

Abstract
Activation of the cAMP receptor protein (CRP) from Escherichia coli is highly specific to its allosteric ligand, cAMP. Ligands such as adenosine and cGMP, which are structurally similar to cAMP, fail to activate wild-type CRP. However, several cAMP-independent CRP variants (termed CRP*) exist that can be further activated by both adenosine and cGMP, as well as by cAMP. This has remained a puzzle because the substitutions in many of these CRP* variants lie far from the cAMP-binding pocket (>10 Å) and therefore should not directly affect that pocket. Here we show a surprising similarity in the altered ligand specificity of four CRP* variants with a single substitution in D53S, G141K, A144T, or L148K, and we propose a common basis for this phenomenon. The increased active protein population caused by an equilibrium shift in these variants is hypothesized to preferentially stabilize ligand binding. This explanation is completely consistent with the cAMP specificity in the activation of wild-type CRP. The model also predicts that wild-type CRP should be activated even by the lower-affinity ligand, adenosine, which we experimentally confirmed. The study demonstrates that protein equilibrium is an integral factor for ligand specificity in an allosteric protein, in addition to the direct effects of ligand pocket residues.
The cyclic AMP (cAMP) receptor protein (CRP) of Escherichia coli is a well-studied global transcriptional regulator whose activation is highly specific to the binding of cAMP (13). In its cAMP-bound form, CRP binds DNA and interacts with RNA polymerase to stimulate transcription of appropriate genes (3, 21). In the absence of cAMP, CRP displays negligible affinity for DNA. The cAMP specificity of wild-type (WT) CRP is consistent with the structure of active CRP, which has been characterized a number of times and shows specific contacts between protein residues and cAMP (27, 29, 33). CRP is a homodimer in which each subunit contains two domains, the cAMP-binding domain and the DNA-binding domain, separated by a hinge region (27). The structure of the inactive form of CRP has never been determined, so the mechanism of allosteric activation by cAMP is conjectural, but plausible models for the conformational change that cAMP effects have been proposed (29, 39).
CRP variants have been found that have altered ligand specificity (1, 5, 9, 14, 19, 23, 40). The substitutions in some of these variants lie in the effector-binding pocket itself and presumably alter the specific interactions between CRP and the ligand (23, 40). Other CRP variants are fundamentally different and more difficult to explain. Specifically, there is a subset of ligand-independent (CRP*) variants that have detectable in vivo activity in the absence of cAMP and also altered ligand specificity. In these cases, the mutationally altered sites lie far from the cAMP-binding pocket, and it is therefore surprising that they alter functional properties of the ligands. A well-studied case is that of A144T CRP, which displays not only a significant activation by adenosine and cGMP but also an apparently improved cAMP affinity for protein activation (14, 38). We hypothesize that the activation by adenosine and the apparently improved affinity for cAMP are related, simply reflecting an improved affinity for related molecules and perhaps bringing the affinity for adenosine to the detectable level. The activation by cGMP must be different, however, because WT CRP binds cGMP with high affinity (11, 24, 36), so mere improvement of cGMP-binding affinity in A144T CRP cannot be the basis for the activation by cGMP. Some other CRP* variants have also been shown to be activated by GMP, at least in vivo (1, 5, 9). Garges and Adhya suggested an explanation for this by positing that cGMP can cause proper alignment of subunits but not proper domain-domain alignment and proposed that domain-domain alignment is caused by the CRP* substitutions (10). While plausible, this model fails to explain other relevant properties of the same A144T CRP* variant: the activation by adenosine and the apparently improved affinity for cAMP.
The conformational transitions caused by ligands in an allosteric protein can be described by the “preexisting equilibrium/conformational selection” model (25). As suggested previously (24, 30, 40), CRP also exists in an equilibrium between active and inactive forms even in the absence of cAMP, and the role of cAMP binding to the protein is to shift the equilibrium toward the active form. Based on this well-known concept, we have applied an equilibrium-shift model that quantitatively connects the equilibrium poise with the ligand specificity in the CRP* variants, as well as WT CRP. We demonstrate here that the particular equilibrium poise of WT CRP is set to respond to physiological cAMP levels but to avoid activation by the physiological levels of other ligands.
MATERIALS AND METHODS
Materials.
The compounds, cAMP, cGMP, adenosine, and AMP were purchased from Sigma (St. Louis, MO).
Strains, plasmids, and recombinant DNA methodology.
Standard methods were used for the isolation and manipulation of DNA (32). Synthetic oligonucleotides were from Integrated DNA Technologies, Inc. (Coralville, IA). Bacterial strains carrying different plasmids were propagated in 1% tryptone, 0.5% yeast extract, and 0.5% NaCl with 15 μg of tetracycline/ml, 25 μg of chloramphenicol/ml, or 50 μg of ampicillin/ml as appropriate.
Cloning of crp, site-directed generation of CRP variants, and in vivo screening for CRP* variants.
E. coli crp was cloned into pEXT20, and then six histidine codons were subsequently added at the 3′ end as described previously (39). WT CRP and CRP variants used in the present study were all His tagged. Site-directed mutagenesis was by PCR amplification with mutagenic primers (6). At CRP positions 53 and 148, D53H and L148R have been known as CRP* variants (1). To examine other substitutions at each position, we randomized each codon (53 or 148) separately and screened the mutagenized plasmid pool for ligand independency using the screening scheme described below. Codon randomization involved similar PCR amplification as described above, but the primers contained completely randomized codons at the desired positions. For screening CRP* activity in vivo, we used a cya crp E. coli strain, UQ3811 (39) with lacZ under the control of the class I CC(−61.5) promoter, and monitored the β-galactosidase accumulation in colonies. The crp genes from selected variants were sequenced to determine the causative residue changes. Based on their intensity of colony color, the clones with D53S and L148K CRP were chosen for further study.
Overexpression and purification of CRP proteins.
Overexpression of his-tagged WT CRP and CRP variants was carried out in the strain UQ3811, and the purification was carried out by using a nickel-nitrilotriacetate column (Novagen, Madison, WI). The detailed procedure is described elsewhere (39). The final purity of the proteins was >95%.
Measurement of in vitro DNA-binding activity of CRP proteins.
