

Abstract
The fusion of human immunodeficiency virus type 1 (HIV-1) to host cells is a dynamic process governed by the interaction between glycoproteins on the viral envelope and the major receptor, CD4, and coreceptor on the surface of the cell. How these receptors organize at the virion-cell interface to promote a fusion-competent site is not well understood. Using single-molecule force spectroscopy, we map the tensile strengths, lifetimes, and energy barriers of individual intermolecular bonds between CCR5-tropic HIV-1 gp120 and its receptors CD4 and CCR5 or CXCR4 as a function of the interaction time with the cell. According to the Bell model, at short times of contact between cell and virion, the gp120-CD4 bond is able to withstand forces up to 35 pN and has an initial lifetime of 0.27 s and an intermolecular length of interaction of 0.34 nm. The initial bond also has an energy barrier of 6.7 kBT (where kB is Boltzmann's constant and T is absolute temperature). However, within 0.3 s, individual gp120-CD4 bonds undergo rapid destabilization accompanied by a shortened lifetime and a lowered tensile strength. This destabilization is significantly enhanced by the coreceptor CCR5, not by CXCR4 or fusion inhibitors, which suggests that it is directly related to a conformational change in the gp120-CD4 bond. These measurements highlight the instability and low tensile strength of gp120-receptor bonds, uncover a synergistic role for CCR5 in the progression of the gp120-CD4 bond, and suggest that the cell-virus adhesion complex is functionally arranged about a long-lived gp120-coreceptor bond.
The fusion of human immunodeficiency virus type 1 (HIV-1) to host cells is a dynamic process governed by the interaction between four key proteins, including two glycoproteins on the viral surface and two main receptors on the surface of the host cell. The viral envelope (Env)-associated complex is a heterodimer consisting of glycoproteins gp41, which is anchored in the viral Env, and gp120, which is noncovalently bound to gp41 and protrudes from the virion (13, 30, 57, 61). These heterodimers are organized in trimer complexes on the surface of the virion (13). The fusion process is initiated by the binding of gp120 to the main host cell receptor CD4 (1, 2, 11, 30, 31). This binding promotes a conformational change in gp120, which produces a binding site for a secondary host cell receptor (51, 52, 64). The most common strains of HIV-1 utilize the seven-transmembrane molecule CCR5 or CXCR4 as a coreceptor (5, 29, 47). CD4 binding to gp120 results in conformational changes in gp41 that expose an N-terminal hydrophobic fusion peptide, which is inserted into the cellular plasma membrane (12). Heptad repeat (HR) regions (1 and 2) of the gp41 trimer subsequently fold in to form a six-helix bundle referred to as a coiled-coil complex (7, 27, 60). The formation of this new complex couples viral and cellular membranes and releases a free energy sufficient to promote their fusion (34). While CD4 binding is sufficient to induce six-helix bundle formation in gp41, coreceptors substantially improve the efficiency of its formation (21).
Current assays cannot probe early fusion dynamics at single-molecule resolution in live cells and in real time. Traditional assays have provided an important mechanistic understanding of the fusion process, which has led to the development of novel viral entry inhibitors. However, static assays, such as crystallographic studies or binding assays with purified proteins, characterize only halted steps of the fusion process. Commonly used infection assays rely on phenotypes developed far downstream from the initial virus-cell interaction (40). Similarly, while membrane fusion assays have been utilized to extract kinetic data, they depend critically on temperature-arresting states (TAS) (14, 34). Initial binding to the target cell is induced during a TAS, and the fusion process is only reinitiated after physiological temperature is restored. This leaves initial complexes, such as gp120-CD4 bonds formed during TAS, unexamined. These issues have begun to be addressed with magnetically synchronized viral attachment at physiological temperatures although only postattachment events are observable (15). Ultimately, assays that rely on downstream effects prevent direct mechanistic insights into the initial interactions between the virus and host cell. Moreover, these assays average kinetic constants in bulk and may overlook “molecular individuality,” i.e., the possibility that subsets of Env glycoproteins and receptors of the same type may respond differently at discrete points in time during fusion.
Here, we develop an assay that directly probes the early interactions between virion and receptors on living host cells at single-molecule resolution. This assay retains the native conformation of both the Env proteins and the receptors in the plasma membrane, while simultaneously preserving the physiological geometries of fusion proteins for infection. Kinetic, mechanical, and thermodynamic properties of the molecular bonds between gp120 and receptors CD4 and CCR5 are computed rigorously, and the time-dependent maturation of these bonds is monitored directly without the use of proxies or downstream phenotypes.
