

Abstract
Development of quantitative PCR (QPCR) assays typically requires extensive screening within and across a given species to ensure specific detection and lucid identification among various pathogenic and nonpathogenic strains and to generate standard curves. To minimize screening requirements, multiple virulence and marker genes (VMGs) were targeted simultaneously to enhance reliability, and a predictive threshold cycle (CT) equation was developed to calculate the number of starting copies based on an experimental CT. The empirical equation was developed with Sybr green detection in nanoliter-volume QPCR chambers (OpenArray) and tested with 220 previously unvalidated primer pairs targeting 200 VMGs from 30 pathogens. A high correlation (R2 = 0.816) was observed between the predicted and experimental CTs based on the organism's genome size, guanine and cytosine (GC) content, amplicon length, and stability of the primer's 3′ end. The performance of the predictive CT equation was tested using 36 validation samples consisting of pathogenic organisms spiked into genomic DNA extracted from three environmental waters. In addition, the primer success rate was dependent on the GC content of the target organisms and primer sequences. Targeting multiple assays per organism and using the predictive CT equation are expected to reduce the extent of the validation necessary when developing QPCR arrays for a large number of pathogens or other targets.
The detection and identification of multiple pathogens requires simultaneously targeting a large number of virulence and marker genes (VMGs) (14, 30a, 34, 42, 49, 53). This is because (i) the use of multiple markers for the same pathogen enhances the specificity and reliability of the assay, (ii) often the allelic variability requires multiple primers for the same gene, (iii) some of the markers may be less specific (i.e., they may also be found in nonpathogens), and (iv) the VMGs for a given pathogen may be unevenly distributed among various strains. Use of a varied number of VMGs to differentiate clinical isolates has been described for many pathogenic organisms, including Staphylococcus aureus (3, 48), Aeromonas spp. (8), Campylobacter jejuni (38), Pseudomonas aeruginosa (13), Vibrio spp. (49), Escherichia coli, and Shigella flexneri (5, 52).
When many pathogens must be screened in parallel with the ability to characterize the uneven distribution of the associated VMGs and their allelic variability, optimization is necessary for high-throughput tools with high sensitivity and specificity, e.g., quantitative PCR (QPCR) (14, 19, 27, 32, 44, 46, 50, 51). This optimization may be cumbersome because it requires the generation of multiple standard curves with the caveat that all primer sets must perform under the same amplification conditions. Approaches that increase the reliability of the primer design or avoid the use of standard curves altogether should be extremely useful in developing such parallel assays. The development of such approaches will undoubtedly depend upon the sequence characteristics (guanine and cytosine [GC] content, melting temperature [Tm], amplicon length, etc.) of primers, amplicons, genome size, amplification conditions, and the matrix in which the target is present. However, the influence of these factors on the performance of primer sets, especially on the threshold cycle (CT) number extensively used in QPCR to predict the abundance of pathogens, has not been explored fully.
This study used the nanoliter-volume BioTrove OpenArray platform (33) to examine the capacity of QPCR for highly parallel diagnostics of human pathogens and to systematically examine the influence of target and primer sequence characteristics on specificity, sensitivity, and CT value. The results were used to establish a predictive CT equation to estimate the number of starting copies without the use of standard curves. The study was performed with Sybr green I and used approximately 220 primer sets targeting 200 VMGs for 30 human pathogens. The performance of the predictive CT equation is examined, and the success rate of previously unvalidated pathogen-targeted primer sets is also presented. These results have significance in developing high-throughput and reliable screening tools for large numbers of pathogens without extensive validation.
MATERIALS AND METHODS
DNA targets.
Multiple organisms were used to develop and validate the predictive CT equation. Genomic DNA (gDNA) from 21 bacterial pathogens (Table 1) was used, including Clostridium perfringens, Enterococcus faecalis, Listeria monocytogenes, Legionella pneumophila, Pseudomonas aeruginosa, Salmonella enterica, Vibrio parahaemolyticus, and Yersinia enterocolitica type strains, which were obtained from the American Type Culture Collection (ATCC; Manassas, VA) and grown as per the protocol provided. For Helicobacter pylori, Campylobacter jejuni, Cryptosporidium parvum, Giardia intestinalis, Staphylococcus aureus, Vibrio cholerae, Mycoplasma genitalium, Haemophilus influenzae, Leptospira interrogans, Bacillus cereus, and Bordetella pertussis, gDNA was obtained directly from the ATCC. Escherichia coli gDNA was kindly provided by Thomas Whittam at Michigan State University. DNA from pure cultures was extracted using Promega's Wizard DNA extraction kit (Promega, Madison, WI).
TABLE 1.