In vitro DNA-binding assays were carried out by using a fluorescence polarization method with a Beacon 2000 fluorescence polarization detector (Invitrogen Corp., Carlsbad, CA). The 26-bp DNA probe containing CCpmelR sequence was tagged with Texas Red (39). Binding assays were performed in 50 mM Tris-HCl (pH 8.0)-50 mM KCl-1 mM EDTA with a probe concentration of 5 nM in the presence of 6.4 μM salmon sperm DNA (nonspecific competitor).
HPLC analysis of cGMP.
High-pressure liquid chromatography (HPLC) analysis of commercial cGMP (Sigma) used a Beckman Coulter HPLC System Gold equipped with a photodiode array detector (Fullerton, CA) and a 250-by-4.6-mm C18 column (Alltech, Deerfield, IL). The elution mode was isocratic using the mobile phase of 20 mM potassium phosphate (pH 5.5) containing 12% methanol at a flow rate of 0.5 ml/min (22).
Subtilisin digestion.
Proteolytic digestion of CRP proteins was carried out by using the procedure described elsewhere (16) with a slight modification. Each CRP (1.12 mg/ml) was digested with various amounts of subtilisin; the ratio of subtilisin concentration versus protein was from 1/100 to 1/3200. When necessary, 100 μM cAMP was used. The reaction was carried out at 25°C for 24 h. Finally, the reaction was stopped by adding 1/9 volume of 10 mM phenylmethylsulfonyl fluoride in 100% ethanol.
Quantitative analysis for ligand binding to CRP proteins.
The proposed coupled equilibrium between protein conformation and ligand-CRP-DNA (L-P-D) ternary interaction was used to quantitatively analyze the titration data. We assumed the following. (i) There are only two conformations (inactive and active in DNA binding) for CRP regardless of the number of ligands bound. (ii) The intrinsic ligand affinity of the inactive form (Pi = CRPinactive) of free CRP is much less than that of active form (Pa = CRPactive), so the population of inactive form of CRP with one or two ligands bound is negligible and is ignored. (iii) DNA-binding constants are the same for all active forms of CRP regardless of the number of ligands bound. (iv) Cooperativity between ligand-binding sites could be ignored as an approximation. The system can then be completely described by the equilibrium constant for conformational change of free CRP (Kc), intrinsic association constant (k) of ligand to the active CRP form and DNA-binding constant (ka) of active forms of CRP with any number of ligands bound (Kc = [Pa]/[Pi], 2k = [PaL]/([Pa][L]) and ka = [PaLnD]/([PaLn][D]), n = 0, 1, or 2).
The equations for total concentrations for all species in the system are as follows.
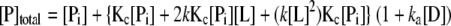 |
(1) |
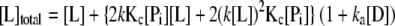 |
(2) |
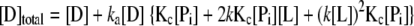 |
(3) |
Once the three equations are solved for [Pi], [L], and [D], the concentrations of all components in the system can be calculated. The detailed procedure is described elsewhere (40). The binding isotherms were analyzed using equation 4 with an assumption that the anisotropy signals are the same for all PaLnD regardless of the number of ligand bound (n).
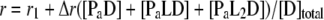 |
(4) |
where r1 is the anisotropy of free DNA and Δr is the anisotropy change relative to free DNA upon binding of PaLn.
Implicit fitting was performed by using the nonlinear regression program NONLIN (17) combined with the numerical algorithm described above.
RESULTS
The activation by other ligands is common among CRP* variants whose alterations lie far from the cAMP pocket.
We assumed that CRP* variants whose substitutions are far (>10 Å) from the cAMP-binding pocket probably exert effects on ligand specificity by an indirect mechanism, which we supposed to be a shift in their protein equilibrium toward the active form. We therefore examined several CRP* variants altered at different residues distant from the pocket, with the prediction that they should behave in roughly similar fashion because all of the variants share the shifted equilibrium. The CRP* variants we studied were D53S, G141K, A144T, and L148K CRP. The CRP* phenotype of G141K and A144T has been published (9, 14, 19), and D53S and L148K were made in our laboratory at positions where other substitutions were known to cause a CRP* phenotype (see Materials and Methods).
We confirmed that all of the variants have detectable CRP* activity in vivo relative to WT CRP when expressed from a plasmid in UQ3811 (39), a cya crp E. coli strain that contains the lacZ gene under the control of the class I CC(−61.5) promoter. This activity was seen as detectably blue colonies under a condition where colonies with WT CRP were white (data not shown). We then purified each of these variants as described in Materials and Methods and measured in vitro DNA affinity in the absence of any ligand, using the fluorescence anisotropy method. The DNA affinity of these variants in the absence of cAMP was increased over that of WT CRP at protein concentrations of 100 nM, but not so significantly enhanced as one would expect (Fig. 1A). We reasoned that ligand-free WT CRP had extremely low DNA-binding activity, and therefore even a significant shift of the equilibrium might not result in detectable increase of DNA affinity. In order to test this, we sought to add substitutions that could shift the equilibrium toward the active form but were not near the original substitution sites. The T127L or S128I substitutions met the above criteria because (i) both substitutions contribute to the high constitutive activity of T127L/S128I CRP variant (39) and (ii) they are not near the sites of the original CRP* substitutions. Therefore, the effect of the additional T127L or S128I substitutions should be independent of the original CRP* substitutions. After introducing either the T127L or the S128I substitution into each CRP* background, including that of WT CRP, we purified these variants and analyzed the DNA affinity. As shown in Fig. 1A, the T127L or S128I CRP substitutions by themselves had no detectable effect on WT CRP, but each dramatically raised the DNA affinity of all of the CRP* variants. The result indicates that the equilibria of the four CRP* variants are shifted toward the active form compared to WT CRP. The rather similar ligand-independent DNA-binding activity in these double variants (Fig. 1A) also suggests that the protein equilibrium shift in the original single CRP* variants is similar.
FIG. 1.

