among adenovirus survival ratios and normalized viral E1A and DNA concentrations of different samples were assessed by a single-factor analysis of variance (ANOVA) test using MS Excel (a one-way, fixed-effects ANOVA). An independent MS Excel two-tailed, two-sample t test assuming unequal variances was used for an analysis comparing normalized E1A protein concentrations with adenovirus survival ratios.
RESULTS
UV inactivation kinetics.
To characterize the kinetics of UV inactivation for adenovirus 2, at a constant pH 8.0 ± 0.2 and ambient temperature (22 ± 2°C), virus replication in cells infected with untreated (C) versus UV-treated virions was assessed by plaque assays. The results of two independent experiments, UV-1 and UV-2, are shown in Fig. 1, with the experimental conditions for these two experiments (starting virus titer and UV exposure time) listed in Table 1. As would be expected, the lack of UV-C treatment did not alter the virus viability, as measured by plaque assays (data not shown). In contrast, treatment of viruses with UV-C resulted in a decreased virus titer (Fig. 1), such that there was a 4-log decrease in infective virions when virions received 167 mJ/cm2. As depicted in Fig. 1, resulting adenovirus survival ratios were very similar. The data obtained from the experiments utilizing UV-treated adenovirus (UV-1 and UV-2 shown in Fig. 1) were fitted simultaneously with the pseudo-first-order expression.
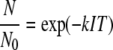 |
(1) |
in which N0 and N are the concentrations of infective viruses (PFU/ml) at the beginning of the experiment and after a certain exposure time, respectively. N/N0 is the survival ratio (dimensionless), k is the first-order inactivation rate constant (cm2/mJ), and IT is the UV light fluence (mJ/cm2) or product of the UV light intensity (mW/cm2) and exposure time (s) (as listed in Table 1). The first-order rate constant obtained from fitting data sets UV-1 and UV-2 in Fig. 1 with equation 1 was k = 0.055 cm2/mJ. The fit line in Fig. 1 was used to calculate the fluences (IT) used for subsequent experiments in which UV-treated virions were utilized to perform molecular experiments (experiments UV-M-1, UV-M-2, and UV-M3). As depicted in Fig. 1, resulting adenovirus survival ratios for experiments UV-M-1, UV-M-2, and UV-M3 were generally consistent with the two more complete data sets within the variability observed.
FIG. 1.

Inactivation kinetics of adenovirus serotype 2 with UV light at pH 8 and room temperature. The continuous line was obtained by simultaneous fitting of UV-1 and UV-2 experiments with equation 1. The initial virus titers for each experiment are listed in Table 1. IT values for UV-C-treated samples S1 and S2 are given in Table 3.
Monochloramine inactivation kinetics.
Similar to the experiments described for measuring the kinetics of UV inactivation, three independent experiments (MN-M-1, MN-M-2, and MN-M-3) were performed to determine the inactivation rate of monochloramine on adenovirus by using plaque assays. Results obtained for these experiments are shown in Fig. 2. No decay in monochloramine concentration was observed within the duration of the experiments, and so the CT values corresponded to the product of the initial monochloramine concentration c0 and contact times, as listed in Table 2. Control experiments, in which adenovirus particles were untreated, showed that there was no significant decrease in virus viability over the duration of the longest experiment, as measured by plaque assays (data not shown).
FIG. 2.

Kinetics of monochloramine inactivation for adenovirus serotype 2 obtained in the present study (star symbols) and those previously reported (39) (other symbols). Continuous lines represent data previously reported for monochloramine treatment at pH 8 and pH 10 (39), while dashed lines represent data obtained from fitting experiments MN-M-1 (black stars), MN-M-2 (white stars), and MN-M-3 (gray stars) performed in the present study (Table 2) with equation 1 using previously reported k (39) and N1/N0 as the fitting parameter.
The extent of the lag phase for experiments MN-M-1 to MN-M-3 was estimated by fitting all three data sets simultaneously with the Delayed Chick-Watson model (34):
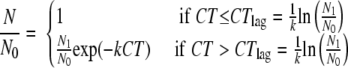 |
(2) |
in which k is the post-lag-phase pseudo-first-order inactivation rate constant (liters/[mg × min]), N1/N0 is the y intercept resulting from extrapolating the pseudo-first-order line, and CTlag is the CT value of the post-pseudo-first-order line at N/N0 = 1. Because preliminary fitting efforts revealed that the simultaneous determination of both fitting parameters (k and N1/N0) would result in significant errors due to the MN-M-1 to MN-M-3 data sets being limited to the first log of inactivation, k = 0.00774 liter/(mg × min) reported elsewhere (39) at 20°C and pH 8 was used. With this simplification, the resulting fitted inactivation curve is shown (dashed line) in Fig. 2, and the fitting parameter was N1/N0 = 3.2. The corresponding CTlag = 150 mg × min/liter was calculated from N1/N0 and k values with the expression given in equation 2.