Pathogens and VMGs targeted with OpenArray plates and organisms used for validationa
| Genus (n = 30) | Species | Marker gene(s) targeted (n = 200) | No. of primer sets (n = 220)
|
Strain(s) tested (ATCC) | |
|---|---|---|---|---|---|
| Designed (n = 200) | From literature (n = 20) | ||||
| Aeromonas | hydrophila | alt, exeF, tapA, arcV, ascT | 5 | 0 | |
| Bacillus | cereus, anthracis | atxA, capA, lef, cerA, clo, hlyIII, mprF, pagA, plcR | 9 | 0 | 10987 |
| Bordetella | pertussis | cyaC, dnt, ptx, tcfA | 4 | 0 | BAA-589D |
| Burkholderia | mallei, pseudomallei | pilA, pilD, putative outer membrane gene, BPSS1407 | 4 | 0 | |
| Campylobacter | jejuni, coli | hipO, racR, mapA, gyrA, cdtC, cdtB, cdtA, ciaB, cfrA, tonB | 8 | 2 | 700819 |
| Clostridium | perfringens, difficile, botulinum | cpe, plc, pfo, cpb2, etx, bontA, ha33, ha70, cdtA, cdtB | 10 | 0 | 12916 |
| Corynebacterium | diphtheriae | dtxR | 1 | 0 | |
| Coxiella | burnetii | dotA, icmP, icmQ, mip | 4 | 0 | |
| Enterococcus | faecalis, faecium | cylA, ace, cylLS, esp, gelE | 6 | 0 | 19433 |
| Escherichia | coli (including Shigella) | uidA, stx1, stx2, eae, papG, lucC, ehxA, sfaA, ipaH, mxiH | 11 | 3 | BAA-460D |
| Francisella | tularensis | acpA, clpB, mglA | |||
| Haemophilus | influenzae | lic2A, tbp1, tbp2 | 3 | 0 | 51907D |
| Helicobacter | pylori | cagA, cagE, ureA, virD4, virB9, virB11, ureI, ureB, flaB | 10 | 2 | 700392 |
| Klebsiella | pneumoniae | nuc, magA, kvgS, kci, kca | 5 | 0 | |
| Legionella | pneumophila | lepB, icmQ, lepA, mip, dotA, icmB, icmR, lspH, lssD | 9 | 1 | 33152 |
| Leptospira | interrogans | hlyB, lipA, chpI, lipL21, ompL1, chpK, etpK | 6 | 1 | 43642 |
| Listeria | monocytogenes | actA, hlyA, inlA, mpl, plcB, lisA, plcA, iap, prfA | 8 | 1 | 15313 |
| Mycobacterium | avium complex, paratuberculosis, tuberculosis, leprae | erp, glnA1, mmpA, mmpB, IS900, mmpC, plcA | 6 | 1 | |
| Mycoplasma | genitalium | hww3, p1, p200, p30 | 4 | 0 | 33530D |
| Proteus | mirabilis | lpp, uca, zapA, zapD | 4 | 0 | |
| Pseudomonas | aeruginosa | exoS, iasA, pcrV, xcpA, popD, popB, las, etA, prfA, pilD | 11 | 10145 | |
| Salmonella | enterica serovars Enterica and Typhimurium | fimA, flicC, fljB, invA, hiA, hiD, invE, prgH, sipB, sipC, spaR, tyv | 15 | 1 | 13311, 19585 |
| Serratia | marcescens | flhDC, hasD, lipA, safA | 4 | 0 | |
| Staphylococcus | aureus | hly, seA, seC, tsst-1, lukE, lukF, lytS, nuc, seA, tst | 10 | 1 | 700699 |
| Streptococcus | pyogenes | ssa, mtsA, ska, speA, speC | 5 | ||
| Vibrio | cholerae, mimicus, parahaemolyticus, vulnificus | ace, ompU, tdh, tdhS, tlh, toxR, ctxA, ctxB, mshA, tcpA, zot, trh1, vpl, vvhA, wp, tagA, trh1 | 14 | 2 | 39315 43996 |
| Yersinia | enterocolitica, pestis, pseudotuberculosis | crp, yopD, yscD, yst, bipA, yadA, yspB, yscN, ail | 9 | 2 | 55075 |
| Cryptosporidium | parvum, hominis | gp40, hsp70, COWP gene, cp23, cgd7 | 7 | 1 | PRA-67 |
| Giardia | intestinalis, lamblia | β-giardin gene, VSPH71, VSP4177, VSP4173, vsp, VSPH7 | 5 | 2 | 30888 A-1 |
The VMGs in bold were only tested in validation sample mixtures prepared to validate the predictive CT equation. Primer sequences are provided in the supplemental material.
Complex background gDNA.
The gDNA from pure cultures was spiked into complex background gDNA extracted from environmental waters. River water was collected from the Red Cedar River in Michigan and activated sludge and tertiary effluent from a wastewater treatment plant in East Lansing, MI. Samples were filtered through 0.45-μm nitrocellulose filters (Millipore, Billerica, MA) immediately after collection. The gDNA was extracted from the filters as instructed with the MegaPrep UltraClean soil DNA kit (Mo Bio Laboratories, Carlsbad, CA). The gDNA was purified further through ethanol precipitation. DNA was quantified and its purity was examined with the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). Prior to the QPCR experiments, background DNA was tested for inhibition by PCR amplification with a universal primer set targeting the 16S rRNA gene.
Design of the PCR primers to establish the predictive CT equation.
A set of primers was designed to target 20 human pathogens and was used to examine relationships between primer sequence characteristics and experimental CT. Prior to the primer design, consensus sequences were generated by aligning sequences for each of 96 VMGs using Kodon (Applied Maths, Austin, TX). The consensus sequence was used for the primer design with Primer Express (Applied Biosystems, Foster City, CA). A majority of the designed amplicons had a maximum length of 150 bases, and primers had a theoretical melting temperature (Tm) of 59°C. Some genes required longer amplicons (<250 bases) for generating acceptable primer sets. From the list of primers provided by Primer Express, optimal primers were selected using a script that automatically highlighted unspecific primers. The script used NCBI BLAST (2) to check specificity against the GenBank database. Specificity was based on the extent of 3′-end perfect matches to nontargeted bacterial sequences. Sequences were selected manually based on the results of the BLAST output. When available, primers described in the literature for successful QPCR were also used. Overall, 110 primer pairs were either designed or extracted from the literature (Table 1; primer sequences are listed in the supplemental material). Including primers previously described in the literature, this primer set targeted 3,687 VMG sequences (determined by BLAST analysis with GenBank, May 2006). Primer sequences and references for primers extracted from literature are listed in the supplemental material.
Design of PCR primers to validate correlations used in the development of the predictive CT equation.
To validate the predictive CT equation and further examine the success rate of the primers, 111 new primers were designed. The new group of primers was designed with the same criteria as the primers designed to establish the predictive CT equation. In addition, the primers were filtered further to select sequences with the lowest possible percentages of GC bases in the 3′ ends of the primers. One nonspecific primer targeting C. parvum was removed from the set due to false-positive observations. Thus, a total of 220 primer sets (109 used to establish the predictive CT equation, and 111 used to validate the predictive CT equation) targeting 200 VMGs for 30 pathogenic bacteria were tested (Table 1; the new VMGs targeted with this primer set are in bold).