Shared activation properties of the CRP* variants. (A) The in vitro DNA affinity of the CRP proteins in the absence of ligand was measured by fluorescence anisotropy at protein levels of 100 nM. The anisotropy values for “no DNA binding” and for “saturated DNA binding” were 0.138 and 0.181 (WT CRP + 100 μM cAMP), respectively. Each CRP* substitution was also made in T127L or S128I CRP backgrounds, and the DNA affinity was also measured as follows: left panel, WT and CRP* variants; center panel, WT and CRP* variants with T127L; right panel, WT and CRP* variants with S128I. (B) The CRP* variants were activated by various ligands, while WT CRP is activated only by cAMP. The DNA affinity of each protein (at 100 nM) was measured by a fluorescence anisotropy method, but with various ligands: 0.1 mM cAMP, 0.1 mM cGMP, 0.4 mM adenosine, and 4 mM AMP.
We then examined the activation of these CRP* variants by various ligands. Under conditions where WT CRP is detectably activated only by cAMP, the four CRP* variants showed strong and roughly similar activations by cAMP, adenosine, and cGMP and a somewhat weaker activation by AMP (Fig. 1B). Moreover, preliminary data suggested that all of the CRP* variants required a lower concentration of cAMP for activation than did WT CRP (data not shown). Since all of these variants possess CRP* activity, we hypothesized a common underlying basis for this phenomenon, which is the alteration in the protein equilibrium poise between the active and inactive forms. We first develop a model for CRP that explains ligand specificity of these variants, as well as WT CRP. We will then return to the response of both WT CRP and CRP* variants to cGMP.
A simple equilibrium-shift model explains the enhanced activation by cAMP and adenosine in the CRP* variants.
CRP exists in an equilibrium between active and inactive forms in the absence of cAMP, and cAMP stabilizes the active form. This notion demands that cAMP-bound CRP should also be in equilibrium between active and inactive forms. Figure 2A shows an equilibrium-shift scheme that is similar to the one devised to analyze the interaction between hemoglobin and oxygen (28). As indicated in Fig. 2A, the process of cAMP binding to CRP involves six different CRP populations: three active populations (CRPactive, cAMP-CRPactive, and cAMP2-CRPactive) and three inactive populations (CRPinactive, cAMP-CRPinactive, and cAMP2-CRPinactive). The cAMP-bound inactive populations are negligible in the presence of cAMP since the active forms will be preferentially stabilized by cAMP. In this scheme, the DNA-binding activity of CRP is proportional to the sum of [CRPactive], [cAMP-CRPactive] and [cAMP2-CRPactive], which is the sum of [CRPactive], [CRPactive]2k[cAMP], and [CRPactive]k2[cAMP]2, in which k is the intrinsic association constant of cAMP to the active CRP form (CRPactive). According to this concept, the activation of the CRP* variants by a much lower cAMP concentration can be achieved when the active CRP population is increased even without higher intrinsic cAMP-binding affinity. This view is well compatible with the surprising fact that the CRP* variants whose substitutions are far from the ligand-binding pocket have apparently increased cAMP affinity for their protein activation. The schematic description of our model is shown in Fig. 2A using the natural ligand, cAMP, but the model can be applied to other ligands as well.
FIG. 2.

Models of the equilibrium of CRP with or without ligands. (A) Simple equilibrium-shift model. CRP exists in an equilibrium between active (CRPactive) and inactive (CRPinactive) forms, but the major form is the inactive one (CRPinactive) in the absence of cAMP. The role of cAMP binding is to shift the equilibrium toward the active form ([cAMP-CRPactive] ≫ [cAMP-CRPinactive] and [cAMP2-CRPactive] ≫ [cAMP2-CRPinactive]). The ligand cAMP can bind both to CRPactive and CRPinactive, with the intrinsic affinities of k and ki, respectively. The term Kc ([CRPactive]/[CRPinactive]) is the population ratio of CRPactive versus CRPinactive. The length of arrows indicates either the protein equilibrium or the strength of ligand affinity in a qualitative way. The scheme in panel A is shown for cAMP, but it can be generalized for other ligands such as adenosine. (B) Modified equilibrium-shift model for cGMP. The cGMP is hypothesized to bind to two different states (A-mode and B-mode) of CRP that are equilibrium linked through CRPinactive, and one cGMP binding mode excludes the other. The A-mode binding of cGMP to CRP is hypothesized to stabilize the active form, similar to the simple equilibrium-shift model described in panel A. The B-mode binding of cGMP to CRP is hypothesized to stabilize the inactive form. In this scheme, the length of the arrows indicates our assumptions about protein equilibrium and cGMP binding affinity, but there is little experimental evidence.
The equilibrium-shift model is supported by extensive analysis of D53S CRP.
The validity of this equilibrium-shift model was then extensively tested with one of the CRP* variants, D53S CRP. We monitored the abilities of various ligands to activate purified D53S and WT CRP by measuring in vitro DNA-binding activity of each protein at various ligand concentrations. As shown in Fig. 3, D53S CRP required much less cAMP (∼100-fold less) than did WT CRP to reach a given level of DNA binding. We hypothesize such a marked difference is primarily the consequence of different Kc values ([active population]/[inactive population] in the absence of ligand) between WT and D53S CRP. It is worth noting that Kc is not necessarily linearly correlated to apparent ligand affinity, because the dimeric nature of the protein and its relationship is dependent upon cooperativity of ligand binding. We then examined the efficacy of adenosine to activate D53S CRP (Fig. 3B) and confirmed that adenosine activates this protein when it binds to the protein. However, the isotherm of D53S CRP shows that adenosine is much poorer ligand than cAMP (∼104-fold in terms of ligand concentration). We reason that the ∼104 difference only reflects the protein's differential intrinsic binding affinity for cAMP and adenosine. If this reasoning is correct, WT CRP is predicted to be activated by adenosine as well at a higher ligand concentration. As shown in Fig. 3A, WT CRP was indeed activated by adenosine at a high ligand concentration (monitoring at concentrations of >6 mM was not tested due to the limited solubility of adenosine). The result clearly shows that WT CRP can be activated by adenosine and suggests that the reason that activation of WT CRP by adenosine has been missed previously is because of its intrinsically poor ligand affinity. The structural basis for poorer adenosine binding has been suggested to be its inability to use Arg82 as stabilizing interaction, unlike cAMP (38). Together, the results are completely consistent with the equilibrium-shift model as shown in Fig. 2.
FIG. 3.