Effect of UV disinfection on E1A protein synthesis and DNA replication.
After observation that UV irradiation of adenovirus decreased the number of functional virions, as measured by plaque assays, an effort was made to investigate how UV irradiation affected the virion by detecting the portion of the virus life cycle that was halted during infection with these treated virions. UV irradiation is known to damage both the genome (5, 27, 33) and capsid proteins (28) of viruses. Thus, E1A protein synthesis was detected by immunoblotting as a measure of assessing the ability of the disinfected viral capsid to enter the host cell and to deliver functional genetic material to the host cell nucleus. Genomic replication, an event that occurs post-E1A production, was measured by slot blotting as a method for detecting whether UV irradiation damaged the DNA genome.
To detect the effect of UV irradiation on the ability of virions to produce E1A proteins in host cells, cells were harvested 12 h p.i. and cytoplasmic extracts were assessed for E1A proteins by immunoblotting. Previous reports observed adenovirus E1A protein expression as early as 8 h p.i. (51). Thus, a 12-h time point, in which E1A protein synthesis was predicted to be easily detected, was chosen. As shown in Fig. 3A, lysates from cells infected with untreated virions contain E1A proteins, as indicated by a doublet that represents the major 12S and 13S polypeptides translated from the E1A transcript. As expected, mock-infected cellular lysates did not produce any bands (data not shown). In contrast to untreated virions, the E1A-containing bands were less intense when lysates from cells infected with UV-treated viruses were probed (S1 and S2; see Table 3 for corresponding IT values). This effect was not due to lower protein contents in lysate samples, as actin levels were similar in each lane (Fig. 3A). Additionally, this was not due to E1A protein migration to the nucleus, since very low amounts of E1A proteins were present in nucleus-extracted proteins (data not shown). Using densitometry, the intensities of the bands representing E1A were compared, with the bands from lanes containing lysates from cells infected with untreated virions arbitrarily set to a value of 1.0.
FIG. 3.

Effect of UV irradiation on either E1A protein (A) or DNA synthesis (B). A-549 cells were mock infected or infected with virions (MOI = 5) that were either untreated or treated with UV-C (S1 and S2). IT values for samples S1 and S2 are given in Table 3.
TABLE 3.
Light fluences, survival ratios, and normalized E1A and DNA concentrations obtained for control (C) and disinfected samples (S1 and S2) for the inactivation of adenovirus serotype 2 with UV lighta
| Expt/sample | IT (mJ/cm2) | N/N0 (n) | Normalized E1A density (n) | Normalized DNA concn (n) | P |
|---|---|---|---|---|---|
| UV-M-1/C | 0.0 | 1.000 (1) | NAb | 1.000 (1) | |
| UV-M-1/S1 | 12.8 | 0.431 (1) | NA | 0.069 (1) | |
| UV-M-1/S2 | 29.8 | 0.100 (1) | NA | 0.034 (1) | |
| UV-M-2/C | 0.0 | 1.000 (1) | 1.000 (2) | 1.000 (2) | |
| UV-M-2/S1 | 12.8 | 0.588 (1) | 0.760 ± 0.047 (2) | 0.084 ± 0.024 (2) | |
| UV-M-2/S2 | 29.8 | 0.140 (1) | 0.360 ± 0.244 (2) | 0.095 ± 0.030 (2) | |
| UV-M-3/C | 0.0 | 1.000 (1) | 1.000 (1) | 1.000 (8) | |
| UV-M-3/S1 | 8.8 | 0.619 (1) | 0.312 (1) | 0.119 ± 0.032 (8) | |
| UV-M-3/S2 | 23.4 | 0.424 (1) | 0.054 (1) | 0.018 ± 0.020 (8) | |
| UV-M-1 to UV-M-3/C | 0.0 | 1.000 (3) | 1.000 (2) | 1.000 (3) | |
| UV-M-1 to UV-M-3/S1 | 11.5 ± 1.3 | 0.546 ± 0.058 (3) | 0.536 ± 0.224 (2) | 0.091 ± 0.015 (3) | 0.0289 |
| UV-M-1 to UV-M-3/S2 | 27.7 ± 2.1 | 0.221 ± 0.102 (3) | 0.207 ± 0.153 (2) | 0.049 ± 0.023 (3) | 0.386 |
Values given are averages for each separate experiment ± standard deviations (UV-M-1, UV-M-2, or UV-M-3) as well as sample composite averages for two (UV-M-2 and UV-M-3 for E1A samples) or all three (UV-M-1, UV-M-2, and UV-M-3 for DNA samples) experiments ± standard errors of the means.
NA, not available.