PCR on BioTrove OpenArray plates.
Primer sets were tested simultaneously on the BioTrove OpenArray plates. Primers were synthesized by Sigma-Aldrich (St. Louis, MO) and preloaded (128 nM for primers designed to establish the predictive CT equation and 400 nM for primers designed to validate the predictive CT equation) into BioTrove OpenArray plates (Woburn, MA) (33, 46). Two to four subarrays (each with 64 wells for 56 separate assays and eight loading controls) were used for each PCR sample. PCR mixtures (5 μl for each sample array) consisted of 1× LightCycler FastStart DNA Master Sybr green I mix (Roche Applied Sciences, Indianapolis, IN), 1.6× Sybr green I, 0.5% glycerol, 0.2% Pluronic F-68, 1 mg per ml bovine serum albumin (New England Biolaboratories, Beverly, MA), 2.5 mM MgCl2, 8% formamide, and a DNA mixture. After the initial enzyme activation at 95°C for 10 min, 36 cycles of the following program were used for amplification: denaturation at 95°C for 10 s, annealing at 53°C for 10 s, and elongation at 72°C for 10 s.
Design of the sample mixtures for development of the predictive CT equation.
Samples were mixed to evaluate the influences of the sequence characteristics of primers, amplicons, genome size, amplification conditions, and the matrix in which the target is present on specificity and sensitivity. To evaluate specificity without a complex background, gDNA from 14 pathogenic organisms (pure cultures; ATCC numbers are listed in Table 1) was tested individually (6 ng in the total sample or 20 pg per reaction well). To develop standard curves and further evaluate specificity and sensitivity within a complex background, gDNA from 14 pathogens was mixed and spiked at various concentrations into gDNA from the wastewater tertiary effluent and river water samples (20 pg, 2 pg, 200 fg, 20 fg, and 2 fg of each of the 14 pathogens mixed together spiked into 66.6 pg of background gDNA per reaction well). The mixture of various concentrations was also examined without a background to serve as a control. The complex background samples were also tested without spiking with gDNA from pure cultures. All of these samples were tested with the set of PCR primers designed to establish the predictive CT equation. These samples were examined further to evaluate the influence of sample inhibition and variation between OpenArray plates. All samples were tested in triplicate.
Design of the validation sample mixtures for validation of the predictive CT equation.
The validation samples were used to further evaluate the effect of characteristics of the template and primer sequences on the primer success rate and to evaluate the predictive CT equation. For the validation samples, gDNA from 21 organisms (ATCC numbers are listed in Table 1; the organisms used solely with these mixtures are in bold) was spiked at various concentrations, for an absolute abundance of approximately 10, 100, and 1,000 genomic copies per reaction well, into gDNA extracted from either river water, tertiary effluent, or activated sludge (0.99 ng per reaction well). In total, 36 validation samples were prepared. All samples were tested in triplicate.
Data analysis.
For all analysis, data was filtered to differentiate true- and false-positive and -negative signals. Amplification was considered positive if the CT was less than 26 for all three replicates and the experimental Tm was consistent. The influence of a primer's 3′-end GC content on specificity was analyzed using the primer (either forward or reverse) with the highest GC content. Primers were grouped based on the number of GC bases within the last 5 bases on the 3′ end. For developing the predictive CT equation, only the primer sets displaying true-positive amplification were considered.
The influence of the GC content of the primers and the target organisms on the success rates of novel primers was examined based on the average sum of the successes (taken from 36 sample mixtures for validating the predictive CT equation) for all assays targeting an organism. Assays were considered successful if they displayed a true-positive or true-negative signal, and an organism was deemed present if two or more assays (targeting one organism) displayed amplification with a CT less than 26 for all three replicates. The average GC content of all primers used for a targeted organism was employed for the analysis examining the influence of primer GC content on the success rate. The three-dimensional plot was generated with a loess smoother and 1.0 sampling proportion. This smoothing was performed to identify characteristics of the population.
For comparison with the predictive CT equation, standard curves were generated using an average slope and intercept from all three replicates in all three backgrounds (control and gDNA spiked with river and tertiary gDNA) from sample mixtures designed to develop a predictive CT equation. PCR efficiency was examined to determine the influence of the sample background on quantitative values and was calculated from the slope of the standard curves with the following equation: PCR efficiency = −1 + 10−1/slope.
Raw results of all the experiments are included in the supplemental material.
RESULTS AND DISCUSSION
Sensitivity and specificity.
The influence of primer and template sequence characteristics on specificity and sensitivity was examined using the primer sets and sample mixtures designed to develop the predictive CT equation. The percentage of assays displaying false-positive/false-negative signals for primer sets tested individually and within a complex background did not change for target concentrations above a range of 1 to 10 copies (Fig. 1, left panel). Within the 1 to 10 copy range (i.e., with 20 fg of target gDNA present per reaction well), approximately half of the targeted assays displayed positive amplification. Depending on the organisms, this corresponds to 1.5 to 11 genomic copies per reaction well. A majority of the assays displaying true-positive amplification at this concentration were for the following organisms: L. monocytogenes (6.3 copies), S. enterica (3.7 copies), V. parahaemolyticus (3.6 copies), H. pylori (11.1 copies), C. jejuni (11.6 copies), and V. cholerae (4.6 copies). Assays targeting all other organisms within this concentration had a detection limit higher than 10 copies per reaction well, implying that those organisms were not detectable at this concentration. In addition, a large majority of the targeted assays not displaying amplification at higher concentrations (Fig. 1, left panel) were for primer sets targeting the same organisms. For example, with 2,000 fg (100 to 1,000 copies) of target gDNA per reaction spiked into 66.6 pg per reaction, the following organisms displayed false-negative signals in at least two of the three replicates: P. aeruginosa (three out of five primer sets), S. aureus (one out of four primer sets), and G. intestinalis (four out of five primer sets). This accounts for all assays (8 of 69) showing false-negative amplification at 100 to 1,000 copies per reaction well. A closer look indicated that the GC content was either very low (37% for S. aureus) or very high (67% for P. aeruginosa). G. intestinalis had the largest genome of all the organisms targeted (12 Mbp).
