

Abstract
Flavobacterium psychrophilum is currently one of the main bacterial pathogens hampering the productivity of salmonid farming worldwide, and its control mainly relies on antibiotic treatments. To better understand the population structure of this bacterium and its mode of evolution, we have examined the nucleotide polymorphisms at 11 protein-coding loci of the core genome in a set of 50 isolates. These isolates were selected to represent the broadest possible diversity, originating from 10 different host fish species and four continents. The nucleotide diversity between pairs of sequences amounted to fewer than four differences per kilobase on average, corresponding to a particularly low level of diversity, possibly indicative of a small effective-population size. The recombination rate, however, seemed remarkably high, and as a consequence, most of the isolates harbored unique combinations of alleles (33 distinct sequence types were resolved). The analysis also showed the existence of several clonal complexes with worldwide geographic distribution but marked association with particular fish species. Such an association could reflect preferential routes of transmission and/or adaptive niche specialization. The analysis provided no clues that the initial range of the bacterium was originally limited to North America. Instead, the historical record of the expansion of the pathogen may reflect the spread of a few clonal complexes. As a resource for future epidemiological surveys, a multilocus sequence typing website based on seven highly informative loci is available.
The bacterium Flavobacterium psychrophilum belongs to the family Flavobacteriaceae, in the phylum Bacteroidetes (5, 7). Until the mid-1980s, F. psychrophilum was known only in North America, where it caused infections in Coho salmon and rainbow trout (9). It was then found in France and Germany in farmed rainbow trout (4, 6, 45). At about the same time, it was also identified in Japan in Coho salmon and ayu, a local salmonoid species (46). It has since been reported from all regions in the world involved in salmonid aquaculture, including Israel, Chile, and Tasmania (5). Flavobacterium psychrophilum is now widely recognized as one of the main sources of disease and economic loss. The two main clinical forms are rainbow trout fry syndrome and bacterial cold-water disease (14, 35). To date, attempts to establish F. psychrophilum-free salmonid farms have proved unsuccessful (I. Dalsgaard, personal communication) and vaccines are still at the experimental stage (2, 16). Hence, disease control still mostly relies on antibiotic administration (14).
Answers to a number of important questions regarding F. psychrophilum are intimately connected with a better knowledge of its population structure. Distribution of F. psychrophilum is currently worldwide, but it is unclear what the initial geographic range of the pathogen was prior to the development of the fish-farming industry and international trade of fish and fish eggs. The range of natural host species also remains unclear: F. psychrophilum has been occasionally isolated from nonsalmonid freshwater fish, such as carp, sturgeon, sea lamprey, and eel (17, 26), but whether these fish species harbor a significant population of the bacterium and may constitute reservoirs for salmonid fish is not known. It also remains to be understood how the respective contributions of the pathogen characteristics (i.e., virulence and host specificity), as opposed to the fish susceptibility, affect the success and severity of the infection. There is experimental evidence for strain-dependent variations of virulence (28). Hence, diffusion of the disease could have resulted from the spreading of some highly virulent strains. The trade of fish eggs could have played a critical role, as the vertical transmission of F. psychrophilum is highly suspected (10, 13, 34).
To gain insight into the population structure of F. psychrophilum and its mode of evolution, we undertook a multilocus sequence analysis taking advantage of the recent determination of the whole-genome sequence of strain JIP 02/86 (15). Here, we report on the analysis of sequence polymorphisms at 11 core genome loci across a collection of 50 isolates selected to represent the broadest possible genetic diversity. This study also aimed at establishing a multilocus sequence typing (MLST) scheme (29, 43) for future epidemiological monitoring as a complement or an alternative to other typing methods, such as random amplification of polymorphic DNA, PCR-restriction fragment length polymorphism, ribotyping, pulsed-field gel electrophoresis, serotyping, and plasmid profiling (3, 11, 12, 23, 27, 37, 41). To our knowledge, MLST data already published for bacteria isolated from diseased fish are limited to a few strains of Yersinia ruckeri and Vibrio vulnificus (8, 25).
MATERIALS AND METHODS
Loci, strains, and experimental protocol.
The 11 loci were selected in the absence of any knowledge of the genetic polymorphism of the F. psychrophilum population. They were distributed along the JIP 02/86 chromosome (15) and located within single-copy protein-coding genes with homologues in the genomes of the other sequenced members of the family Flavobacteriaceae. They can be considered typical core genome genes whose polymorphisms are likely relatively neutral (i.e., not experiencing frequent adaptive selection). Individually, each of these genes had already been used in MLST studies of other bacterial species.
The 50 F. psychrophilum strains used in this study were isolated from all over the world over a period of more than 20 years, between 1981 and 2003, except for the type strain NCIMB 1947T, whose year of isolation is unknown but which was deposited in culture collections long before 1980. They represented six different geographical areas (North America, Europe, Israel, Chile, Tasmania, and Japan) and 10 different fish species (rainbow trout [Oncorhynchus mykiss], cutthroat trout [Oncorhynchus clarki], Coho salmon [Oncorhynchus kisutch], Chinook salmon [Oncorhynchus tshawytscha], ayu [Plecoglossus altivelis], Atlantic salmon [Salmo salar], brown trout [Salmo trutta], European eel [Anguilla anguilla], tench [Tinca tinca], and carp [Cyprinus carpio]). Most strains had already been included in previous typing studies performed in our laboratory (11, 12).