To measure viral genomic replication, cellular and viral DNA was isolated from virus-infected cell lysates at 24 h p.i. Previous reports observed adenovirus genomic DNA replication as early as 12 h p.i. (51). Thus, a 24-h time point was chosen to ensure the detection of viral DNA replication. Next, DNA was cross-linked to a nylon membrane, and the membrane was incubated with a probe specific for adenovirus DNA. If viral DNA was present, as would be expected for cells infected with untreated virions, then a signal such as that seen in Fig. 3B, lane C, would be present. In contrast, if viral DNA was absent, such as in DNA from mock-infected cells, then no signal would be observed (data not shown). In comparison to DNA levels from cells infected with untreated virions, the density of the band representing genomic DNA decreased when cells were instead infected with viruses that were treated previously with UV-C (S1 and S2; see Table 3 for corresponding IT values). Similar to what was described above, the densities of DNA bands for cells infected with UV-treated virions (S1 and S2) were measured and normalized to nontreated virions (C), whose value was set to 1 (Fig. 3B).
Normalized E1A protein and DNA concentrations were obtained by averaging results similar to those shown in Fig. 3A and 3B for n replicates of each sample (Table 3). No data for E1A protein synthesis were obtained for samples derived from experiments UV-M-1 and UV-M-3 due to weak signal problems. Similarly, no data for viral genomic replication were reported for samples derived from experiment UV-M-1, due to background interference. Sample composite average normalized E1A protein and DNA concentrations were obtained for the control and S1 and S2 samples by averaging the corresponding results of experiments UV-M-2 and UV-M-3 for E1A protein and experiments UV-M-1, UV-M-2, and UV-M-3 for DNA. The composite average values obtained are summarized in the three bottom rows of Table 3 and plotted in Fig. 4A together with the corresponding adenovirus survival ratios.
FIG. 4.

Sample composite averages and standard errors of normalized viral E1A protein (12 h p.i.) and DNA (24 h p.i.) concentrations and survival ratios for either untreated control (C) or virions treated (S1 and S2) with either UV light (A) or monochloramine (B). Average IT and CT values for samples S1 and S2 are given in Tables 3 and 4, respectively.
Performance of one-way ANOVA tests of the results shown in Fig. 4A showed significant differences (P = 0.03 < 0.05) among the adenovirus survival ratio and corresponding normalized viral E1A protein density and DNA concentration for sample S1. The lack of significant difference between the normalized viral DNA, E1A protein, and PFU/ml values for sample S2 (P = 0.39 > 0.05) might have resulted from the DNA signal having a relative large standard deviation as a result of having approached the detection limit of the slot blotting assay. Independent two-tailed, two-sample t tests assuming unequal variances showed no significant difference between normalized E1A density and adenovirus survival ratio for samples S1 (P = 0.94) and S2 (P = 0.97). In summary, the decrease observed in normalized E1A density at 12 h p.i. was statistically the same as the corresponding decrease in survival ratio. In contrast, the disinfected sample (S1) with a more robust DNA concentration signal had a significantly lower normalized DNA concentration than the corresponding survival ratio.
Effect of monochloramine disinfection on viral E1A protein synthesis and DNA replication.
Monochloramine disinfection also decreased the numbers of infectious viruses, as measured by plaque assays. Chemical disinfectants such as monochloramine have been reported to damage both the viral genome (22, 23, 25, 27) and capsid proteins (22, 28, 42). To assess which mechanism was more likely for monochloramine inactivation, monochloramine-disinfected virus samples were further analyzed for E1A protein synthesis and DNA replication, as a method to assess the ability of the disinfected viral capsid to enter the host cell and to deliver genetic material to the host cell nucleus and as a measure of DNA damage by monochloramine disinfection, respectively.
As would be expected, E1A protein production was readily detected in lysates from cells infected with untreated virions (Fig. 5A). These proteins were also present, although to a lesser extent, in lysates from cells infected with monochloramine-treated virions (Fig. 5A). Since the levels of cellular actin were detected in equal amounts from all three samples, the decreasing E1A band density in samples S1 and S2 is not due to lower protein contents in lysate samples. The E1A band intensities were measured and normalized to the intensity from the C sample, whose value was set to 1.
FIG. 5.

Effect of monochloramine disinfection on either E1A protein (A) or DNA synthesis (B). A-549 cells were mock infected or infected with virions (MOI = 5) that were either untreated (C) or treated with monochloramine (S1 and S2). CT values for samples S1 and S2 are given in Table 4.
The effect of monochloramine disinfection on viral DNA replication was also assessed, and the results obtained for experiment MN-M-3 are shown in Fig. 5B. As before, viral DNA replication was easily detected from lysates of cells infected with untreated virions (C). In contrast, treatment of virions with monochloramine rendered virions that were unable to mount a productive infection, as indicated by less-intense DNA-containing bands (Fig. 5B). Similar to before, the densities of these bands were measured and normalized to the control sample (C), whose value was set to 1 (Fig. 5B).