FIG. 1.

Impact of gDNA from various environmental water samples and primer stability on specificity and sensitivity. The left panel shows the percentages of targeted and nontargeted primer assays displaying amplification at various dilutions of organisms spiked in gDNA from environmental samples. Error bars represent the standard deviations between replicates performed on three plates. The right panel shows the sum of the GC bases on the terminal 3′ end (various-size circles) versus the percentages of primer sets displaying false-positive amplification when targets are spiked into background gDNA and not spiked into background gDNA. bkg, background.
Previous PCR studies of organisms with high GC content have found similar influences and suggest that the effect is due to a stabilizing secondary structure influencing polymerase extension or primer-template annealing, causing false-negative observations (16, 24, 36). Solutions to minimize the effect include using higher denaturation and annealing temperatures or using additives such as glycerol, betaine, formamide, or dimethyl sulfoxide. The false-negative signal observed with S. aureus was likely due to the low GC content of the organism, requiring the length of the primers to be increased to maintain suitable stability. The reverse primer for the S. aureus assay displaying a false-negative signal was longer (32 bp) than any other primer tested in this experiment. Internal complements within longer primers may reduce annealing to target sequences (39).
The false-negative signal observed with G. intestinalis may be due to lower relative abundance caused by having a larger genome and a potential reduction in the availability of the target. The influence of genome size on amplification potential has been described previously (11, 15) and may be due to a decrease in the relative abundance of the template over the nontarget DNA. Garner proposed that in addition to a decreased relative abundance, there is an increased chance of the nonspecific annealing of primers to nontarget regions, diminishing the annealing of primers to the target strand (15). Optimizing the PCR cycle conditions or the concentration of the reagents may alleviate false-negative signals (40); however, changing these parameters may influence the specificity and sensitivity of the targeted assays that behaved well.
Since the primers were designed to have the same theoretical Tm, the terminal 3′ end of the primers was also examined. The GC content, Tm, and binding energy within the terminal 7, 5, 3, and 2 bases on the terminal 3′ end of the primers were considered for all false-positive signals. Correlations (provided in the supplemental material) were used to determine that both the Tm of the last 7 bases and the GC content of the last 5 bases had the highest influence on false-positive signals (considering targets alone and spiked into a background). To demonstrate this, primer sets were grouped based on the GC content within the last 5 bases of the terminal 3′ end (Fig. 1, right panel). Two out of seven primer sets (28%) with five GCs within the last 5 bp at the 3′ end of the primer displayed false-positive amplification. The percentage of false-positive signals decreased with the amount of GC in the 3′ ends of the primers. The influence of the 3′ end of a primer on the specificity of the amplification has been described previously (31). As a result, many primer design software programs now analyze the 3′ ends of potential primers, while Primer3 emphasizes the stability of 5-base segments of the terminal 3′ end (37).
It should be noted that the results obtained with the OpenArray nanoliter-volume reactions are comparable to conventional microliter-volume QPCR. Low-volume PCR has been optimized in nanoliter-volume reactions by adjusting surface chemistry (33), ramping rates and decreased annealing temperatures (46), and adjusting the PCR master mix composition with extra Sybr green I, bovine serum albumin, and formamide added to the standard PCR mixture. Cross-platform comparisons between the PCR performed with the microplate format (10- and 20-μl-volume reactions in the 7900HT) and with the OpenArray platform have shown high similarities in PCR efficiencies and detection limits (E. Ortenberg and D. Roberts, unpublished data; 7). The comparison also demonstrated a high correlation (between the two platforms) of specific gene regulation patterns between experimental (diseased heart) and control (normal adult heart) tissues. The PCR efficiency observed with the experiments in this study (described below) also demonstrates the success of PCR primers with the OpenArray environment.
Development of the predictive CT equation.
An empirical equation was developed using the sequence-specific results observed with the primers designed to develop the predictive CT equation. Multiple parameters were considered for the predictive CT equation, including size of the genome, GC content of the genome, and primer binding energy. Since all of the primers were designed with the same theoretical Tm (for simultaneous amplification of all primer sets on the OpenArray plate), the terminal 3′ ends of the primers were also considered. This included the binding energy, position of the G and C bases, Tm, and GC content for the terminal 7, 5, 3, and 2 bases on the 3′ ends of the primers. Correlations (Table 1; see the supplemental material) were used to determine which parameters had a greater influence on the CT value. The inclusion of parameters other than those chosen either had no effect or decreased the correlation between the predictive and experimental CTs. In addition, using the general linear model requires that all variables are independent; thus, only the top independent parameters were used. For example, parameters such as binding energy are not entirely independent of the Tm of the terminal 7 bases, as a primer with a high binding energy may have a higher Tm on the terminal 3′ end. Five parameters were identified that influenced the CT of each primer set. These were (i) the genome size of the targeted organism, (ii) the target organism concentration, (iii) the GC content of the targeted organism, (iv) amplicon length, and (v) the theoretical Tm of the last 7 bases on the primer's 3′ end. The levels of influence of these variables on the CT were in the order listed (i.e., the genome size of the targeted organism had the greatest influence, and the Tm of the last 7 bases on the primer's 3′ end had the least).
A multiple-parameter linear regression curve was used to place a weighted influence on each of these parameters, and the following equation was developed:
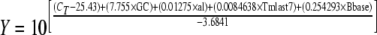 |
where GC is the GC content (e.g., for an organism with 30%, the GC is 0.3), al is the amplicon length (bases long), CT is the experimental CT, Y is the number of starting copies, Bbase is the size of the genome (Mbp), and Tmlast7 is the Tm calculated for the seven terminal bases on the primer's 3′ end, the lowest temperature (°C) of the two primers with the base stacking calculation (41).