Activation of WT CRP and CRP* variants by various concentrations of ligands. The DNA affinities of wild-type CRP (A), D53S CRP (B), and D53S/R82G CRP (C) were measured in various concentrations of the ligands cAMP (○), cGMP (⋄), and adenosine (▵). The in vitro DNA binding was measured by using a fluorescence anisotropy method. The concentration of each protein used was 100 nM. The scales of x axis (ligand concentration) and y axis (anisotropy value) are the same in the three panels. The solid lines in each panel indicate the best fits for the isotherms using the scheme in Fig. 2A and equations detailed in Materials and Methods.
We wanted to rule out the possibility that a trace amount of contaminant cAMP in the commercial adenosine is the activating ligand. The observed D53S activation by adenosine would result from only 0.01% cAMP contamination, so the direct detection of that would not be technically trivial. In order to indirectly test this, we took advantage of the fact that Arg82 contributes to the binding affinity of cAMP (2, 12) but not to that of adenosine (38). An R82G substitution was introduced into D53S CRP with the expectation that this substitution would have a substantial effect on apparent cAMP affinity but not much effect on apparent adenosine affinity. The obtained results were that adenosine affinity in D53S/R82G CRP is unaffected relative to that of D53S CRP but that cAMP affinity is dramatically reduced (Fig. 3B and C). The result conclusively shows that both D53S CRP and WT CRP are activated by adenosine.
Quantitative analysis for the activation WT and D53S CRP by cAMP and adenosine.
As described above, the difference in the amount of the active population between WT and D53S CRP is the key to the quantitative analysis of the equilibrium-shift model. We therefore tried first to estimate Kc (Kc = [CRPactive]/[CRPinactive]) in both WT and D53S CRP by measuring the DNA affinities of the proteins in the absence of ligand. Although the DNA affinity of WT CRP was not measurable in our assay, D53S CRP showed some ligand-independent DNA affinity at a high protein concentration in a titration experiment (data not shown), revealing a Kc estimate of 4.0 × 10−3 (Table 1). We then used this Kc value to fit the adenosine isotherm of D53S CRP using the nonlinear fitting method described in Materials and Methods. As shown in Fig. 3B, the adenosine activation isotherm was well fitted and the kadenosine (intrinsic association constant of adenosine for the active CRP population) of D53S was extracted to be 1.1 × 105 M−1 (Table 2). Using the same kadenosine, we obtained a reasonable fit for the adenosine isotherm of WT CRP (Fig. 3A) and obtained a Kc value of 1.1 × 10−6 (Table 2). This Kc value of WT CRP corresponds to ∼100 M−1 of Kobs (apparent DNA-binding affinity) in the absence of ligand. We note that this Kobs is within the range of 6 to 300 M−1 obtained by another analysis (36). We then applied the WT CRP Kc value of 1.1 × 10−6 to the nonlinear fitting of the cAMP isotherm of WT CRP and extracted kcAMP (the intrinsic association constant of cAMP for the active CRP population) of 4.3 × 108 M−1 (Table 2). The fitting quality was again visually and statistically reasonable (Fig. 3A). The intrinsic affinity ratio of ∼3,900 (kcAMP/kadenosine = 4.3 × 108/1.1 × 105) from our analysis is also consistent with the 3-orders-of-magnitude difference between the two ligands measured in WT CRP by another method (31). Our equilibrium-shift model demands that the cAMP isotherm of D53S should be described by two parameters: the Kc of D53S and the kcAMP obtained from the WT-cAMP result described above. As predicted, the isotherm is reasonably fitted (Fig. 3B), although not quite as well as the others. Varying the Kc or kcAMP (or both) resulted in apparently improved fitting, although still imperfect (data not shown). When we simulated using a wide range of kcAMP values above 9.3 × 109 M−1, we obtained surprisingly invariant Kc (2.1 × 10−3 to 3.5 × 10−3), which is close to the initial value of 4.0 × 10−3 determined by an independent experiment (Table 1). Although we do not fully understand the basis for the slight deviation of the D53S-cAMP isotherm from the simulation (fitting) based on the model, this might suggest that the cooperativity of cAMP binding is different from that of adenosine binding. Nonetheless, we did not incorporate cooperativity between ligand-binding sites into our model for several reasons: (i) the data for WT CRP was well fitted by our equilibrium-shift model even without the factor of cooperativity, (ii) the cooperativity of D53S CRP is not necessarily identical to that of WT CRP, and (iii) there is disagreement about the direction and magnitude of cooperativity in CRP (13, 24, 30, 36).
TABLE 1.
Kc values of D53S and D53S/R82G estimated from in vitro DNA-binding analysis
| CRP | Parameter
|
||
|---|---|---|---|
| Kobs (M−1)a | ka (M−1)b | Kcc | |
| WT | <5.0 × 104 | 1.0 × 108 | <5.0 × 10−4 |
| D53S | 4.0 × 105 | 1.0 × 108 | 4.0 × 10−3 |
| D53S/R82G | 4.5 × 105 | 1.0 × 108 | 4.5 × 10−3 |
Kobs, calculated as ka Kc/(1 + Kc), is the observed DNA-binding constant for each protein without any ligand, which was experimentally determined except for WT CRP.
The intrinsic DNA-binding constant was estimated by measuring the DNA-binding activity of WT CRP at a saturating concentration of cAMP (100 μM), and this value (108) was used for the extraction of Kc values for D53S and D53S/R82G CRP.
That is, [CRPactive]/[CRPinactive].
TABLE 2.
Ligand-binding parameters for CRP proteins obtained by the equilibrium-shift model
| CRP | Kc | Kc ratio | Intrinsic affinity (M−1)a
|
|
|---|---|---|---|---|
| cAMP | Adenosine | |||
| WT | 1.1 × 10−6 | 1 | 4.3 × 108 | 1.1 × 105* |
| D53S | 4.0 × 10−3 | 3.6 × 103 | 4.3 × 108 | 1.1 × 105* |
| D53S/R82G | 4.5 × 10−3 | 4.1 × 103 | 4.9 × 105 | 6.8 × 104 |
*, The intrinsic affinities of the WT and D53S CRPs for adenosine are assumed to be the same.