Strains were cultivated on modified Anacker and Ordal agar at 18°C as described previously (33). PCR amplifications were performed with 10 ng of purified genomic DNA extracted using a Wizard genomic DNA purification kit (Promega). The following primers were designed from regions showing high degrees of conservation within the genus Flavobacterium: trpB (forward, AAGATTATGTAGGCCGCCC; reverse, TGATAGATTGATGACTACAATATC), gyrB (GTTGTAATGACTAAAATTGGTG and CAATATCGGCATCACACAT), glyA (AAAGATAGACAAATTCACGG and GGTGATTTATCATCAAAAGG), dnaK (AAGGTGGAGAAATTAAAGTAGG and CCACCCATAGTTTCGATACC), tuf (GAAGAAAAAGAAAGAGGTATTAC and CACCTTCACGGATAGCGAA), rplB (TCAAATTAAGAAAGCTGTTGA and TAATGAACGTGCCATATCTT), fumC (CCAGCAAACAAATACTGGGG and GGTTTACTTTTCCTGGCATGAT), ftsQ (TTACGATAGGTGCGGGGGATAT and GTGCTGACCCGCAATACCTAC), murG (TGGCGGTACAGGAGGACATAT and GCATTCTTGGTTTGATGGTCTTC), recA (TAGACAAGACTTACGGCAAAGG and TCCGTAGCTAAACCACGATCC), and atpA (CTTGAAGAAGATAATGTGGG and TGTTCCAGCTACTTTTTTCAT). PCR amplifications were performed with a 20-μl reaction volume by using recombinant Taq polymerase (Invitrogen) and the following conditions: 94°C for 3 min, followed by 35 cycles at 94°C for 0.5 min, 52°C for 0.5 min, and 72°C for 2 min, and a final extension at 72°C for 8 min. Two microliters of the PCR products was resolved on a 1% agarose gel to check amplification. One microliter of the PCR products was purified by using exonuclease I (Biolabs)-alkaline phosphatase (USB) for 1 h at 37°C, followed by enzyme inactivation for 5 min at 94°C. One-tenth of the purified PCR products was sequenced on both strands, using the same primers as for the PCR and a BigDye Terminator version 3.1 sequencing kit (Applied Biosystems). The resulting products were analyzed with an Applied Biosystems 3730 automated sequencer. All chromatograms were manually verified to ensure high sequence quality. Sequences of strain CSF 259-93 were retrieved from the whole-genome sequencing project USDA-ARS 1930-32000-002-03 (G. Wiens, personal communication).
Data analysis.
According to MLST standards (43), arbitrary numbers were used for unambiguous identification of the allele types (ATs; particular alleles at particular loci) and sequence types (STs; unique combinations of ATs at the different loci).
A neighbor-joining tree with a simple Jukes-Cantor model of sequence evolution was constructed using the neighbor program of PHYLIP (version 3.6; J. Felsenstein, Department of Genome Sciences, University of Washington, Seattle, WA) for the graphical representation of the overall sequence divergence between isolates. Bootstrap analysis was based on 1,000-locus resampling.
For a more appropriate network representation of the relationships between STs, E-burst version 3 with default settings (19) and a new allele-sharing network representation were used. The latter involved bidimensional projection of the distance matrix between STs, defined as dij = −log[(Lij + 1)/(L + 1)], where Lij is the number of loci with identical ATs in ST i and ST j, and L is the total number of loci (L = 11). The projection was carried out with the nonmetric multidimensional scaling method implemented in the Sammon function of the R statistical language (39, 44).
Statistical analysis of the population structure (host-genotype association) was performed using the analysis of molecular variance method with simple Euclidean distance between STs (dij = 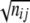 , where nij is the number of differences at the nucleotide level) and a permutation-based, nonparametric estimate of statistical significance (18).
, where nij is the number of differences at the nucleotide level) and a permutation-based, nonparametric estimate of statistical significance (18).
The detection of recombination within and between loci involved the homoplasy test (31) and computation of the Hudson and Kaplan lower bound on the minimal number of recombination events in an infinite site model, Rmin (22). The minimal number of apparent homoplasies (h) was computed on the most parsimonious tree found with DNApars (PHYLIP). For the purpose of statistical testing, the fraction of effective sites was set to 18%, and h was recomputed on biallelic sites only. Rmin was computed on biallelic sites by using LDhat (32). A quantitative estimate of the contribution of recombination versus that of mutation in short-term divergence between strains was obtained by examining the relationships between single-locus-variant (SLV) STs according to the method described by Feil et al. (20, 21).
Nucleotide sequence accession numbers.
Nucleotide sequences have been deposited in GenBank (accession numbers EU428196 to EU428795).
RESULTS
Pattern of polymorphism: low nucleotide diversity and pervasive traces of recombination.
Sequence comparisons revealed 136 single-nucleotide polymorphisms (SNPs) across the 9,578 bp surveyed. A summary of the main characteristics of the polymorphisms and their repartitions among the 11 loci is presented in Table 1. Overall, 1.42% of the positions showed variations and the pairs of sequences differed on average at 0.39% of the positions (nucleotide diversity π). The numbers of SNPs and the nucleotide diversities differed between loci, from 2 SNPs and 0.02% pairwise diversity (at locus ftsQ) to 26 SNPs and 0.95% pairwise diversity (at locus atpA). The vast majority of the SNPs were biallelic (four were triallelic), as expected given the small fraction of polymorphic sites. Only 16 SNPs corresponded to nonsynonymous variations, suggesting that most of the polymorphisms reported here are selectively neutral or almost neutral. The numbers of distinct alleles ranged from 3 (at locus ftsQ) to 18 (at locus tuf), with 17 at locus atpA. The combination of the ATs at the 11 loci allowed 33 different STs to be distinguished among the 50 isolates.
TABLE 1.
Summary of polymorphisms
| Gene | Value for:
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Locus
|
Polymorphism
|
Recombination
|
||||||
| Position (kbp)a | Length (bp) | Sb | Snonsync | No. of typesd | π (bp−1)e | Rminf | hg | |
| trpB | 616 | 933 | 11 | 2 | 11 | 0.0022 | 2 | 3* |
| gyrB | 639 | 1,149 | 21 (1) | 0 | 19 | 0.0071 | 7 | 21** |
| glyA | 804 | 930 | 7 | 0 | 8 | 0.0018 | 3 | 3* |
| dnaK | 994 | 897 | 10 | 2 | 15 | 0.0026 | 2 | 7** |
| tuf | 1374 | 939 | 21 (2) | 1 | 18 | 0.0059 | 5 | 14** |
| rplB | 1521 | 822 | 5 | 1 | 9 | 0.0018 | 2 | 3** |
| fumC | 1823 | 849 | 6 | 1 | 9 | 0.0016 | 1 | 2* |
| ftsQ | 2319 | 720 | 2 | 0 | 3 | 0.0002 | 0 | 0 |
| murG | 2321 | 825 | 23 (1) | 9 | 15 | 0.0055 | 6 | 12** |
| recA | 2573 | 756 | 4 | 0 | 9 | 0.0015 | 2 | 4** |
| atpA | 2820 | 978 | 26 | 0 | 17 | 0.0094 | 9 | 36** |
| Total | ||||||||
| Sum | 9,798 | 136 (4) | 16 | 0.0039 | 39 | 105** | ||
| Concatenation | 9,798 | 136 (4) | 16 | 33 | 0.0039 | 49 | 294** | |
Chromosomal position on the complete genome of strain JIP 02/86.
Number of segregating sites (number of tri-allelic sites in parentheses).
Number of nonsynonymous SNPs.
Number of distinct types (either AT or ST).
Average level of pairwise nucleotide diversity per site.