Normalized E1A protein and DNA concentrations obtained by averaging results similar to those shown in Fig. 5A and B for n replicates of each sample are shown in Table 4. No data on the detection of E1A proteins were obtained for samples from experiment MN-M-1 due to weak signal problems, and data measuring genomic DNA levels for a duplicate of experiment MN-M-2 could not be resolved due to background interference. The average values of normalized E1A protein and DNA concentrations were obtained for the control and S1 and S2 samples by averaging the corresponding results of experiments MN-M-2 and MN-M-3 for E1A protein levels and experiments MN-M-1, MN-M-2, and MN-M-3 for viral genomic DNA levels. The composite average values obtained are summarized in the three bottom rows of Table 4 and plotted in Fig. 4B together with the corresponding adenovirus survival ratios.
TABLE 4.
Disinfectant exposures, survival ratios, and normalized E1A and DNA concentrations obtained for control (C) and disinfected samples (S1 and S2) for the inactivation of adenovirus serotype 2 with monochloraminea
| Expt/sample | CT (mg × min/liter) | N/N0 (n) | Normalized E1A density (n) | Normalized DNA concn (n) | P |
|---|---|---|---|---|---|
| MN-M-1/C | 0 | 1.000 (1) | NAb (0) | 1 (3) | |
| MN-M-1/S1 | 212 | 0.508 (1) | NA (0) | 0.353 ± 0.083 (3) | |
| MN-M-1/S2 | 312 | 0.226 (1) | NA (0) | 0.231 ± 0.089 (3) | |
| MN-M-2/C | 0 | 1.000 (1) | 1.000 (3) | 1.000 (1) | |
| MN-M-2/S1 | 218 | 0.545 (1) | 0.557 ± 0.140 (3) | 0.498 (1) | |
| MN-M-2/S2 | 315 | 0.213 (1) | 0.307 ± 0.081 (3) | 0.304 (1) | |
| MN-M-3/C | 0 | 1.000 (1) | 1.000 (3) | 1.000 (3) | |
| MN-M-3/S1 | 174 | 0.786 (1) | 0.388 ± 0.089 (3) | 0.345 ± 0.036 (3) | |
| MN-M-3/S2 | 298 | 0.319 (1) | 0.086 ± 0.034 (3) | 0.054 ± 0.037 (3) | |
| MN-M-1 to MN-M-3/C | 0 | 1.000 (3) | 1.000 (2) | 1.000 (3) | |
| MN-M-1 to MN-M-3/S1 | 201 ± 14 | 0.613 ± 0.087 (3) | 0.472 ± 0.084 (2) | 0.398 ± 0.050 (3) | 0.190 |
| MN-M-1 to MN-M-3/S2 | 308 ± 5 | 0.253 ± 0.033 (3) | 0.196 ± 0.110 (2) | 0.197 ± 0.074 (3) | 0.802 |
Values given are averages for each separate experiment ± standard deviations (MN-M-1, MN-M-2, or MN-M-3) as well as sample composite averages for two (MN-M-2 and MN-M-3 for E1A samples) or all three (MN-M-1, MN-M-2, and MN-M-3 for DNA samples) experiments ± standard errors of the means.
NA, not available.
One-way ANOVA tests of the results shown in Fig. 4B showed no significant differences among the adenovirus survival ratios and corresponding normalized viral E1A protein densities and DNA concentrations for both sample S1 (P = 0.19 > 0.05, Table 4) and sample S2 (P = 0.80 > 0.05, Table 4). In summary, the decreases observed in normalized E1A levels at 12 h p.i. and normalized DNA concentration at 24 h p.i. were statistically the same as the corresponding decrease in survival ratio.
DISCUSSION
Adenovirus virions that have been exposed to UV-C light or monochloramine in the laboratory may either remain infectious, resulting in the production of progeny virions, or become inactive, thereby no longer replicating in host cells. To detect the percentage of virions that remain activated after a treatment, we utilized plaque assays, comparing the numbers of infectious virus particles in untreated and treated virion samples. However, plaque assays do not provide information about the inactivated particles, particularly how a treatment may affect different stages of the virus life cycle (entry, viral gene expression, viral genome replication, or viral morphogenesis) to yield a nonproductive infection. For this information, molecular techniques, such as immunoblotting or slot blotting, would give information as to how a treatment is limiting the virus life cycle. Finding this limiting step might elucidate the molecular target of a treatment and might aid in developing more-effective means to control adenovirus in drinking water. To understand how UV and monochloramine treatment affects adenovirus, two viral life cycle events were investigated in this study: E1A protein synthesis, which was analyzed by immunoblotting at 12 h p.i., and viral DNA replication, which was analyzed by slot blotting at 24 h p.i. The E1A protein and viral genomic DNA were not determined at the same time because a previous report showed that the DNA was detected at as early as 12 h p.i. while E1A protein was detected at as early as 8 h p.i. (51). Therefore, analyzing both E1A and DNA at the same time could result in signals that might not be quantifiable because of being too low or too high. Also the DNA replication step follows the E1A synthesis step; therefore, DNA analysis was performed at a later time than the E1A protein assay was.