Finally, the intrinsic association constants of D53S/R82G CRP for both cAMP and adenosine were also obtained and are listed in Table 2. This variant was highly perturbed in terms of cAMP binding (by ∼900-fold) but much less so in adenosine binding (only by 1.6-fold), indicating genuine adenosine activation of D53S/R82G CRP, as well as of D53S and WT CRP.
A different type of analysis also supports the validity of the equilibrium-shift model. Even without initial input of Kc value for D53S CRP, the curve-fitting resulted in reasonable Kc and ligand affinity values that are very similar to those listed in Table 2. In this analysis of ligand titrations, both Kc and kligand are the variable fitting parameters. Even though we utilized the “ligand-free” DNA-binding isotherm to estimate Kc and to use it in subsequent analyses of ligand titrations, one can vary (or fit) both Kc and kligand to get an unique set of those values for a specific condition. When Kc is sufficiently large (>10−3 in this analysis for D53S and D53S/R82G CRP variants), the active form of free CRP can be significantly populated at a low ligand concentration ([ligand]). Since we assume that all active forms of CRP can bind DNA, free CRP contributes to the anisotropy signal for the condition mentioned above (large Kc and low [ligand]). The feature of relatively higher and increasing anisotropy signal of D53S and D53S/R82G CRP variants at the very beginning of titration is a reflection of that aspect and enabled us to uniquely determine Kc because the primary parameter contributing to the early phase is Kc. However, for very low values of Kc (≪10−3 in this analysis for WT CRP), the populations contributing to DNA binding are primarily the active form of CRP with one or two ligands bound. In this case, binding signal or anisotropy signal always reflects the composite effect of Kc and kligand, which cannot be determined uniquely. Varying both Kc and kligand in the analysis of variants (except the D53S-cAMP result) generated fitting results that were fairly consistent with those listed in Table 2, which were obtained by fixing Kc at the estimated values from ligand-free DNA-binding assays. For example, in the analysis of the D53S-adenosine interaction, we obtained a Kc of 4.8 × 10−3 and a kadenosine of 9.5 × 104. Alternatively, we tried a wide range of Kc values (10−1 to 10−7) for D53S CRP to see the fitting quality and to test the impact on the KcVariants/KcWT and the kcAMP/kadenosine. Although the fitting qualities for variants got worse when Kc deviated from the best-fit value that is listed in Table 2, all of those relations still hold: the value of KcVariants/KcWT is about 3,600, and the values of kcAMP/kadenosine are about 4,000 for WT and D53S CRP and about 6 for D53S/R82G CRP, respectively. In short, this analysis indicates that the parameters listed in Table 2 are somewhat invariant as long as the measured Kc value of D53S CRP (or D53S/R82G CRP) is <0.1.
Protease sensitivity experiments suggest a complicated molecular ensemble of CPR proteins.
We intended to use another method to corroborate the estimated population distribution (active and inactive) in both D53S and WT CRP (Table 2). However, the Kc value for D53S CRP is 4.0 × 10−3, meaning that only 0.4% is in the active conformation in the absence of ligand. One might assume a similar percentage of active population in our other CRP* variants and WT CRP has an even lower active population in the absence of ligand. Most biophysical and/or chemical methods are unable to quantitatively (even qualitatively) access such small populations in the absence of any ligand. We therefore tried an accumulative assay utilizing the protease sensitivity of active CRP, using the procedure reported previously (16). All of the CRP* variants (D53S, G141K, A144T, and L148K) showed higher subtilisin sensitivity than did WT CRP in the absence of any ligand (Fig. 4), a finding consistent with the proposal that all of the CRP* variants have higher proportions of the active form in the absence of ligand. Surprisingly, three CRP* variants (G141K, A144T, and L148K) revealed remarkably high subtilisin sensitivity in the absence of ligand compared to the level seen with cAMP-treated WT CRP (Fig. 4). This is in contrast to the fact that their ligand-independent DNA-binding activities are at least ∼100-fold weaker than that of cAMP-bound WT CRP (data not shown). This apparent discrepancy indicates that protease sensitivity only partially correlates with the active DNA-binding conformation of CRP protein, as has been noted previously (8). This might suggest that there are more than two simple states (active and inactive) in the molecular ensemble of CRP* variants and possibly WT CRP.
FIG. 4.

Proteolytic digestion of WT CRP and CRP* variants by subtilisin. Each CRP (1.12 mg/ml) was digested with various amounts of subtilisin; the black wedge indicates twofold serial dilution of subtilisin, and the first lane contained 11.2 μg of subtilisin/ml (CRP/subtilisin, 1:100). An asterisk indicates the control lane showing each CRP treated identically but without subtilisin. Arrows indicate the uncut CRP band. The concentration of cAMP used for WT CRP was 100 μM. The reaction was carried out at 25°C for 24 h.
Despite this complication, it remains true that modeling based on the equilibrium-shift concept using only two states (active and inactive) is sufficient to explain the altered ligand specificity of CRP* variants and cAMP-specific behavior of WT CRP.
The activation of D53S CRP by cGMP can be explained by a modified equilibrium-shift model.
D53S CRP is activated by cGMP at an ∼10 μM ligand concentration, whereas WT CRP shows no response (Fig. 3A and B), and this phenomenon cannot be explained by the simple equilibrium-shift model, as explained earlier. Again, we considered the possibility of cAMP contamination in the cGMP sample, since ∼1% cAMP contaminant in the commercial cGMP would result in the cGMP activation of D53S CRP. This explanation seemed doubtful because such contamination should also activate WT CRP at high levels of commercial cGMP, which was not seen (Fig. 3A). The commercial cGMP was directly analyzed by HPLC; essentially no absorbing material was seen at the retention time of bona fide cAMP and certainly not the 1% contamination level necessary for the effect (data not shown). We further purified cGMP by HPLC and confirmed that it continued to activate D53S CRP (data not shown). These results indicate that cGMP is the actual ligand activating D53S CRP and presumably other CRP* variants.