A high correlation (R2 = 0.816) between the CT predicted with the predictive CT equation and the experimental CT was observed (Fig. 2). Since the number of gene copies will vary based on the targeted organisms and gene, the predictive CT equation solely predicts starting copies per reaction. The accuracy of the equation was tested and compared with standard curves using validation sample mixtures with the primer set designed to validate the empirical CT equation.
FIG. 2.

Experimental versus predicted CT using predictive CT equation. The results obtained are with the primer designed to establish the predictive CT equation. The equation is derived using amplicon length, starting genomic copies, number of base pairs in the target organism's genome, GC content of the target organism's genome, and theoretical Tm of the last 7 bases of the primer's 3′ end. Errors bars represent the standard deviations of experimental CT between replicates on three plates.
It is likely that constants in the predictive CT equation will change based on the chosen system, reaction conditions, and reagents from different vendors. Therefore, some validation is necessary for setting up novel, highly parallel assays. BioTrove suggests using the same reaction conditions for all Sybr-based diagnostics on OpenArray plates. Therefore, users of BioTrove for diagnostics of microbial communities can readily apply the predictive CT equation. Development of a predictive CT equation for a new assay will consist of validating with organisms from targeted genera (with various target characteristics) and establishing a multiple-parameter linear regression curve to place a weighted influence on parameters. To normalize for the stability of the reagent (a fresh one versus a less fresh one), the lot number between reagents, sample loading, and inhibition within various samples, interassay normalization with an internal control can be used with the predictive CT equation. Internal controls can consist of DNA strands absent in the diagnostic target sample (1, 9, 30) and/or a universal and stable unregulated endogenous standard (28, 29, 35).
Validation of an organism's sequence characteristics to determine primer success rate.
Validation sample mixtures were tested with primers designed to validate the predictive CT equation to determine the success rate of novel primers. The influence of primer and targeted organism GC content on the success of the primers was observed (Fig. 3). Results show that the sequence of organisms with extreme GC content (low or high) had lower success rates. A study by Housley et al. (18) observed similar results concerning the influence of GC content and the success of the primers, describing success rates of 56.9% for primers designed to target an amplified region with GC content greater than 50% and 74.2% for primers designed to target an amplified region with GC content less than 50%. Targeted organisms with high GC content frequently tend to give weak signals in amplification (due to secondary structure and template-template annealing), and primers with high GC content will amplify nontargeted regions (due to high stability). In addition, an organism with a low GC content in the genome will have a higher rate of false-negative signals. The number of assays required for confidently determining the presence or absence of an organism is dependent on the success rates of designed primers. Therefore, targets with extreme GC content will require more assays for determining the presence and absence of organisms.
FIG. 3.

Success rate (%) of all assays targeting an organism. Success rate is defined as the sum of all true-positive and -negative assays divided by the sum of all assays targeting a single organism. The success rate was taken as an average of all 36 validation sample mixtures prepared to validate the predictive CT equation.
These results suggest that validation requirements can be reduced if multiple primer sets are assayed simultaneously, overcoming issues in specificity and sensitivity observed with organisms and genes with extreme GC content. Typically, amplification reactions can be optimized to overcome issues with false-positive or -negative signals. However, large-scale PCR diagnostics such as the platform described here require broad conditions to satisfy suitable reactions for the majority of targeted assays. The use of redundant probes to increase confidence in determining the presence and absence of organisms has been described for other high-throughput platforms, such as microarrays (30a, 53). It should be noted that these observations may not be observed using methods that provide greater differentiation between true-positive and false-positive amplification, such as TaqMan QPCR, dissociation analysis, or ligation-based methods. However, the cost and time to complete the diagnostics significantly increased with these approaches, especially for high-throughput diagnostics.
Validation of the predictive CT equation.
The predictive CT equation was examined and compared with standard curves (obtained from dilution experiments performed with primers designed to develop the predictive CT equation) by predicting the starting copy number in the 36 validation samples tested with the primers designed to validate the predictive CT equation. A distribution of the predicted values shows a high distribution of predicted starting copies around 20, 100, and 1,000, which is close to the actual starting copy numbers of 10, 100, and 1,000 spiked into the validation samples (Fig. 4). Comparing the predicted starting copies using the Wilcoxon signed-rank test showed that the standard curve tended to predict higher starting copies than using the predictive CT equation. Using the predictive CT equation made differentiating between assays spiked at 10 and 100 starting copies unclear.
FIG. 4.

Distribution of predicted starting copy numbers using predictive CT equation and standard curves for validation samples and primers designed to validate the predictive CT equation. Note that the x axes of the three panels have different scales. The templates of all the organisms were spiked at either 10, 100, or 1,000 genomic copies per reaction well (indicated by the dotted lines). Error bars represent the standard deviations between three replicates performed on the same OpenArray plates.
The large distribution observed with both quantitative measures may be due to differences in reaction constitute lot number (between experiments performed to establish and validate the predictive CT equation), the manual mixing of samples, and the chosen calibration parameters. The influences of reagent formulation and calibration parameters (e.g., replication and serial n-fold dilution) on the precision of standard curves have been documented previously (12, 25). Inaccuracies in quantifying gDNA using UV absorbance may have also occurred. The correlation of the predictive starting copies calculated using the standard curve and predictive CT equation was R = 0.87. This suggests that the error between the predicted and actual starting copies may have been due to inaccuracies in mixing the validation samples and/or variations caused by reagents and not the predictive methods themselves.