UV disinfection.
As shown in Fig. 1, the kinetics of low-pressure UV disinfection obtained in this study was found to be within 60% of those previously reported for adenovirus serotypes 2 (1, 2, 13, 24, 37, 41), 1 and 6 (30), 5 (2), 15 (41), 40 (26, 43), and 41 (2, 19, 26) and only 11% lower than the average value adopted in the LT2ESWTR (45) based on an analysis of previous data sets. Reasons for the variability among data sets could be a decrease in resistance to UV associated with differences in freeze-thawing procedures (13, 30, 43), stock storage temperature and age (30), and interference by organic matter present in natural waters or associated with virus propagation (10, 43).
When host cell levels of E1A protein and viral DNA were measured after infection with UV-treated virions, it was observed that the normalized E1A protein signal in infected cells was not significantly different from the corresponding survival ratios. This observation could imply that UV irradiation damaged the surface proteins of the virions, decreasing the number of virions that can attach to and penetrate host cells and thereby inhibiting any of the life cycle steps that occur after attachment and entry (Fig. 6). These steps include viral protein synthesis and genomic DNA replication. Alternatively, decreased E1A protein synthesis could occur if UV irradiation affected DNA to the extent that the genome was unable to be transcribed or if the transcript from damaged DNA was unstable, truncated, or severely mutated (14). Since it has been shown that adenovirus serotype 2 was still able to attach and internalize in host cells after being exposed to UV at fluences as high as 4,000 mJ/cm2 (33), the current favored hypothesis is that UV irradiation's effects are due to the mutation of the input virus's genomes.
FIG. 6.

Replication cycle of adenovirus in host cell showing steps taking place prior to E1A protein synthesis that could have been inhibited by monochloramine and UV disinfection.
When viral genome levels in cells infected with UV-treated virions were studied, the normalized DNA levels were lower than the corresponding normalized E1A protein levels and survival ratio (Fig. 4A). These results were surprising, given that the viral survival ratio was expected to be limited by viral DNA replication. One possible reason for these results is that genomic DNA replication in cells infected with UV-treated virions initially occurred at a lower rate than that for untreated virions but eventually increased over time. This is supported by data that showed that genomic DNA levels in cells infected with UV-treated virions at 48 h p.i., obtained by following a protocol identical to that used at 24 h p.i. except for the longer incubation time before harvesting of the viruses, were more than 10 times higher than those at 24 h p.i. (data not shown). For plaque assays, plaque formation was observed daily, with no measurable lag in the formation of plaques detected for UV-treated samples compared to controls that were monitored daily from days 3 to 10 (data not shown). This indicates that the delay in DNA replication was significantly shorter than a 24-h period. Such a delay in DNA synthesis could be due to the repair of UV-damaged DNA. Interestingly, nuclear excision repair has been reported to be involved by human fibroblast host cells and human KB cells on UV-irradiated adenovirus serotype 2 (8, 32, 33), suggesting that there are host cellular mechanisms that may correct damaged viral DNA. Indeed, because adenovirus has been shown to have the lowest rate of inactivation by UV light among all other waterborne viral, bacterial, and protozoan pathogens (13, 17, 26, 29, 38, 41, 43, 50), the repair mechanism has been, in fact, suggested to be a reason for the high resistance to UV of adenovirus (15).
Monochloramine disinfection.
Several differences were observed when the effects of monochloramine versus UV disinfection were studied. Temperature and pH have strong effects on the inactivation kinetics of monochloramine (39) but have no effect on UV neutralization. For monochloramine, the pH effect on its inactivation kinetics was illustrated when the kinetic curves at pH 8 versus pH 10 were compared (39) (Fig. 2). Because the mechanistic investigation of monochloramine was performed with virions treated with monochloramine at pH 8, it is important to recognize that the mechanism for inactivation might be different under different temperature and pH conditions.
For adenovirus disinfected with monochloramine at two different CT levels, the normalized levels of both E1A protein and viral DNA were not significantly different from the corresponding survival ratios measured by plaque assay (Fig. 4B). Consequently, the fraction of virions that were unable to complete their life cycle were also unable to synthesize E1A protein and replicate DNA. Similarly to UV disinfection, monochloramine inhibited E1A synthesis of the inactivated fraction either by reacting with key proteins involved in virion attachment and internalization, escape from endosome, translocation toward the cell nucleus, or nuclear delivery of DNA or by damaging the viral E1A genes (Fig. 6). Unlike UV disinfection, the viral DNA replication was not delayed after treatment with monochloramine.