With the strategic use of entry-inhibiting small molecules and the controlled expression of various cellular receptors, individual specific binding events between host cell receptors and virion ligands can be monitored. We find that, unlike the relatively stable gp120-CCR5 bond, the gp120-CD4 bond becomes rapidly unstable. We also observe that the coreceptor CCR5 enhances this instability. To decipher the mechanism driving these unstable intermolecular interactions, we performed assays in the presence of small-molecule entry inhibitors. Together, these approaches provide new and important mechanistic insight into the initial interactions of HIV-1 with the surface of living cells and the effects of viral entry inhibitors.
MATERIALS AND METHODS
Cell culture.
GHOST (3) parental (CD4+ CCR5−) cells (developed by V. Kewal Ramani and D. Littman) were grown in Dulbecco's modified Eagle's medium (ATCC, Manassas, VA) supplemented with 10% fetal calf serum (ATCC), 500 μg ml−1 G418 (Cell-Gro), and 100 μg ml−1 penicillin-streptomycin (Sigma, St. Louis, MO) for the parental cells or 1.0 μg ml−1 puromycin for the coreceptor encoding HOS.CCR5 (CD4−/CCR5+) cells. GHOST (3) Hi-5 (CD4+/CCR5+) cells (developed by N. Landau) were grown in Dulbecco's modified Eagle's medium with 10% fetal calf serum and 1.0 μg ml−1 puromycin (37). Cells were passaged every 2 or 3 days in a humidified 5% CO2-95% air incubator maintained at 37°C. Cells were washed with Hanks medium (Sigma) and treated with 0.25% trypsin-EDTA for 7 min at 37°C and then split 1 to 10. Prior to single-molecule force measurements, 200 μl of 1 × 106 cells ml−1 was added to a 60-mm tissue culture dish containing 5 ml of culture medium and was incubated overnight in 5% CO2 at 37°C to allow for cell spreading and restoration of normal cell morphology. Immediately before an experiment, the medium was changed to serum-free medium containing HEPES (Invitrogen, Carlsbad, CA) to stabilize the pH while cells were outside the incubator environment.
Purification of the virus.
The viral vector pNL4-3-EGFP-ΔE was used to encode the core of a single-cycle infectious pseudovirus by replacing the sequence that encodes HIV-1 env with that of the enhanced green fluorescence protein (EGFP) (44). Virus particles with the Env of the HIV-1, CCR5-tropic, YU-2 strain were subsequently generated by cotransfecting 30 × 106 293T cells in a T150 flask with 20 μg of the pNL4-3-EGFP-ΔE vector and 10 μg of an expression vector encoding the env of YU-2. The host cell protein furin can cleave the precursor glycoprotein gp160 into infectious gp41/gp120 units (16). Therefore, pseudovirus with uncleaved gp160 env glycoproteins was also produced by simultaneously transfecting cells with 10 μg of the pCI.neo.PDX expression vector, which encodes the furin inhibitor alpha 1-PDX and G418 resistance (3, 16). Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Five hours later, the transfection medium was replaced by cell culture medium and, for pCI.neo.PDX transfections, supplemented with 250 μg/ml G418 to ensure furin inhibition. The supernatant containing pseudovirus was collected 48 h after cell transfection. Cell debris was removed from the supernatant by centrifugation at 470 × g at 4°C for 5 min and subsequent filtration through a 0.22-μm-pore-size filter. The supernatant was concentrated by ultracentrifugation at 112,000 × g at 4°C for 1.75 h through a sucrose cushion. Twenty percent (wt/vol) sucrose was prepared in TNE buffer (20 mM Tris [pH 8.0], 150 mM NaCl, and 2 mM EDTA) and was loaded beneath the infectious supernatant in the ultracentrifugation tube with a 1:10 volume ratio of sucrose cushion to viral supernatant. Monodispersed virions were purified using an OptiPrep density gradient (Sigma) according to the manufacturer's instructions. The pseudovirus was further purified by ultracentrifugation at 112,000 × g at 4°C for 1.75 h.