Inhibitions within various environmental samples may have also influenced quantification. The distribution of PCR efficiencies shifted when targets were spiked into gDNA from different environmental waters (Fig. 5, left panel), as observed by testing primers and samples designed to validate the predictive CT equation. The tertiary effluent background shifted the distribution of PCR efficiency above 1, and the river water shifted the distribution below 1. An analysis of variance showed significant inequality involving the mean PCR efficiencies between the control and the two backgrounds. Approximately 83% of the targeted assays with 1,000 to 10,000 starting copies had a CT standard deviation equal to or less than 0.35, as did 80% of the assays with 100 to 1,000 starting copies, 33% with 10 to 100 starting copies, and 15% with 1 to 10 copies. A CT standard deviation of 0.35 corresponds to a coefficient of variation of 25% for the estimated starting copies (assuming a standard curve slope of −3.3), which has been described in previous reports as a cutoff for acceptable precision in QPCR diagnostics (4, 20, 43). A correlation matrix was used to examine whether primer characteristics influenced assays with high standard deviations (data not shown). Analysis showed that the size of the genome had the highest weighted influence on the standard deviation of the CT, followed by the number of starting copies and the GC content of the target genome. Other studies have observed an influence of GC content on primer success. A study by Vanichanon et al. (47) found reduced repeatability with primer sets with high GC content. Thus, primers with GC content closer to 50% will be ideal for maintaining high reproducibility (10, 21, 23). This fluctuation in efficiency could be due to the designed assays, enzyme instability, and sample-dependent inhibitions (22).
FIG. 5.

Factors potentially influencing quantitative inaccuracies as observed with primers and samples designed to establish the predictive CT equation. In the left panel, a box plot shows the distribution of PCR efficiency for target organisms spiked into gDNA from complex environmental waters and a control (no background). The right panel shows the cumulative frequency distributions of the standard deviations of CT determined between three separate OpenArray plates with various transcript concentrations.
Random microscale defects of the interior polymer surface coating of OpenArray plates, as suggested by Morrison et al. (33), may have also contributed to differences between the predicted and actual numbers of starting copies. Intra-assay reproducibility was examined with three replicates performed on three separate OpenArray plates run simultaneously with primers and samples designed to establish the predicted CT equation. The results illustrate that 93% of the assays with 1,000 to 10,000 starting copies displayed a coefficient of variation equal to or less than 25% between OpenArray plates, as did 93% of assays with 100 to 1,000 starting copies, 70% with 10 to 100 starting copies, and 33% with 1 to 10 starting copies (Fig. 5, right panel). The results suggest greater deviation was observed between the sample backgrounds than between the plates, and the accumulation of both aspects may have influenced the distribution between the predicted and actual starting copies.
Future diagnostics will include both an exogenous internal positive control and a universal assay targeting the 16S rRNA gene to allow normalization for the potential influence of sample inhibitions, plate variation, lot constitutes, and assays on CT (17, 26, 51). Cumulatively, the large distributions observed with predicting starting copies with standard curves along with the cumbersome requirement of validation make using the predictive CT equation an attractive potential alternative. One other alternative strategy to quantify starting copies without the generation of standard curves has been described using competitive PCR with a fluorescence quenching (45). This alternative has the potential to quantify DNA within the presence of inhibitions, as tested with high concentrations of gDNA from pure cultures spiked into humic acid; however, the accuracy and precision of the quantification and the influence of inhibitors were not described with samples containing less than 1,000 starting copies.
Conclusion.
The influences of the sequence characteristics of primers, amplicons, target genome size, amplification conditions, and the matrix in which the target is present on CT were explored. A predictive CT equation was experimentally established by examining PCR-based assays targeting multiple VMGs and may be an efficient alternative to generating standard curves. These results are valuable when developing high-throughput and reliable screening tools for a large number of pathogens without extensive validation.
Supplementary Material
Acknowledgments
We are grateful for the help from Elen Ortenberg, with a proof-of-principal study with BioTrove.
This work was supported by the National Institutes of Health (grant R01 RR018625-01), Environmental Protection Agency (RD83162801-0 and RD83301001), and 21st Century Michigan Economic Development Corporation (GR-476 PO 085P3000517).
Footnotes
Published ahead of print on 18 April 2008.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Abdulmawjood, A., S. Roth, and M. Bulte. 2002. Two methods for construction of internal amplification controls for the detection of Escherichia coli O157 by polymerase chain reaction. Mol. Cell. Probes 16:335-339. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker, K., A. W. Friedrich, G. Peters, and C. von Eiff. 2004. Systematic survey on the prevalence of genes coding for staphylococcal enterotoxins SEIM, SEIO, and SEIN. Mol. Nutr. Food Res. 48:488-495. [DOI] [PubMed] [Google Scholar]
- 4.Behets, J., P. Declerck, Y. Delaedt, L. Verelst, and F. Ollevier. 2007. A duplex real-time PCR assay for the quantitative detection of Naegleria fowleri in water samples. Water Res. 41:118-126. [DOI] [PubMed] [Google Scholar]
- 5.Blanco, J., M. Blanco, J. E. Blanco, A. Mora, E. A. Gonzalez, M. I. Bernardez, M. P. Alonso, A. Coira, A. Rodriguez, J. Rey, J. A. Alonso, and M. A. Usera. 2003. Verotoxin-producing Escherichia coli in Spain: prevalence, serotypes, and virulence genes of O157:H7 and non-O157 VTEC in ruminants, raw beef products, and humans. Exp. Biol. Med. (Maywood) 228:345-351. [DOI] [PubMed] [Google Scholar]
- 6.Reference deleted.