These results indicate that treating virions either with monochloramine or with UV-C renders them unable to complete an infectious life cycle, specifically halting steps prior to viral early protein synthesis. This implies that the early portion of the life cycle is being compromised when either disinfectant is used. However, with differences in the inactivation kinetics and the presence of pH dependence in monochloramine but not in UV disinfection, it indicates that UV and monochloramine could inhibit different steps of the early portion in the adenovirus replication cycle (Fig. 6). Increasing applications of UV and monochloramine are predicted in response to the release of LT2ESWTR and the Stage 2 DBPR. However, adenovirus is not effectively controlled in drinking water by either UV or monochloramine, and therefore, it has emerged as a waterborne pathogen of public health concern. With the primary goal of water disinfection to control adenovirus and other waterborne pathogens in drinking water, a better understanding of these two disinfection methods is indispensable. Future efforts to further characterize the mechanisms of adenovirus inactivation with UV and monochloramine should focus on assessing which of the steps in the early portion of the virus life cycle (Fig. 6) is limiting by assessing potential damage to key proteins and E1A genes. Once the inactivation targets/mechanisms are known, it will facilitate further development of novel disinfection materials or more effective disinfection schemes for drinking water applications.
Acknowledgments
The Royal Thai Government is acknowledged for financial support, provided in the form of a fellowship to the lead author. This work was partially supported by the WaterCAMPWS, a Science and Technology Center of Advanced Materials for the Purification of Water with Systems, under National Science Foundation agreement number CTS-0120978.
We thank Sandra McMasters from the University of Illinois at Urbana-Champaign Cell Media Facility and Daniel R. Dahling from the U.S. Environmental Protection Agency National Risk Management Research Laboratory, Cincinnati, OH, for providing training and assistance on the cell culture technique during the early phase of this study.
Footnotes
Published ahead of print on 18 April 2008.
REFERENCES
- 1.Ballester, N. A., and J. P. Malley, Jr. 2004. Sequential disinfection of adenovirus type 2 with UV-chlorine-chloramine. J. Am. Water Works Assoc. 96:97-103. [Google Scholar]
- 2.Baxter, C. S., R. Hofmann, M. R. Templeton, M. Brown, and R. C. Andrews. 2007. Inactivation of adenovirus types 2, 5, and 41 in drinking water by UV light, free chlorine, and monochloramine. J. Environ. Eng. 133:95-103. [Google Scholar]
- 3.Berk, A. J. 2007. Adenoviridae: the viruses and their replication, p. 2355-2394. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA.
- 4.Bolton, J. R., and K. G. Linden. 2003. Standardization of methods for fluence (UV dose) determination in bench-scale UV experiments. J. Environ. Eng. 129:209-215. [Google Scholar]
- 5.Cameron, K. R. 1973. Ultraviolet irradiation of herpes simplex virus: action spectrum for the survival of infectivity in relation to the small-plaque effect. J. Gen. Virol. 18:51-54. [DOI] [PubMed] [Google Scholar]
- 6.Chroboczek, J., E. Gout, A. Favier, and R. Galinier. 2003. Novel partner proteins of adenovirus penton, p. 37-55. In W. Doerfler and P. Bohm (ed.), Adenoviruses: model and vectors in virus-host interactions. Springer-Verlag, Berlin, Germany. [DOI] [PubMed]
- 7.Crabtree, K. D., C. P. Gerba, J. B. Rose, and C. N. Haas. 1997. Waterborne adenovirus: a risk assessment. Water Sci. Technol. 35:1-6. [Google Scholar]
- 8.Day, R. S., III. 1974. Studies on repair of adenovirus 2 by human fibroblasts using normal, xeroderma pigmentosum, and xeroderma pigmentosum heterozygous strains. Cancer Res. 34:1965-1970. [PubMed] [Google Scholar]
- 9.Eaton, A. L., L. S. Clesceri, E. W. Rice, and A. E. Greenberg. 2005. Standard methods for the examination of water and wastewater, 21st ed. American Public Health Association, Washington, DC.