What could be the basis for the activation of the CRP* variants by cGMP? This remains paradoxical to us because cGMP can bind both WT CRP and the CRP* variants, but with functionally different effects where there is no direct change in the cAMP-binding pocket by the substitutions. Although we have no clear answer to this paradox, nor is it clear how to address it experimentally, we suggest that a generalization of the equilibrium-shift model could explain the cGMP behavior in both WT CRP and the CRP* variants. We posit that there are two nonidentical modes of cGMP binding in CRP, which are mutually exclusive (Fig. 2B). The cGMP binding to one mode (A-mode in Fig. 2B) stabilizes the active form of CRP and the binding to the other (B-mode in Fig. 2B) stabilizes the inactive form. Now, this consideration requires three populations in equilibrium in the absence of ligand: CRPactive, CRPinactive, and CRP′inactive (Fig. 2B). CRPactive is the active population that becomes stabilized by cGMP binding. Both CRPinactive and CRP′inactive are the inactive populations, but CRP′inactive is the species that can bind cGMP with higher affinity. We hypothesize that the protein equilibrium of WT CRP is poised such that cGMP binds predominantly to the CRP′inactive form, resulting in the inactive cGMP-bound form. In the CRP* variants, the increase of active CRP form (CRPactive) would result in the increase of [CRPactive]/[CRP′inactive] because all three populations (CRPactive, CRPinactive, and CRP′inactive) are equilibrium linked. We hypothesize that this collateral equilibrium shift would lead to a predominance of cGMP binding to the CRPactive form, resulting in detectable DNA affinity in the CRP* variants. One critical prediction of this three-state model is that there must be some active cGMP-bound population even in the WT CRP in the presence of cGMP, although the level might be very low. In this regard, it is worth noting that many different research groups have reported the in vivo activation of WT CRP by cGMP (1, 9, 37). We interpret this to mean that (i) there is some active cGMP-bound population in WT CRP in the presence of cGMP and (ii) the extreme sensitivity of the in vivo assay allowed them to detect it.
Finally, we hypothesize that the physical location for A-mode cGMP-binding for protein activation (see Fig. 2B) is identical to that of cAMP. This is based on the fact that the additional R82G substitution in D53S CRP functionally perturbs both cAMP and cGMP affinities at a similar level (Fig. 3B and C). The physical location for B-mode cGMP-binding in CRP could be the same as that for A-mode, and therefore that of cAMP binding, but there is no supporting evidence for this.
DISCUSSION
The present study demonstrates that the specific protein equilibrium dynamic (between active and inactive forms) of WT CRP has important physiological consequences because it sets the proper activation window for cAMP but avoids activation by other potential ligands. In the CRP* variants, the windows are changed by the equilibrium-shift mechanism, causing physiological challenges for E. coli. First, they can respond to the physiological adenosine level, a behavior that would be quite inappropriate for E. coli. Based on the 130 μM Km value for adenosine deaminase (20), the physiological adenosine level in E. coli is thought to be 100 to 200 μM, which is certainly the responsive level for the CRP* variants. Indeed, the apparently increased affinity of CRP* variants for adenosine means that all published results on their CRP* activity (the in vivo activity of such variants in the absence of cAMP) are suspect; we simply cannot tell the degree to which the observed activity is actually due to ligand free-CRP or instead to adenosine-bound CRP. Second, the regulation of CRP activity by cAMP would be altered. The protein equilibrium poise of WT CRP allows its activity to be lost when cAMP levels fall modestly. This proper regulation would not occur in the CRP* variants because they have a much higher apparent binding affinity for cAMP. Perhaps this is the reason why various CRP* variants often cause growth problems in the presence of cAMP (1, 14, 19), given the fact that too much unregulated CRP activity is detrimental to E. coli (39). Although not tested here, a protein equilibrium shift toward the inactive form would be also problematic since it would require a higher level of cAMP for normal CRP activity in E. coli. One might argue that lessening the intrinsic affinity for cAMP can compensate for the equilibrium shift in the CRP* variants and yield proper CRP function. Indeed, such a compensatory substitution has been described that confers cAMP-specific activation on a CRP* variant (10). We note, however, that in this case the efficacy of CRP regulation by cAMP will be perturbed due to the increased level of active population in the absence of ligand. CRP appears to have evolved to optimally sense cAMP with its high intrinsic ligand affinity and highly shifted protein equilibrium poise toward the inactive form.
Importantly in our view, a change in the protein equilibrium poise of the CRP* variants does not change the relative ligand specificity between cAMP and adenosine of WT CRP: the intrinsic ligand affinity ratio of cAMP to adenosine (3,900) is the same for D53S and WT CRP (Table 2). The case for cGMP is less clear, but the activation by cGMP in the CRP* variants can be also explained without changing the relative ligand specificity of WT CRP in our model (Fig. 2B). This view is in high contrast to the conventional one that the CRP* variants actually have relaxed ligand specificity (13). We propose that the physical binding site for cAMP, adenosine, and cGMP for protein activation is the same cAMP pocket. This notion is suggested by the effect of R82G substitution on the activation of D53S by the ligands and is consistent with the general observation in allosteric proteins (26). The cGMP-binding mode leading to the inactive form (B-mode) can also be in the cAMP-binding pocket, but perhaps with a different cGMP form, the “syn” cGMP binding. There are many examples of proteins that bind cGMP with both ligand conformations, “syn” or “anti” (35). Finally, the possibility of the binding of ligands to the secondary cAMP-binding site in CRP is highly unlikely to be relevant to the present study, based on the previously reported requirement for much higher ligand concentrations for binding there (24, 34). Taken together, the preserved ligand specificity and shared ligand-binding site in the CRP* variants are internally consistent with the original assumption that the cAMP-binding pocket is not directly altered by CRP* substitutions.
The various CRP* variants analyzed here have similar equilibrium-shift properties resulting in a CRP* phenotype and altered ligand response, but the mechanistic basis for the equilibrium shift in each variant is probably different. For example, the D53S substitution may stabilize a Phe136 position that is compatible with the active form of CRP (35). In the crystallized CRP structures, Leu148 is exposed to the surface, so the L148K substitution might reduce the energetic penalty by favoring the solvent exposure of that residue. The G141K or A144T substitutions may stabilize the topology of the hinge region for CRP activation, as proposed earlier (9, 19). Because a shifted CRP* protein equilibrium could result from any substitution stabilizing the active form or destabilizing the inactive form, we believe that there are many more CRP* variants to be found with such a property. L195R CRP appears to be one of them, based on the published data (15). However, the CRP* variants that directly perturb cAMP pocket, such as S62F and T127L/S128I CRP (7, 39), should be treated differently. In these cases, correct analysis would require the separation of the intertwined effects of the substitutions on both protein equilibrium shift and the intrinsic affinity for ligand.