Hudson and Kaplan's lower bound for the number of recombination.
Minimal number of apparent homoplasies (*, P = 0.05; **, P = 0.005).
The presence of recombination in the genealogy of the sequences was investigated using two summary statistics, Rmin (22) and h (31), whose values are reported in Table 1. Both statistics revealed pervasive traces of recombination within and between loci. Rmin is a lower bound on the minimal number of recombination events when a single mutation per polymorphic site is assumed (reasonable for most sites, given the small fraction of polymorphic sites). Rmin was greater than 0 for all loci, with the exception of ftsQ, where the extremely low genetic diversity obliterated the chance of detecting recombination. The lower bound on the minimal number of recombinations within the 11 loci was 39, as given by the sum of 11 independent Rmin values. When computed on the concatenation of 11 loci, the Rmin value was found to be 49 (39 + 10), indicative of additional recombinations between the 10 pairs of consecutive loci.
The second statistic, h, is the minimal number of apparent homoplasies obtained as the difference between the number of visible polymorphisms and the minimal number of mutations on a tree for explaining the sequences. This number is 0 in the absence of recurrent mutations and recombinations. Here, h was greater than 0 for all loci except ftsQ and reached 36 for atpA. Statistical testing showed that each of the 10 positive values of h was significantly greater than expected as a consequence of recurrent mutations alone, indicative of pervasive recombination within loci. Furthermore, the value of h obtained on the concatenated sequence was much higher than the sum of 11 independent values of h (294 versus 105), suggesting additional recombination between loci.
The level of polymorphism at locus ftsQ seemed unexpectedly low compared to those at the 10 other loci. Somewhat surprisingly, comparison with the sequences of two other members of the genus Flavobacterium, F. johnsoniae and F. columnare, indicated that locus ftsQ tended to evolve faster than the other loci (data not shown). Therefore, the low diversity is unlikely to result from a particularly low rate of molecular evolution. A possibility would be that the nucleotide diversity was recently swept out as a consequence of fixation of a beneficial mutation (30).
Population structure: clonal complexes, host association, and quantification of recombination.
The relationships between isolates can be represented in several ways. Although recombination precludes the reconstruction of a phylogenetic tree for the 50 isolates, Fig. 1 shows a tree aimed at reflecting the genetic distances between isolates. The limited value of this tree in terms of sequence genealogy is well illustrated by the low bootstrap support of most internal branches. The shape of the tree is well balanced in the sense that none of the isolates stands as “atypical.” This gives another indication of the homogeneity of the species already supported by the low level of nucleotide divergence. Examination of the host fish species and the geographical origins of the isolates clearly shows an association between the genotype of the strain and the host fish species. In particular, 17 STs were sampled more than once but each ST always occurred in a unique fish species, sometimes in very distant regions of the world, for instance, ST 10 (rainbow trout) in North America and Europe, ST 12 (rainbow trout) in Chile and Europe, and ST 9 (Coho salmon) in North America and Chile. Conversely, very different STs coexisted in the same geographical area in association with different hosts. For instance, at least four very different STs coexisted in Japan: ST 17 (rainbow trout), ST 13 and ST 30 (Coho salmon), and ST 5 (ayu). The bootstrap analysis further suggested the existence of five groups of STs with low levels of divergence within groups, each group being preferentially associated with a particular fish species. The association between host fish species and genotype was quantitatively assessed by an analysis of molecular variance (18). The fish species accounted for 51.3% of the total molecular variance of the sample in terms of nucleotide differences. This association was highly statistically significant: the quantile associated with the P value 10−3 was estimated at 15% of the variance by random permutation of the fish labels.
FIG. 1.

Tentative tree representation of the relationships between the 50 Flavobacterium psychrophilum isolates. For each isolate, the host fish species, geographical area, and ST number are reported. Internal branches supported by more than 700 out of 1,000 bootstrap replicates are indicated by plain lines, and bootstrap values are provided. The bootstrap support associated with the cluster of strains highlighted with a vertical bar (*) was above 700/1,000 only when ST 6 and ST 21 were excluded from the analysis (the positions of these two sequences in the tree were unstable). The tree is unrooted. Abbreviations of fish names: Rbt, rainbow trout; BrT, brown trout; CuT, cutthroat trout; AtS, Atlantic salmon; ChS, Chinook salmon; CoS, Coho salmon; Ten, tench; Car, carp. Abbreviations of geographical areas: EU, Europe; ISR, Israel; NA, North America; CHL, Chile; AUS, Australia; JPN, Japan.
Global genetic distance between isolates cannot reflect the variation of genetic relatedness across loci that results from recombination. Table 2 reports the 11 ATs and the ST of each isolate together with basic information on its origin. The table clearly shows that identical ATs occurred in very different genetic contexts as a result of genetic exchanges across the population. To some extent, the amount of exchange made the 33 STs look more like mosaics of ATs than sequences at terminal leaves of a single tree.
TABLE 2.