- 10.Enriquez, C. E., C. J. Hurst, and C. P. Gerba. 1995. Survival of the enteric adenoviruses 40 and 41 in tap, sea, and wastewater. Water Res. 29:2548-2553. [Google Scholar]
- 11.Evans, J. D., and P. Hearing. 2003. Distinct roles of the adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J. Virol. 77:5295-5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fong, T., D. W. Griffin, and E. K. Lipp. 2005. Molecular assays for targeting human and bovine enteric viruses in coastal waters and their application for library-independent source tracking. Appl. Environ. Microbiol. 71:2070-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerba, C. P., D. M. Gramos, and N. Nwachuku. 2002. Comparative inactivation of enteroviruses and adenovirus 2 by UV light. Appl. Environ. Microbiol. 68:5167-5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harm, W. 1980. Biological effects of ultraviolet radiation. Cambridge University Press, Melbourne, Australia.
- 15.Hijnen, W. A. M., E. F. Beerendonk, and G. J. Medema. 2006. Inactivation credit of UV radiation for viruses, bacteria and protozoan (oo)cysts in water: a review. Water Res. 40:3-22. [DOI] [PubMed] [Google Scholar]
- 16.Howitt, J., C. W. Anderson, and P. Freimuth. 2003. Adenovirus interaction with its cellular receptor CAR, p. 331-364. In W. Doerfler and P. Bohm (ed.), Adenoviruses: model and vectors in virus-host interactions. Springer-Verlag, Berlin, Germany. [DOI] [PubMed]
- 17.Jacangelo, J., P. Loughran, B. Petrik, D. Simpson, and C. McIlroy. 2003. Removal of enteric viruses and selected microbial indicators by UV irradiation of secondary effluent. Water Sci. Technol. 47(9):193-198. [PubMed] [Google Scholar]
- 18.Jiang, S. C. 2006. Human adenoviruses in water: occurrence and health implications: a critical review. Environ. Sci. Technol. 40:7132-7140. [DOI] [PubMed] [Google Scholar]
- 19.Ko, G., T. L. Cromeans, and M. D. Sobsey. 2005. UV inactivation of adenovirus type 41 measured by cell culture mRNA RT-PCR. Water Res. 39:3643-3649. [DOI] [PubMed] [Google Scholar]
- 20.LaBarre, D. D., and R. J. Lowy. 2001. Improvements in methods for calculating virus titer estimates from TCID50 and plaque assays. J. Virol. Methods 96:107-126. [DOI] [PubMed] [Google Scholar]
- 21.Lee, S., and S. Kim. 2002. Detection of infectious enteroviruses and adenoviruses in tap water in urban areas in Korea. Water Res. 36:248-256. [DOI] [PubMed] [Google Scholar]
- 22.Li, J. W., Z. T. Xin, X. W. Wang, J. L. Zheng, and F. H. Chao. 2004. Mechanisms of inactivation of hepatitis A virus in water by chlorine dioxide. Water Res. 38:1514-1519. [DOI] [PubMed] [Google Scholar]
- 23.Li, J. W., Z. T. Xin, X. W. Wang, J. L. Zheng, and F. H. Chao. 2002. Mechanisms of inactivation of hepatitis A virus by chlorine. Appl. Environ. Microbiol. 68:4951-4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linden, K. G., J. Thurston, R. Schaefer, and J. P. Malley, Jr. 2007. Enhanced UV inactivation of adenoviruses under polychromatic UV lamps. Appl. Environ. Microbiol. 73:7571-7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maillard, J. Y. 1999. Viricidal activity of biocides. D. Mechanisms of viricidal action, p. 207-221. In A. D. Russell, W. B. Hugo, and G. A. J. Ayliffe (ed.), Principles and practice of disinfection, preservation, and sterilization. Blackwell Science, Oxford, England.
- 26.Meng, Q. S., and C. P. Gerba. 1996. Comparative inactivation of enteric adenoviruses, poliovirus, and coliphages by ultraviolet irradiation. Water Res. 30:2665-2668. [Google Scholar]
- 27.Nuanualsuwan, S., and D. O. Cliver. 2003. Infectivity of RNA from inactivated poliovirus. Appl. Environ. Microbiol. 69:1629-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nuanualsuwan, S., and D. O. Cliver. 2003. Capsid functions of inactivated human picornaviruses and feline calicivirus. Appl. Environ. Microbiol. 69:350-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nwachuku, N., and C. P. Gerba. 2004. Emerging waterborne pathogens: can we kill them all? Curr. Opin. Biotechnol. 15:175-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nwachuku, N., C. P. Gerba, A. Oswald, and F. D. Mashadi. 2005. Comparative inactivation of adenovirus serotypes by UV light disinfection. Appl. Environ. Microbiol. 71:5633-5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pina, S., M. Puig, F. Lucena, J. Jofre, and R. Girones. 1998. Viral pollution in the environment and in shellfish: human adenovirus detection by PCR as an index of human viruses. Appl. Environ. Microbiol. 64:3376-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rainbow, A. J. 1980. Reduced capacity to repair irradiated adenovirus in fibroblasts from xeroderma pigmentosum heterozygotes. Cancer Res. 40:3945-3949. [PubMed] [Google Scholar]
- 33.Rainbow, A. J., and S. Mak. 1973. DNA damage and biological function of human adenovirus after U.V. irradiation. Int. J. Radiat. Biol. 24:59-72. [DOI] [PubMed] [Google Scholar]
- 34.Rennecker, J. L., B. J. Mariñas, J. H. Owens, and E. W. Rice. 1999. Inactivation of Cryptosporidium parvum oocysts with ozone. Water Res. 33:2481-2488. [Google Scholar]
- 35.Seiradake, E., H. Lortat-Jacob, O. Billet, E. J. Kremer, and S. Cusack. 2006. Structural and mutational analysis of human Ad37 and canine adenovirus 2 fiber heads in complex with the D1 domain of coxsackie and adenovirus receptor. J. Biol. Chem. 281:33704-33716. [DOI] [PubMed] [Google Scholar]
- 36.Sharp, P. A. 1984. Adenovirus transcription, p. 173-204. In H. S. Ginsberg (ed.), The adenoviruses. Plenum Press, New York, NY.