The equilibrium-shift model presented here is unlike the previous one (10) that is fundamentally identical to an induced-fit model. In that model, WT CRP is completely homogeneous in its inactive form when a ligand is absent and ligand binding to the inactive form induces the conformation change of CRP from inactive to active form for DNA binding. The model can be described as follows if the stoichiometry of ligand binding is not considered.
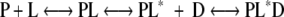 |
(5) |
Here, both P and PL are inactive forms, whereas PL* and PL*D are active forms. The observed DNA-binding constant is given by Kobs = ka (kKc[L])/(1+k[L]+kKc[L]), where k, Kc, and ka represent the equilibrium constants of the first (ligand binding), second (conformational change), and third (DNA binding) steps, respectively. The advantage of this induced-fit model is that paradoxical cGMP-binding behavior (cGMP inhibits WT CRP; cGMP activates CRP* variants) may be explained rather simply. By this induced-fit model, the free energy (Kc) for a conformational change can be assumed to be dependent upon the identity of ligand, and therefore the perturbation of Kc induced by CRP* mutation can be assumed to be different for different ligands. Nonetheless, the increase in the Kc for cGMP by CRP* mutation should be much larger than for cAMP and adenosine. The disadvantage of this model is that CRP* variants are now totally different from WT CRP in terms of ligand response, and therefore their ligand behaviors cannot be understood in the context of WT CRP. We see this as problematic because the substitution sites in the CRP* variants are far from the ligand-binding pockets. Thus, while we cannot completely rule out the induced-fit model, we favor the equilibrium-shift model because the behaviors of CRP* variants can be understood in the context of those existing in WT CRP, in terms of both active/inactive structures and ligand response.
In summary, the analysis presented here for the enhanced adenosine and cAMP activation in the CRP* variants allows us to properly interpret the enigmatic CRP* behavior with an emphasis on the importance of the specific equilibrium poise of WT CRP. Our analysis of the cGMP response in WT CRP and/or the CRP* variants is less robust, but we have clarified the inherent paradox and provided a context for future analysis. Finally, the study is highly consistent with current views about allosteric proteins (4, 18, 25), and the equilibrium-shift model presented here can be therefore expanded to explain the ligand response of any allosteric protein.
Acknowledgments
This study was supported by the College of Agricultural and Life Sciences at UW-Madison and by NIH GM53228 (to G.P.R.) and NIH GM23467 (to M. T. Record, Jr.).
We thank Robert Kerby and Jason Leduc for critical reading of the manuscript, Minseok Cha for HPLC analysis, and Jose Serate for technical assistance.
Footnotes
Published ahead of print on 2 May 2008.
REFERENCES
- 1.Aiba, H., T. Nakamura, H. Mitani, and H. Mori. 1985. Mutations that alter the allosteric nature of cAMP receptor protein of Escherichia coli. EMBO J. 43329-3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belduz, A. O., E. J. Lee, and J. G. Harman. 1993. Mutagenesis of the cyclic AMP receptor protein of Escherichia coli: targeting positions 72 and 82 of the cyclic nucleotide binding pocket. Nucleic Acids Res. 211827-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busby, S., and R. H. Ebright. 1999. Transcription activation by catabolite activator protein (CAP). J. Mol. Biol. 293199-213. [DOI] [PubMed] [Google Scholar]
- 4.Changeux, J.-P., and S. J. Edelstein. 2005. Allosteric mechanisms of signal transduction. Science 3081424-1428. [DOI] [PubMed] [Google Scholar]
- 5.Cheng, X., and J. C. Lee. 1994. Absolute requirement of cyclic nucleotide in the activation of the G141Q mutant cAMP receptor protein from Escherichia coli. J. Biol. Chem. 26930781-30784. [PubMed] [Google Scholar]
- 6.Chiang, L. W., I. Kovari, and M. M. Howe. 1993. Mutagenic oligonucleotide-directed PCR amplification (Mod-PCR): an efficient method for generating random base substitution mutations in a DNA sequence element. PCR Methods Appl. 2210-217. [DOI] [PubMed] [Google Scholar]
- 7.Dai, J., S. H. Lin, C. Kemmis, A. J. Chin, and J. C. Lee. 2004. Interplay between site-specific mutations and cyclic nucleotides in modulating DNA recognition by Escherichia coli cyclic AMP receptor protein. Biochemistry 438901-8910. [DOI] [PubMed] [Google Scholar]
- 8.Ebright, R. H., S. F. Le Grice, J. P. Miller, and J. S. Krakow. 1985. Analogs of cyclic AMP that elicit the biochemically defined conformational change in catabolite gene activator protein (CAP) but do not stimulate binding to DNA. J. Mol. Biol. 18291-107. [DOI] [PubMed] [Google Scholar]
- 9.Garges, S., and S. Adhya. 1985. Sites of allosteric shift in the structure of the cyclic AMP receptor protein. Cell 41745-751. [DOI] [PubMed] [Google Scholar]
- 10.Garges, S., and S. Adhya. 1988. Cyclic AMP-induced conformational change of cyclic AMP receptor protein (CRP): intragenic suppressors of cyclic AMP-independent CRP mutations. J. Bacteriol. 1701417-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorshkova, I., J. L. Moore, K. H. McKenney, and F. P. Schwarz. 1995. Thermodynamics of cyclic nucleotide binding to the cAMP receptor protein and its T127L mutant. J. Biol. Chem. 27021679-21683. [DOI] [PubMed] [Google Scholar]
- 12.Gronenborn, A. M., R. Sandulache, S. Gartner, and G. M. Clore. 1988. Mutations in the cyclic AMP binding site of the cyclic AMP receptor protein of Escherichia coli. Biochem. J. 253801-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harman, J. G. 2001. Allosteric regulation of the cAMP receptor protein. Biochim. Biophys. Acta 15471-17. [DOI] [PubMed] [Google Scholar]