Strains and sequence types
| Straina,b | ATc | ST | Origin | Host | Tissue | Yr | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FPC 837 | 5 | 4 | 1 | 4 | 4 | 1 | 4 | 2 | 2 | 3 | 4 | 5 | Japan | Ayu | Kidney | 1987 |
| FPC 840 | 5 | 4 | 1 | 4 | 4 | 1 | 4 | 2 | 2 | 3 | 4 | 5 | Japan | Ayu | Kidney | 1987 |
| DPIF 91/4043-8 | 4 | 5 | 4 | 4 | 5 | 3 | 3 | 2 | 4 | 4 | 5 | 7 | Tasmania | Atlantic salmon | Eroded fin | 1991 |
| DPIF 91/4043-4 | 4 | 5 | 4 | 4 | 5 | 3 | 3 | 2 | 4 | 4 | 5 | 7 | Tasmania | Atlantic salmon | Eroded fin | 1991 |
| DPIF 91/4043-17 | 4 | 5 | 4 | 4 | 5 | 3 | 3 | 2 | 4 | 4 | 5 | 7 | Tasmania | Atlantic salmon | Eroded fin | 1991 |
| OSU THC02-90 | 4 | 7 | 4 | 6 | 5 | 5 | 5 | 2 | 6 | 5 | 4 | 9 | Oregon | Coho salmon | Kidney | 1990 |
| MHC 1759sc | 4 | 7 | 4 | 6 | 5 | 5 | 5 | 2 | 6 | 5 | 4 | 9 | Chile | Coho salmon | Spinal cord | 2001 |
| OSU 84-254 | 4 | 7 | 4 | 6 | 5 | 5 | 5 | 2 | 6 | 5 | 4 | 9 | British Columbia | Coho salmon | Kidney | 1984 |
| FPC 830 | 4 | 7 | 4 | 6 | 8 | 5 | 5 | 2 | 6 | 5 | 4 | 13 | Japan | Coho salmon | Kidney | 1990 |
| NCIMB 1947T | 4 | 7 | 4 | 6 | 11 | 5 | 5 | 2 | 6 | 5 | 4 | 19 | Washington | Coho salmon | Kidney | |
| LVDJ G2215 | 10 | 14 | 6 | 12 | 14 | 6 | 8 | 2 | 10 | 1 | 14 | 25 | France | Brown trout | Kidney | 1997 |
| OSU SRCh F8-81 | 8 | 17 | 7 | 14 | 17 | 9 | 9 | 2 | 13 | 8 | 16 | 29 | Oregon | Chinook salmon | Kidney | 1981 |
| JIP 16/00 | 4 | 3 | 3 | 3 | 3 | 2 | 3 | 1 | 3 | 2 | 3 | 4 | France | Rainbow trout | 2000 | |
| OSU CCC6-86 | 3 | 16 | 3 | 13 | 16 | 8 | 9 | 2 | 12 | 7 | 15 | 28 | Oregon | Cutthroat trout | Kidney | 1986 |
| IVP CH8/93 | 3 | 19 | 3 | 13 | 16 | 8 | 9 | 2 | 12 | 7 | 15 | 31 | Switzerland | Rainbow trout | Spleen | 1993 |
| JIP 06/98 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | France | Rainbow trout | Kidney | 1998 |
| SVS 910611-1 | 1 | 13 | 1 | 15 | 7 | 4 | 2 | 1 | 1 | 1 | 1 | 32 | Denmark | Rainbow trout | Spleen | 1991 |
| LVDI 5/I | 4 | 6 | 2 | 5 | 6 | 4 | 3 | 2 | 5 | 3 | 6 | 8 | France | Carp | Gills | 1992 |
| LVDJ XP189 | 4 | 6 | 2 | 9 | 6 | 4 | 3 | 2 | 5 | 1 | 6 | 18 | France | Tench | Kidney | 1992 |
| LFNW 131/89 | 7 | 11 | 4 | 8 | 9 | 6 | 7 | 2 | 9 | 1 | 9 | 15 | Germany | Eel | Kidney | 1987 |
| LVDJ H9480 | 7 | 11 | 4 | 8 | 9 | 6 | 7 | 2 | 9 | 1 | 9 | 15 | France | Eel | Kidney | 1998 |
| LVDJ D2172 | 1 | 12 | 3 | 10 | 12 | 3 | 3 | 2 | 3 | 5 | 11 | 22 | France | Tench | 1995 | |
| LFNW 27/90(1) | 1 | 12 | 3 | 10 | 12 | 3 | 3 | 2 | 3 | 5 | 11 | 22 | Germany | Tench | Liver | 1990 |
| LFNW 25/90 | 1 | 12 | 3 | 10 | 12 | 3 | 3 | 2 | 3 | 5 | 11 | 22 | Germany | Tench | Spleen | 1990 |
| OSU AD 10.1.1 | 6 | 9 | 5 | 7 | 5 | 6 | 3 | 2 | 7 | 6 | 7 | 11 | Oregon | Atlantic salmon | Kidney | 1990 |
| LFNW 16/90 | 5 | 10 | 3 | 8 | 1 | 1 | 6 | 2 | 8 | 4 | 8 | 14 | Germany | Carp | Spleen | 1990 |
| CVLO 730/96-1 | 9 | 13 | 4 | 11 | 13 | 7 | 7 | 2 | 9 | 1 | 12 | 23 | Norway | Rainbow trout | Kidney | 1996 |
| FPC 831 | 11 | 18 | 8 | 7 | 18 | 1 | 5 | 2 | 14 | 9 | 17 | 30 | Japan | Coho salmon | Peduncle lesion | 1990 |
| UCD 95-74 | 1 | 1 | 1 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 6 | California | Rainbow trout | Kidney | 1995 |
| JIP 02/86 | 8 | 8 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 20 | France | Rainbow trout | Kidney | 1986 |
| FC 1285/96 | 2 | 8 | 2 | 2 | 7 | 1 | 2 | 2 | 2 | 1 | 2 | 12 | Chile | Rainbow trout | Spleen | 1996 |
| VM F15/2.95 | 2 | 8 | 2 | 2 | 7 | 1 | 2 | 2 | 2 | 1 | 2 | 12 | Germany | Rainbow trout | Spleen | 1995 |
| UCD R3/8/95 | 2 | 8 | 2 | 2 | 15 | 1 | 2 | 2 | 11 | 1 | 2 | 27 | California | Rainbow trout | 1995 | |
| BZ 01 | 2 | 8 | 2 | 2 | 3 | 1 | 2 | 2 | 2 | 1 | 2 | 16 | Israel | Rainbow trout | Spleen | 1998 |
| OSU Ana 9.1.1 | 2 | 8 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 10 | Oregon | Rainbow trout | Kidney | 1991 |
| MLA MT1368 | 2 | 8 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 10 | Scotland | Rainbow trout | Spleen | 1993 |
| LVDL 1829/91 | 2 | 8 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 10 | Spain | Rainbow trout | Liver | 1991 |
| FPC 813 | 2 | 8 | 2 | 2 | 10 | 1 | 2 | 2 | 2 | 1 | 2 | 17 | Japan | Rainbow trout | Kidney | 1992 |
| FPC 814 | 2 | 8 | 2 | 2 | 10 | 1 | 2 | 2 | 2 | 1 | 2 | 17 | Japan | Rainbow trout | Kidney | 1992 |
| CSF 259-93 | 2 | 8 | 2 | 2 | 2 | 1 | 2 | 3 | 15 | 1 | 2 | 33 | Idaho | Rainbow trout | Spleen | 1993 |
| LFNW 123/89 | 2 | 15 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 26 | Germany | Rainbow trout | liver | 1989 |
| JIP 07/99 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 2 | France | Rainbow trout | 1999 | |
| SVS 911209-1 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 2 | Denmark | Rainbow trout | Spleen | 1990 |
| JIP 08/99 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 2 | France | Rainbow trout | Kidney | 1999 |
| JIP P29-98 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 2 | France | Rainbow trout | Dorsal lesion | 1998 |
| IVP CH7/93 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 2 | Switzerland | Rainbow trout | Spleen | 1993 |
| LPAA P03/88 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 2 | France | Rainbow trout | Spleen | 1988 |
| JIP 05/03 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 13 | 24 | France | Brown trout | Mouth lesion | 2003 |
| JIP 10/99 | 3 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 3 | France | Rainbow trout | Spleen | 1999 |
| MHC 1710K | 2 | 2 | 2 | 2 | 7 | 1 | 2 | 2 | 2 | 1 | 10 | 21 | Chile | Coho salmon | Kidney | 2001 |
Bacterial strains are listed according to their order in the tree representation (Fig. 1).