- 7.Brenan, C., and T. Morrison. 2005. High throughput, nanoliter quantitative PCR. Drug Discov. Today Technol. 2:247-253. [DOI] [PubMed] [Google Scholar]
- 8.Chacon, M. R., M. J. Figueras, G. Castro-Escarpulli, L. Soler, and J. Guarro. 2003. Distribution of virulence genes in clinical and environmental isolates of Aeromonas spp. Antonie van Leeuwenhoek 84:269-278. [DOI] [PubMed] [Google Scholar]
- 9.Cubero, J., J. van der Wolf, J. van Beckhoven, and M. M. Lopez. 2002. An internal control for the diagnosis of crown gall by PCR. J. Microbiol. Methods 51:387-392. [DOI] [PubMed] [Google Scholar]
- 10.Dieffenbach, C. W., T. M. J. Lowe, and G. S. Dveksler. 1993. General concepts for PCR primer design. PCR Methods Appl. 3:S30-S37. [DOI] [PubMed] [Google Scholar]
- 11.Farrelly, V., F. A. Rainey, and E. Stackebrandt. 1995. Effect of genome size and rrn gene copy number on PCR amplification of 16S rRNA genes from a mixture of bacterial species. Appl. Environ. Microbiol. 61:2798-2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farriol, M., and X. Orta. 2005. Influence of reagent formulation on mRNA quantification by RT-PCR using imported external standard curves. Acta Biochim. Pol. 52:845-848. [PubMed] [Google Scholar]
- 13.Feltman, H., G. Schulert, S. Khan, M. Jain, L. Peterson, and A. R. Hauser. 2001. Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology 147:2659-2669. [DOI] [PubMed] [Google Scholar]
- 14.Fukushima, H., Y. Tsunomori, and R. Seki. 2003. Duplex real-time SYBR green PCR assays for detection of 17 species of food- or waterborne pathogens in stools. J. Clin. Microbiol. 41:5134-5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garner, T. W. J. 2002. Genome size and microsatellites: the effect of nuclear size on amplification potential. Genome 45:212-215. [DOI] [PubMed] [Google Scholar]
- 16.Grassi, M., E. Volpe, V. Colizzi, and F. Mariani. 2006. An improved, real-time PCR assay for the detection of GC-rich and low abundance templates of Mycobacterium tuberculosis. J. Microbiol. Methods 64:406-410. [DOI] [PubMed] [Google Scholar]
- 17.Hartman, L. J., S. R. Coyne, and D. A. Norwood. 2005. Development of a novel internal positive control for Taqman based assays. Mol. Cell. Probes 19:51-59. [DOI] [PubMed] [Google Scholar]
- 18.Housley, D. J. E., Z. A. Zalewski, S. E. Beckett, and P. J. Venta. 2006. Design factors that influence PCR amplification success of cross-species primers among 1147 mammalian primer pairs. BMC Genomics 7:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu, C. F., T. Y. Tsai, and T. M. Pan. 2005. Use of the duplex TaqMan PCR system for detection of Shiga-like toxin-producing Escherichia coli O157. J. Clin. Microbiol. 43:2668-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang, H. Y., and T. M. Pan. 2004. Detection of genetically modified maize MON810 and NK603 by multiplex and real-time polymerase chain reaction methods. J. Agric. Food Chem. 52:3264-3268. [DOI] [PubMed] [Google Scholar]
- 21.Innis, M. A., and D. H. Gelfand. 1990. Optimization of PCRs, p. 3-12. In M. A. Innis, D. H. Gelfand, J. J. Sninksy, and T. J. White (ed.), PCR protocols: a guide to methods and application. Academic Press, Inc., New York, NY.
- 22.Kainz, P. 2000. The PCR plateau phase—towards an understanding of its limitations. Biochim. Biophys. Acta 1494:23-27. [DOI] [PubMed] [Google Scholar]
- 23.Kim, H. S., and O. Smithies. 1988. Recombinant fragment assay for gene targetting based on the polymerase chain reaction. Nucleic Acids Res. 16:8887-8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kureishi, A., and L. E. Bryan. 1992. Pre-boiling high GC content, mixed primers with 3′ complementation allows the successful PCR amplification of Pseudomonas aeruginosa DNA. Nucleic Acids Res. 20:1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai, K. K. Y., L. Cook, E. M. Krantz, L. Corey, and K. R. Jerome. 2005. Calibration curves for real-time PCR. Clin. Chem. 51:1132-1136. [DOI] [PubMed] [Google Scholar]
- 26.Layton, A., L. McKay, D. Williams, V. Garrett, R. Gentry, and G. Sayler. 2006. Development of Bacteroides 16S rRNA gene TaqMan-based real-time PCR assays for estimation of total, human, and bovine fecal pollution in water. Appl. Environ. Microbiol. 72:4214-4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lebuhn, M., M. Effenberger, A. Gronauer, P. A. Wilderer, and S. Wuertz. 2003. Using quantitative real-time PCR to determine the hygienic status of cattle manure. Water Sci. Technol. 48:97-103. [PubMed] [Google Scholar]
- 28.Lee, C., S. Lee, S. G. Shin, and S. Hwang. 2008. Real-time PCR determination of rRNA gene copy number: absolute and relative quantification assays with Escherichia coli. Appl. Microbiol. Biotechnol. 78:371-376. [DOI] [PubMed] [Google Scholar]
- 29.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 30.Malorny, B., J. Hoorfar, C. Bunge, and R. Helmuth. 2003. Multicenter validation of the analytical accuracy of Salmonella PCR: towards an international standard. Appl. Environ. Microbiol. 69:290-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30a.Miller, S. M., D. M. Tourlousse, R. D. Stedtfeld, S. W. Baushke, A. B. Herzog, L. M. Wick, J. M. Rouillard, E. Gulari, J. M. Tiedje, and S. A. Hashsham. 2008. In situ-synthesized virulence and marker gene biochip for detection of bacterial pathogens in water. Appl. Environ. Microbiol. 74:2200-2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miura, F., C. Uematsu, Y. Sakaki, and T. Ito. 2005. A novel strategy to design highly specific PCR primers based on the stability and uniqueness of 3′-end subsequences. Bioinformatics 21:4363-4370. [DOI] [PubMed] [Google Scholar]