- 37.Shin, G., K. G. Linden, and M. D. Sobsey. 2005. Low pressure ultraviolet inactivation of pathogenic enteric viruses and bacteriophages. J. Environ. Eng. Sci. 4:7-11. [Google Scholar]
- 38.Shin, G., K. G. Linden, and M. D. Sobsey. 2001. Low pressure UV inactivation of pathogenic enteric viruses and bacteriophages. In Proceedings of the 2001 American Water Works Association Water Quality Technology Conference. American Water Works Association, Denver, CO.
- 39.Sirikanchana, K., J. L. Shisler, and B. J. Mariñas. 2008. Inactivation kinetics of adenovirus serotype 2 with monochloramine. Water Res. 42:1467-1474. [DOI] [PubMed] [Google Scholar]
- 40.Smith, P. K., R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson, and D. C. Klenk. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150:76-85. [DOI] [PubMed] [Google Scholar]
- 41.Thompson, S. S., J. L. Jackson, M. Suva-Castillo, W. A. Yanko, Z. E. Jack, J. Kuo, C. Chen, F. P. Williams, and D. P. Schnurr. 2003. Detection of infectious human adenoviruses in tertiary-treated and ultraviolet-disinfected wastewater. Water Environ. Res. 75:163-170. [DOI] [PubMed] [Google Scholar]
- 42.Thurman, R. B., and C. P. Gerba. 1988. Molecular mechanisms of viral inactivation by water disinfectants. Adv. Appl. Microbiol. 33:75-105. [DOI] [PubMed] [Google Scholar]
- 43.Thurston-Enriquez, J., C. N. Haas, J. Jacangelo, K. Riley, and C. P. Gerba. 2003. Inactivation of feline calicivirus and adenovirus type 40 by UV radiation. Appl. Environ. Microbiol. 69:577-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.United States Environmental Protection Agency. 2006. 40 CFR parts 9, 141, and 142. National primary drinking water regulations: stage 2 disinfectants and disinfection byproducts rule, final rule. Fed. Regist. 71:388-493. [Google Scholar]
- 45.United States Environmental Protection Agency. 2006. 40 CFR parts 9, 141, and 142. National primary drinking water regulations: long term 2 enhanced surface water treatment rule, final rule. Fed. Regist. 71:653-702. [PubMed] [Google Scholar]
- 46.United States Environmental Protection Agency. 2005. Drinking water contaminant candidate list 2, final notice. Fed. Regist. 70:9071-9077. [Google Scholar]
- 47.United States Environmental Protection Agency. 1987. US EPA manual of methods for virology, chapter 10. EPA/600/4-84/013 (R-10). http://www.epa.gov/microbes/chapt10.pdf.
- 48.van Heerden, J., M. M. Ehlers, and W. O. K. Grabow. 2005. Detection and risk assessment of adenoviruses in swimming pool water. J. Appl. Microbiol. 99:1256-1264. [DOI] [PubMed] [Google Scholar]
- 49.Vantarakis, A., and M. Papapetropoulou. 1999. Detection of enteroviruses, adenoviruses and hepatitis A viruses in raw sewage and treated effluents by nested-PCR. Water Air Soil Pollut. 114:85-93. [Google Scholar]
- 50.Yates, M. V., J. Malley, P. Rochelle, and R. Hoffman. 2006. Effect of adenovirus resistance on UV disinfection requirements: a report on the state of adenovirus science. J. Am. Water Works Assoc. 98:93-106. [Google Scholar]
- 51.Zhang, W., and M. J. Imperiale. 2003. Requirement of the adenovirus IVa2 protein for virus assembly. J. Virol. 77:3586-3594. [DOI] [PMC free article] [PubMed] [Google Scholar]