Abbreviations of culture collections: BZ, strain provided by A. Eldar, Dept. of Poultry and Fish Diseases, The Kimron Veterinary Institure, Bet Dagan, Israel; CSF, sequences provided by G. Wiens, National Center for Cool and Cold Water Aquaculture, Kearneysville, WV; CVLO, strain provided by S. Høie, Central Veterinary Laboratory, Oslo, Norway; DPIF, strains provided by J. Carson, Department of Primary Industries, Fish Health Unit, Kings Meadows, Tasmania; FC and MHC, strains provided by J. Montaña, Fish Diseases Laboratory, Marine Harvest Chile S.A., Puerto Montt, Chile; FPC, strains provided by H. Wakabayashi, Department of Aquatic Bioscience, University of Tokyo, Japan; IVP, strains provided by T. Wahli, Universität Bern, Zentrum Fisch und Wildtiermedizin, Bern, Switzerland; JIP, Culture Collection of Unité de Virologie et Immunologie Moléculaires, Institut National de la Recherche Agronomique, Jouy-en-Josas, France; LFNW, strains provided by D. Mock, Landesanstalt fũr Fischerei Nordrhein-Westfalen, Kirchhundem-Albaum, Germany; LPAA, strain provided by F. Baudin-Laurencin, Laboratoire de Pathologie des Animaux Aquatiques, Agence Française de Sécurité Sanitaire des Aliments, Plouzané, France; LVDI, strain provided by J.-L. Bind, Laboratoire Vétérinaire Départemental d'Indre-et-Loire, Tours, France; LVDJ, strains provided by M. Morand, Laboratoire Vétérinaire Départemental du Jura, Lons-le-Saunier, France; LVDL, strain provided by P. Nougayrède, Laboratoire Vétérinaire Départemental des Landes, Mont de Marsan, France; MLA, strain provided by D. Bruno, FRS Marine Laboratory, Aberdeen, Scotland; NCIMB, National Collection of Industrial and Marine Bacteria, Aberdeen, United Kingdom; OSU, strains provided by M. Whipple and R. A. Holt, Oregon State University, Corvallis, OR; SVS, strains provided by E. Lorenzen, DFVF, Århus, Denmark; UCD, strains provided by R. Hedrick, University of California, Davis, CA; and VM, strain provided by M. Schaumburg, Veterinäruntersuchungsamt Mittelhessen, Giessen, Germany.
Loci are listed according to their positions on the chromosome of strain JIP 02/86. Boldface highlights the loci retained in the MLST scheme.
Figure 2 is intended to shed light on the pattern of allele sharing reported in Table 2. Figure 2a is the E-burst MLST diagram that highlights the differences between closely related STs (19): lines connect pairs of STs that differed at a single AT (SLVs). In the MLST context, SLVs are usually believed to have a recent common ancestor and to result from the progressive diversification of a single clone. Groups of STs connected by SLVs are thus often referred to as clonal complexes. Three such clonal complexes were identified here (CC1, CC2, and CC3). The E-burst diagram further attempts to retrace the evolutionary process of clone diversification on the basis of the number of SLVs of each ST (STs with high numbers of SLVs are considered ancestral). The new representation used in Fig. 2b relaxes the stringent SLV criterion to highlight the similarities between STs. Each ST is connected to all other STs that share at least one AT, and the amount of AT sharing is reflected in the line type. Remarkably, the 33 STs formed a single network component, strengthening the hypothesis of a single, cohesive population of bacteria. Five network subcomponents whose members are connected through the sharing of at least four ATs were also identified.
FIG. 2.

Network representations. (a) eBURST diagram. The three clonal complexes identified as groups of STs connected by SLVs are highlighted. (b) Allele-sharing network. Line styles reflect the number of ATs shared by a pair of STs: >3 ATs, plain black lines; 2 or 3 ATs, plain gray lines; and 1 AT, dotted gray lines. The major AT at locus ftsQ was not considered here, because of its overwhelming preponderance in the population.
Clonal complexes in Fig. 2a and network subcomponents in Fig. 2b enrich the tree representation (Fig. 1) and will be briefly discussed here. The first subcomponent in terms of number of STs was almost exclusively associated with rainbow trout infections and was particularly well represented in our collection of strains due to the predominance of rainbow trout farms in France and neighboring countries. E-burst analysis identified clonal complex CC1 as the core of this group. Two distantly related STs that formed a distinct cluster in the tree (ST 1 and ST 32) and were also associated with rainbow trout were connected to this first group via ST 6. The second subcomponent corresponded to ST 9, ST 13, and ST 19, found exclusively in Coho salmon, and was identified with E-burst as clonal complex CC2. The three other subcomponents were each represented by only two STs. One component corresponded to ST 28 and ST 31, which formed clonal complex CC3, associated with infections in cutthroat trout and rainbow trout. Another component consisted of ST 8 and ST 18, isolated from two cyprinids (tench and carp) and differing by two ATs. The last component connected the relatively distant STs ST 15 and ST 23, which shared only four ATs, isolated from very different types of fish (eel and rainbow trout). Interestingly, the last group may reflect a recombination event affecting several adjacent loci: four adjacent ATs, spanning more than 700 kb, were shared (from fumC to recA), whereas six out of the seven other ATs differed. The connection between ST 6 and ST 1 might also arise from such a large recombination event: the ATs were identical at three adjacent loci, spanning about 200 kb (from trpB to glyA), whereas they differed at six out of the eight other loci.
A simple quantitative estimate of the contribution of recombination to the generation of new genotypes can be obtained from the study of the events that participated in the diversification process of clonal complexes (20, 21). A total of 11 SLV pairs were examined. The E-burst diagram served as a reference to select the eight SLV pairs and to orient the genetic events in CC1. Three additional SLV pairs belonged to CC2. E-burst inferred that ST 9 was ancestral to this clonal complex, given the number of isolates that ST 9 represented, but we preferred to consider ST 19 ancestral: ST 19 corresponded to the oldest isolate in our collection (i.e., the species type strain). This choice was also more parsimonious in terms of sequence evolutionary changes (data not shown). For the last SLV pair (ST 28 and ST 31), the inferred genetic event was independent of the choice of the ancestral ST. Nine out of the 11 SLV pairs were better explained by recombination events and 2 by mutation events. At the nucleotide level, 52 changes were apparently due to recombinations and only 2 to mutations.