- 32.Molenkamp, R., A. van der Ham, J. Schinkel, and M. Beld. 2007. Simultaneous detection of five different DNA targets by real-time Taqman PCR using the Roche LightCycler480: application in viral molecular diagnostics. J. Virol. Methods 141:205-211. [DOI] [PubMed] [Google Scholar]
- 33.Morrison, T., J. Hurley, J. Garcia, K. Yoder, A. Katz, D. Roberts, J. Cho, T. Kanigan, S. E. Ilyin, D. Horowitz, J. M. Dixon, and C. J. H. Brenan. 2006. Nanoliter high throughput quantitative PCR. Nucleic Acids Res. 34:e123. [DOI] [PMC free article] [PubMed]
- 34.Pannucci, J., H. Cai, P. E. Pardington, E. Williams, R. T. Okinaka, C. R. Kuske, and R. B. Cary. 2004. Virulence signatures: microarray-based approaches to discovery and analysis. Biosens. Bioelectron. 20:706-718. [DOI] [PubMed] [Google Scholar]
- 35.Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raychaudhuri, A., and P. A. Tipton. 2004. Protocol for amplification of GC-rich sequences from Pseudomonas aeruginosa. BioTechniques 37:752, 754, 756. [DOI] [PubMed] [Google Scholar]
- 37.Rozen, S., and H. Skaletsky. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:365-386. [DOI] [PubMed] [Google Scholar]
- 38.Rozynek, E., K. Dzierzanowska-Fangrat, P. Jozwiak, J. Popowski, D. Korsak, and D. Dzierzanowska. 2005. Prevalence of potential virulence markers in Polish Campylobacter jejuni and Campylobacter coli isolates obtained from hospitalized children and from chicken carcasses. J. Med. Microbiol. 54:615-619. [DOI] [PubMed] [Google Scholar]
- 39.Rychlik, W., and R. E. Rhoads. 1989. A computer program for choosing optimal oligonucleotides for filter hybridization, sequencing and in vitro amplification of DNA. Nucleic Acids Res. 17:8543-8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sahdev, S., S. Saini, P. Tiwari, S. Saxena, and K. S. Saini. 2007. Amplification of GC-rich genes by following a combination strategy of primer design, enhancers and modified PCR cycle conditions. Mol. Cell. Probes 21:303-307. [DOI] [PubMed] [Google Scholar]
- 41.SantaLucia, J., Jr. 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 95:1460-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sergeev, N., M. Distler, S. Courtney, S. F. Al-Khaldi, D. Volokhov, V. Chizhikov, and A. Rasooly. 2004. Multipathogen oligonucleotide microarray for environmental and biodefense applications. Biosens. Bioelectron. 20:684-698. [DOI] [PubMed] [Google Scholar]
- 43.Shindo, Y., H. Kuribara, T. Matsuoka, S. Futo, C. Sawada, J. Shono, H. Akiyama, Y. Goda, M. Toyoda, and A. Hino. 2002. Validation of real-time PCR analyses for line-specific quantitation of genetically modified maize and soybean using new reference molecules. J. AOAC Int. 85:1119-1126. [PubMed] [Google Scholar]
- 44.Skanseng, B., M. Kaldhusdal, and K. Rudi. 2006. Comparison of chicken gut colonisation by the pathogens Campylobacter jejuni and Clostridium perfringens by real-time quantitative PCR. Mol. Cell. Probes 20:269-279. [DOI] [PubMed] [Google Scholar]
- 45.Tani, H., T. Kanagawa, S. Kurata, T. Teramura, K. Nakamura, S. Tsuneda, and N. Noda. 2007. Quantitative method for specific nucleic acid sequences using competitive polymerase chain reaction with an alternately binding probe. Anal. Chem. 79:974-979. [DOI] [PubMed] [Google Scholar]
- 46.van Doorn, R., M. Szemes, P. Bonants, G. A. Kowalchuk, J. F. Salles, E. Ortenberg, and C. Schoen. 2007. Quantitative multiplex detection of plant pathogens using a novel ligation probe-based system coupled with universal, high-throughput real-time PCR on OpenArrays™. BMC Genomics 8:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanichanon, A., N. K. Blake, J. M. Martin, and L. E. Talbert. 2000. Properties of sequence-tagged-site primer sets influencing repeatability. Genome 43:47-52. [PubMed] [Google Scholar]
- 48.von Eiff, C., A. W. Friedrich, G. Peters, and K. Becker. 2004. Prevalence of genes encoding for members of the staphylococcal leukotoxin family among clinical isolates of Staphylococcus aureus. Diagn. Microbiol. Infect. Dis. 49:157-162. [DOI] [PubMed] [Google Scholar]
- 49.Vora, G. J., C. E. Meador, M. M. Bird, C. A. Bopp, J. D. Andreadis, and D. A. Stenger. 2005. Microarray-based detection of genetic heterogeneity, antimicrobial resistance, and the viable but nonculturable state in human pathogenic Vibrio spp. Proc. Natl. Acad. Sci. USA 102:19109-19114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang, L. X., Y. Li, and A. Mustaphai. 2007. Rapid and simultaneous quantitation of Escherichia coli O157:H7, Salmonella, and Shigella in ground beef by multiplex real-time PCR and immunomagnetic separation. J. Food Prot. 70:1366-1372. [DOI] [PubMed] [Google Scholar]
- 51.Welti, M., K. Jaton, M. Altwegg, R. Sahli, A. Wenger, and J. Bille. 2003. Development of a multiplex real-time quantitative PCR assay to detect Chlamydia pneumoniae, Legionella pneumophila and Mycoplasma pneumoniae in respiratory tract secretions. Diagn. Microbiol. Infect. Dis. 45:85-95. [DOI] [PubMed] [Google Scholar]
- 52.Wick, L. M., W. Qi, D. W. Lacher, and T. S. Whittam. 2005. Evolution of genomic content in the stepwise emergence of Escherichia coli O157:H7. J. Bacteriol. 187:1783-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilson, W. J., C. L. Strout, T. Z. DeSantis, J. L. Stilwell, A. V. Carrano, and G. L. Andersen. 2002. Sequence-specific identification of 18 pathogenic microorganisms using microarray technology. Mol. Cell. Probes 16:119-127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.