- 14.Harman, J. G., K. McKenney, and A. Peterkofsky. 1986. Structure-function analysis of three cAMP-independent forms of the cAMP receptor protein. J. Biol. Chem. 26116332-16339. [PubMed] [Google Scholar]
- 15.Harman, J. G., A. Peterkofsky, and K. McKenney. 1988. Arginine substituted for leucine at position 195 produces a cyclic AMP-independent form of the Escherichia coli cyclic AMP receptor protein. J. Biol. Chem. 2638072-8077. [PubMed] [Google Scholar]
- 16.Heyduk, T., and J. C. Lee. 1989. Escherichia coli cAMP receptor protein: evidence for three protein conformational states with different promoter binding affinities. Biochemistry 286914-6924. [DOI] [PubMed] [Google Scholar]
- 17.Johnson, M. L., and S. G. Frasier. 1985. Nonlinear least-squares analyses. Methods Enzymol. 117301-342. [Google Scholar]
- 18.Kern, D., and E. R. Zuiderweg. 2003. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 13748-757. [DOI] [PubMed] [Google Scholar]
- 19.Kim, J., S. Adhya, and S. Garges. 1992. Allosteric changes in the cAMP receptor protein of Escherichia coli: hinge reorientation. Proc. Natl. Acad. Sci. USA 899700-9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koch, A. L., and G. Vallee. 1959. The properties of adenosine deaminase and adenosine nucleoside phosphorylase in extracts of Escherichia coli. J. Biol. Chem. 2341213-1218. [PubMed] [Google Scholar]
- 21.Kolb, A., S. Busby, H. Buc, S. Garges, and S. Adhya. 1993. Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 62749-795. [DOI] [PubMed] [Google Scholar]
- 22.Krstulovic, A. M., R. A. Hartwick, and P. R. Brown. 1979. Reversed-phase liquid chromatographic separation of 3′,5′-cyclic ribonucleotides. Clin. Chem. 25235-241. [PubMed] [Google Scholar]
- 23.Lee, E. J., J. Glasgow, S. F. Leu, A. O. Belduz, and J. G. Harman. 1994. Mutagenesis of the cyclic AMP receptor protein of Escherichia coli: targeting positions 83, 127 and 128 of the cyclic nucleotide binding pocket. Nucleic Acids Res. 222894-2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin, S. H., and J. C. Lee. 2002. Communications between the high-affinity cyclic nucleotide binding sites in Escherichia coli cyclic AMP receptor protein: effect of single site mutations. Biochemistry 4111857-11867. [DOI] [PubMed] [Google Scholar]
- 25.Ma, B., S. Kumar, C. J. Tsai, and R. Nussinov. 1999. Folding funnels and binding mechanisms. Protein Eng. 12713-720. [DOI] [PubMed] [Google Scholar]
- 26.Ma, B., M. Shatsky, H. J. Wolfson, and R. Nussinov. 2002. Multiple diverse ligands binding at a single protein site: a matter of preexisting populations. Protein Sci. 11184-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKay, D. B., and T. A. Steitz. 1981. Structure of catabolite gene activator protein at 2.9 Å resolution suggests binding to left-handed B-DNA. Nature 290744-749. [DOI] [PubMed] [Google Scholar]
- 28.Monod, J., J. Wyman, and J.-P. Changeux. 1965. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1288-118. [DOI] [PubMed] [Google Scholar]
- 29.Passner, J. M., S. C. Schultz, and T. A. Steitz. 2000. Modeling the cAMP-induced allosteric transition using the crystal structure of CAP-cAMP at 2.1 Å resolution. J. Mol. Biol. 304847-859. [DOI] [PubMed] [Google Scholar]
- 30.Popovych, N., S. Sun, R. H. Ebright, and C. G. Kalodimos. 2006. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 13831-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren, Y. L., S. Garges, S. Adhya, and J. S. Krakow. 1990. Characterization of the binding of cAMP and cGMP to the CRP* 598 mutant of the Escherichia coli cAMP receptor protein. Nucleic Acids Res. 185127-5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 33.Schultz, S. C., G. C. Shields, and T. A. Steitz. 1991. Crystal structure of a CAP-DNA complex: the DNA is bent by 90 degrees. Science 2531001-1007. [DOI] [PubMed] [Google Scholar]
- 34.Scott, S.-P., and S. Jarjous. 2005. Proposed structural mechanism of Escherichia coli cAMP receptor protein cAMP-dependent proteolytic cleavage protection and selective and nonselective DNA binding. Biochemistry 448730-8748. [DOI] [PubMed] [Google Scholar]
- 35.Scott, S.-P., R. W. Harrison, I. T. Weber, and J. C. Tanaka. 1996. Predicted ligand interactions of 3′,5′-cyclic nucleotide-gated channel binding sites: comparison of retina and olfactory binding site models. Protein Eng. 9333-344. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi, M., B. Blazy, and A. Baudras. 1989. Ligand-modulated binding of a gene regulatory protein to DNA: quantitative analysis of cyclic-AMP induced binding of CRP from Escherichia coli to nonspecific and specific DNA targets. J. Mol. Biol. 207783-796. [DOI] [PubMed] [Google Scholar]
- 37.Tomlinson, S. R., Y. Tutar, and J. G. Harman. 2006. CRP subunit association and hinge conformation changes in response to cAMP binding: analysis of C-helix cysteine-substituted CRP. Biochemistry 4513438-13446. [DOI] [PubMed] [Google Scholar]
- 38.Vaney, M.-C., G. L. Gilliland, J. G. Harman, A. Peterkofsky, and I. T. Weber. 1989. Crystal structure of a cAMP-independent form of catabolite gene activator protein with adenosine substituted in one of two cAMP-binding sites. Biochemistry 284568-4574. [DOI] [PubMed] [Google Scholar]
- 39.Youn, H., R. L. Kerby, M. Conrad, and G. P. Roberts. 2006. Study of highly constitutively active mutants suggests how cAMP activates cAMP receptor protein. J. Biol. Chem. 2811119-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Youn, H., R. L. Kerby, J. Koh, and G. P. Roberts. 2007. A C-helix residue, Arg-123, has important roles in both the active and inactive forms of the cAMP receptor protein. J. Biol. Chem. 2823632-3639. [DOI] [PubMed] [Google Scholar]