Scheme for future MLST.
MLST is now recognized as a reference method for the typing of many bacterial pathogens. For MLST to be effective, the sequences of a few loci have to provide enough information to discriminate a high number of STs. The pattern of sequence polymorphism reported in this study shows that this is indeed the case for F. psychrophilum. It also allows the most informative loci to be selected for this purpose. As the result of a balance between cost and resolving power, most MLST schemes rely on seven loci. We propose that future MLST surveys of F. psychrophilum use the seven loci with the largest amounts of polymorphism as reflected in number of ATs: trpB, gyrA, dnaK, tuf, fumC, murG, and atpA. As shown in Table 2, the combination of ATs at these seven loci captures the 33 distinct STs. The data for the 50 strains at the seven loci have been deposited in a dedicated MLST database (24) hosted at the Institut Pasteur (http://www.pasteur.fr/mlst/).
DISCUSSION
The 11 loci were selected in the absence of any knowledge of the genetic polymorphism in F. psychrophilum, and the results presented here therefore provide an unbiased view of the nucleotide polymorphism of the core genome of this pathogen. The pattern of polymorphism appears characterized by a low level of diversity and a high recombination rate. Both aspects contribute to high species cohesiveness. Another indication of the species homogeneity comes from the overall balanced divergence between STs, visible in the tree representation. Indeed, our analysis did not reveal evident substructuring of the species population above the clonal complex level. Rather, on the time scale of the genealogy of the species population, the pattern of polymorphism seems compatible with a near-panmictic situation where recombination is usually sufficient to effectively shuffle the alleles across the whole species.
Remarkably, the amount of nucleotide polymorphism as measured by the population mutation parameter (θ = 2Nu, here estimated by π) reported in this study for F. psychrophilum is lower than that reported in the MLST datasets for 16 other bacterial pathogens (36). This low level of polymorphism may reflect a small effective-population size, as there is no sign that the diversity is limited by the recent origin of the pathogen. Indeed, Tajima's D statistic tends here to be positive rather than negative (data not shown) (42). This might indicate a relatively narrow original range of host species or a limited range of geographical origin for the pathogen in wild fish. References are lacking, however, as this is the first study on the nucleotide polymorphism of a fish pathogen and of a cold-living freshwater bacterium. Furthermore, almost nothing is known on the variability of mutation rates in the bacterial world, and the low diversity observed in this study could also reflect a low mutation rate.
The biological mechanism responsible for the high rate of recombination in F. psychrophilum remains to be elucidated. Natural competence cannot be excluded but has never been reported in the literature for this bacterium. Conjugative plasmids and transposons that might mediate DNA transfer between strains have, however, been found (1, 11, 15). To quantify the amount of recombination, we computed that 9 out of 11 pairs of STs differing at one locus (SLV pairs) were best explained by recombination (a ratio of 4.5:1 at the allele level). At the nucleotide level, this translated into 52 changes apparently due to recombination and only 2 changes due to mutation (a ratio of 26:1 at the nucleotide level). Although there is statistical uncertainty associated with those estimates of the contribution of recombination to short-term divergence, they can be compared with values reported in the literature for other bacteria. They are much higher than that for a mildly recombinogenic bacterium, such as Escherichia coli (allele level ratio, 0.84:1; nucleotide level ratio, 5.18:1) (38). They are somewhat lower than those reported in the literature for Streptococcus pneumoniae (allele level ratio, 8.9:1; nucleotide level ratio, 61:1) and Neisseria meningitidis (allele level ratio, 4.75:1; nucleotide level ratio, 100:1), both considered highly recombinogenic bacterial species (20, 38). It is, however, important to note that these values reflect not only the recombination rate but also the average divergence time between the sequences that recombine: the greater this evolutionary time, the more each recombination event alters the sequence (see next paragraph). For S. pneumoniae and N. meningitidis, the average genetic diversities (π) computed from the data of reference 20 are 0.012 and 0.040, respectively. These two species thus harbor higher genetic diversity than F. psychrophilum. Unless this is a simple consequence of higher mutation rates, intraspecific divergence times are also longer. Each recombination event may thus induce more allele and nucleotide changes in S. pneumoniae and N. meningitidis than in F. psychrophilum.
A more explicit description of the relationships between genetic diversity and the proportion of nucleotide change due to recombination is instructive. The expected number of nucleotide changes in a sequence of length L in a short evolutionary time t decomposes into L × t × r × T × u changes due to recombination and L × t × u changes due to mutation, where r and u are the rates of recombination and mutation, respectively, at each nucleotide position per unit of time and T is the average evolutionary time that separates the two sequences that recombine. The ratio between the numbers of nucleotide changes apparently due to recombination and mutation therefore depends not on r alone but rather on the composite parameter r × T. Direct information on T is lacking, but T is likely to be correlated with genetic diversity (π), namely, T = π/u, in a panmictic model. The threefold difference in the proportion of nucleotide change due to recombination in S. pneumoniae versus that for F. psychrophilum might thus reflect the threefold difference in genetic diversity rather than a higher recombination rate.
In keeping with a number of anterior typing surveys, the analysis showed a strong relationship between certain types of isolates and their host fish species (see, for instance, references 3 and 11). The sequence data presented here allowed this relationship to be quantified and revealed that it stems from the existence of clonal complexes with marked preferential association with particular host fish species. Although highly significant from a statistical standpoint, the association was not absolute and representatives of the same clonal complex were occasionally found in different host species. Evolutionary and epidemiological hypotheses could explain this association, and it would be premature to draw any conclusion on the relative contributions of these two lines of explanation. The association between ST and host fish species might reflect adaptive niche specialization, but it could also be merely random and maintained by preferential routes of transmission. The occurrence of identical STs on the same farmed fish species in distant regions of the world (Europe and North America, North America and Chile or Japan, and Europe and Chile) revealed by our data indeed indicates a probable role for the international trade of brood fish and fish eggs in the spread of certain STs. The association between ST and host fish species was not limited to farmed species. For instance, ST 22 (three isolates from a 5-year period) was found only in tench and ST 15 (two isolates from a 9-year period) was found only in eel.
Interestingly, this study provides little support for the hypothesis of a global spread of the pathogen from North America. Such a scenario could hardly explain the diversity of strains found in Europe, particularly those found only on wild, nonsalmonid fish. Instead, the pattern of expansion provided by historical records may reflect the spread of two main clonal complexes (CC1 and CC2) by human activities.
An important objective for future research will be to delineate the respective roles of epidemiology and evolution in the association between clonal complexes and host species. The data collected here allowed the design of an efficient MLST scheme based on seven loci that could distinguish the 33 different STs. The sequences of these loci are available through an MLST database that should provide a powerful tool for future surveys needed to better understand the epidemiology and population structure of F. psychrophilum. In particular, the data collected here will allow the results of in-depth studies of any particular collection of strains to be assigned within the broader context of species diversity. Evolutionary niche adaptation may depend on the presence/absence of specific genes, and experimental evidence of the variability of gene repertoires within the species already exists (40). Our understanding of possible adaptive niche specialization will therefore be considerably improved by additional complete genome sequences. The basic knowledge of the population structure acquired in this study will help interpret the forthcoming genome sequences and prioritize the isolates to be sequenced.
Acknowledgments
We thank Brigitte Kerouault, Christian Michel, and Isabelle Gonçalves for their help and their comments on the manuscript. We also thank Greg Wiens (National Center for Cool and Cold Water Aquaculture, USDA-ARS) for providing us with the sequences of strain CSF 259-93 and Sylvain Brisse (Platform Genotyping of Pathogens and Public Health, Institut Pasteur) for handling the MLST database. We are also indebted to all those individuals who kindly contributed to the strain collection.
This study was supported by the INRA-AIP séquençage program and ANR-07-GMGE-004 FLAVOPHYLOGENOMICS.
Footnotes
Published ahead of print on 18 April 2008.
REFERENCES
- 1.Alvarez, B., P. Secades, M. J. McBride, and J. A. Guijarro. 2004. Development of genetic techniques for the psychrotrophic fish pathogen Flavobacterium psychrophilum. Appl. Environ. Microbiol. 70:581-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoki, M., M. Kondo, Y. Nakatsuka, K. Kawai, and S. Oshima. 2007. Stationary phase culture supernatant containing membrane vesicles induced immunity to rainbow trout Oncorhynchus mykiss fry syndrome. Vaccine 25:561-569. [DOI] [PubMed] [Google Scholar]
- 3.Arai, H., Y. Morita, S. Izumi, T. Katagiri, and H. Kimura. 2007. Molecular typing by pulse-field gel electrophoresis of Flavobacterium psychrophilum isolates derived from Japanese fish. J. Fish Dis. 30:345-355. [DOI] [PubMed] [Google Scholar]
- 4.Bernardet, J.-F., and B. Kerouault. 1989. Phenotypic and genomic studies of Cytophaga psychrophila isolated from diseased rainbow trout Oncorhynchus mykiss in France. Appl. Environ. Microbiol. 55:1796-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernardet, J.-F., and J. P. Bowman. 2006. The genus Flavobacterium, p. 481-531. In M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (ed.), The prokaryotes, a handbook on the biology of bacteria, vol. 7. Springer-Verlag, New York, NY. [Google Scholar]
- 6.Bernardet, J.-F., F. Baudin-Laurencin, and G. Tixerant. 1988. First identification of “Cytophaga psychrophila” in France. Bull. Eur. Assoc. Fish Pathol. 8:104-105. [Google Scholar]
- 7.Bernardet, J.-F., P. Segers, M Vancanneyt, F. Berthe, K. Kersters, and P. Vandamme. 1996. Cutting a Gordian knot: emended classification and description of the genus Flavobacterium, emended description of the family Flavobacteriaceae, and proposal of Flavobacterium hydatis nom. nov. (basonym, Cytophaga aquatilis Strohl and Tait 1978). Int. J. Syst. Bacteriol. 46:128-148. [Google Scholar]
- 8.Bisharat, N., D. I. Cohen, M. C. Maiden, D. W. Crook, T. Peto, and R. M. Harding. 2007. The evolution of genetic structure in the marine pathogen, Vibrio vulnificus. Infect. Genet. Evol. 7:685-693. [DOI] [PubMed] [Google Scholar]
- 9.Borg, A. F. 1948. Studies on myxobacteria associated with diseases in salmonid fishes. Ph.D. thesis. University of Washington, Seattle.
- 10.Brown, L. L., W. T. Cox, and R. P. Levine. 1997. Evidence that the causal agent of bacterial cold-water disease Flavobacterium psychrophilum is transmitted within salmonid eggs. Dis. Aquat. Organ. 29:213-218. [Google Scholar]
- 11.Chakroun, C., F. Grimont, M. C. Urdaci, and J.-F. Bernardet. 1998. Fingerprinting of Flavobacterium psychrophilum isolates by ribotyping and plasmid profiling. Dis. Aquat. Organ. 33:167-177. [DOI] [PubMed] [Google Scholar]
- 12.Chakroun, C., M. C. Urdaci, D. Faure, F. Grimont, and J.-F. Bernardet. 1997. Random amplified polymorphic DNA analysis provides rapid differentiation among isolates of the fish pathogen Flavobacterium psychrophilum and among Flavobacterium species. Dis. Aquat. Organ. 31:187-196. [Google Scholar]
- 13.Cipriano, R. C. 2005. Intraovum infection caused by Flavobacterium psychrophilum among eggs from captive Atlantic salmon broodfish. J. Aquat. Anim. Health 17:275-283. [Google Scholar]
- 14.Cipriano, R. C., and R. A. Holt. 2005. Flavobacterium psychrophilum, cause of bacterial cold-water disease and rainbow trout fry syndrome. Fish disease leaflet no. 86. U.S. Department of the Interior. U.S. Geological Service, National Fish Health Research Laboratory, Kearneysville, WV.
- 15.Duchaud, E., M. Boussaha, V. Loux, J.-F. Bernardet, C. Michel, B. Kerouault, S. Mondot, P. Nicolas, R. Bossy, C. Caron, P. Bessières, J.-F. Gibrat, S. Claverol, F. Dumetz, M. Le Hénaff, and A. Benmansour. 2007. Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat. Biotechnol. 25:763-769. [DOI] [PubMed] [Google Scholar]
- 16.Dumetz, F., E. Duchaud, S. E. LaPatra, C. Le Marrec, S. Claverol, M. C. Urdaci, and M. Le Henaff. 2006. A protective immune response is generated in rainbow trout by an OmpH-like surface antigen (P18) of Flavobacterium psychrophilum. Appl. Environ. Microbiol. 72:4845-4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elsayed, E. E., A. E. Eissa, and M. Faisal. 2006. Isolation of Flavobacterium psychrophilum from sea lamprey, Petromyzon marinus L., with skin lesions in Lake Ontario. J. Fish Dis. 29:629-632. [DOI] [PubMed] [Google Scholar]
- 18.Excoffier, L., P. E. Smouse, and J. M. Quattro. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feil, E. J., B. C. Li, D. M. Aanensen, W. P. Hanage, and B. G. Spratt. 2004. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186:1518-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feil, E. J., E. C. Holmes, D. E. Bessen, M. S. Chan, N. P. Day, M. C. Enright, R. Goldstein, D. W. Hood, A. Kalia, C. E. Moore, J. Zhou, and B. G. Spratt. 2001. Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc. Natl. Acad. Sci. USA 98:182-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feil, E. J., M. C. Enright, and B. G. Spratt. 2000. Estimating the relative contributions of mutation and recombination to clonal diversification: a comparison between Neisseria meningitidis and Streptococcus pneumoniae. Res. Microbiol. 151:465-469. [DOI] [PubMed] [Google Scholar]
- 22.Hudson, R. R., and N. L. Kaplan. 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111:147-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izumi, S., F. Aranishi, and H. Wakabayashi. 2003. Genotyping of Flavobacterium psychrophilum using PCR-RFLP analysis. Dis. Aquat. Organ. 56:207-214. [DOI] [PubMed] [Google Scholar]
- 24.Jolley, K. A., M. S. Chan, and M. C. Maiden. 2004. mlstdbNet—distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics 5:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kotetishvili, M., A. Kreger, G. Wauters, J. G. Morris, Jr., A. Sulakvelidze, and O. C. Stine. 2005. Multilocus sequence typing for studying genetic relationships among Yersinia species. J. Clin. Microbiol. 43:2674-2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann, J., D. Mock, F. J. Sturenberg, and J.-F. Bernardet. 1991. First isolation of Cytophaga psychrophila from a systemic disease in eel and cyprinids. Dis. Aquat. Organ. 10:217-220. [Google Scholar]
- 27.Madetoja, J., I. Dalsgaard, and T. Wiklund. 2002. Occurrence of Flavobacterium psychrophilum in fish-farming environments. Dis. Aquat. Organ. 52:109-118. [DOI] [PubMed] [Google Scholar]
- 28.Madsen, L., and I. Dalsgaard. 1999. Reproducible methods for experimental infection with Flavobacterium psychrophilum in rainbow trout Oncorhynchus mykiss. Dis. Aquat. Organ. 36:169-176. [DOI] [PubMed] [Google Scholar]
- 29.Maiden, M. C., J. A. Bygraves, E. Feil, G. Morelli, J. E. Russell, R. Urwin, Q. Zhang, J. Zhou, K. Zurth, D. A. Caugant, I. M. Feavers, M. Achtman, and B. G. Spratt. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 95:3140-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maynard Smith, J., and J. Haigh. 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23:23-35. [PubMed] [Google Scholar]
- 31.Maynard Smith, J., and N. H. Smith. 1998. Detecting recombination from gene trees. Mol. Biol. Evol. 15:590-599. [DOI] [PubMed] [Google Scholar]
- 32.McVean, G., P. Awadalla, and P. Fearnhead. 2002. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160:1231-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michel, C., D. Antonio, and R. P. Hedrick. 1999. Production of viable cultures of Flavobacterium psychrophilum: approach and control. Res. Microbiol. 150:351-358. [DOI] [PubMed] [Google Scholar]
- 34.Michel, C., D. Elliott, E. Jansson, I. Dalsgaard, M. Urdaci, and P. Midtlyng. 2005. Broodfish testing for bacterial infections. Workpackage 4 report. EC project VESO-1601, Fish Egg Trade. VESO, Oslo, Norway.
- 35.Nematollahi, A., A. Decostere, F. Pasmans, and F. Haesebrouck. 2003. Flavobacterium psychrophilum infections in salmonid fish. J. Fish Dis. 26:563-574. [DOI] [PubMed] [Google Scholar]
- 36.Pérez-Losada, M., E. B. Browne, A. Madsen, T. Wirth, R. P. Viscidi, and K. A. Crandall. 2006. Population genetics of microbial pathogens estimated from multilocus sequence typing (MLST) data. Infect. Genet. Evol. 6:97-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramsrud, A. L., S. A. LaFrentz, B. R. LaFrentz, K. D. Cain, and D. R. Call. 2007. Differentiating 16S rRNA alleles of Flavobacterium psychrophilum using a simple PCR assay. J. Fish Dis. 30:175-180. [DOI] [PubMed] [Google Scholar]
- 38.Salerno, A., A. Delétoile, M. Lefevre, I. Ciznar, K. Krovacek, P. Grimont, and S. Brisse. 2007. Recombining population structure of Plesiomonas shigelloides (Enterobacteriaceae) revealed by multilocus sequence typing. J. Bacteriol. 189:7808-7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sammon, J. W. 1969. A non-linear mapping for data structure analysis. IEEE Trans. Comput. 18:401-409. [Google Scholar]
- 40.Soule, M., K. Cain, S. LaFrentz, and D. R. Call. 2005. Combining suppression subtractive hybridization and microarrays to map the intraspecies phylogeny of Flavobacterium psychrophilum. Infect. Immun. 73:3799-3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soule, M., S. LaFrentz, K. Cain, S. LaPatra, and D. R. Call. 2005. Polymorphisms in 16S rRNA genes of Flavobacterium psychrophilum correlate with elastin hydrolysis and tetracycline resistance. Dis. Aquat. Organ. 65:209-216. [DOI] [PubMed] [Google Scholar]
- 42.Tajima, F. 1989. The effect of change in population size on DNA polymorphism. Genetics 123:597-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Urwin, R., and M. C. Maiden. 2003. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 11:479-487. [DOI] [PubMed] [Google Scholar]
- 44.Venables, W. N., and B. D. Ripley. 2002. Modern applied statistics with S, 4th ed. Springer-Verlag, New York, NY.
- 45.von Weis, J. 1987. Über das Vorkommen einer Kaltwasserkrankheit bei Regenbogenforellen, Salmo gairdneri. Tieraerztl. Umsch. 42:575-577. [Google Scholar]
- 46.Wakabayashi, H., M. Horiuchi, T. Bunya, and G. Hoshiai. 1991. Outbreaks of cold-water disease in Coho salmon in Japan. Fish Pathol. 26:211-212. [Google Scholar]