We note that not all Env gp120 trimer complexes on the viral surface are functional or infectious (9, 32). Our assay probes all trimer units that are able to specifically bind the target cell without selection for infectious over noninfectious complexes. By specifically binding the virion to the host cell, a noninfectious gp120 unit can ultimately aid infection by stabilizing the adhesion interface in which the infectious unit may act.
Analysis of viral infection.
A total of 1 × 106 GHOST (3) Hi-5 (CD4+/CCR5+) cells were plated in a 60-mm dish and allowed to adhere at 37°C for 6 h. After spreading, cells were washed with Hanks medium (Sigma) and infected with the specified virus for 6 h. Cells were washed with Hanks medium (Sigma) and treated with 0.25% trypsin-EDTA for 7 min at 37°C and fixed with 3% formaldehyde in phosphate-buffered saline (PBS) for 30 min. Induced expression of GFP was examined by flow cytometry 48 h after infection and performed by using a FACSCalibur fluorescent cell sorter (BD Biosciences).
Functionalization of the virus for attachment to the cantilever.
Solutions of the pseudovirus were incubated with succinimidyl-4-[N-maleimidomethyl]cyclohexane-1-carboxy-[6-amidocaproate] (LC-SMCC; Pierce Biotechnology, Rockford, IL) in a 10-fold molar excess of the gp120 molecules for 2 h at 4°C. The molar concentration of gp120 was calculated assuming an average of 15 gp120 viral Env trimer complexes per virion (9). LC-SMCC-functionalized virions were separated from excess LC-SMCC by dilution with PBS, pelleted by ultracentrifugation at 112,000 × g at 4°C, and resuspended in PBS. Taking into account that 99% of HIV-1 virions are replication defective (6) and that those able to productively fuse with their host cell require only one functional gp120 Env trimer (62, 63), each cantilever was incubated with 1 × 109 virions in 50-μl aliquots to achieve a Poisson distribution of adhesion events. To determine if LC-SMCC treatment affected viral infection, CD4+ CCR5+ cells were infected with our GFP-encoding pseudovirus that was either treated or not treated with LC-SMCC. The LC-SMCC treatment did not have a noticeable effect on infection (see Fig. S1 in the supplemental material).
Functionalization of the cantilevers.
Atomic force microscopy cantilevers (Veeco Instruments, Santa Barbara, CA) were cleaned by successive 1-min incubations in 70% ethanol-10% HCl, ultrapure water, and 100% ethanol at room temperature. The cantilevers were then silanized for 30 s in 2% 3-(aminopropyl)triethoxysilane (Sigma) in acetone and then functionalized with thiol groups using 2 mg/ml Traut's reagent (Sigma) in PBS at pH 8.0 supplemented with 2 mM EDTA for 1 h. The cantilevers were subsequently washed three times with PBS and incubated with LC-SMCC-labeled virions for 3 h at 4°C. The cantilevers coated with virions were washed three times in cold PBS, incubated for 1 h at 37°C in 1% bovine serum albumin (Sigma) in PBS, and washed again in warm PBS. Finally, the cantilevers were immersed into serum-free Dulbecco's modified Eagle's medium containing HEPES just prior to use.
Recombinant protein and monoclonal antibodies.
The soluble recombinant human CD4 (sCD4; Pharmacia sCD4-183) used here is composed of the first two extracellular domains of human CD4. This protein is reactive with HIV-1 gp120 and anti-CD4 monoclonal and polyclonal antibodies. Monoclonal anti-human CCR5 antibody used in some control experiments was selected for its ability to react specifically with human CCR5 transfectants but not the parental cell line (as assessed by fluorescence-activated cell sorting analysis) (R&D Systems). Cell surface CD4 complex monoclonal B4 (United Biomedical, Inc., Hauppauge, NY) used in control experiments exerts a broad neutralizing activity against several HIV genotypes and clades by blocking access to the CD4 cell surface complex (59). The above reagents were obtained from the AIDS Research and Reference Reagent Program (National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD).
Small-molecule inhibitors.
When experiments were performed in the presence of small-molecule inhibitors, functionalized cantilevers were incubated with each inhibitor in PBS at 37°C for 30 min. The inhibitor was then added to the serum-free medium immediately prior to experimentation. The inhibitor BMS-806 was used at a concentration of 1 μM; T20 was used at a concentration of 100 μg ml−1. BMS-806 was generously donated by Ernesto Freire, Department of Biology, Johns Hopkins University. T20, a fusion inhibitor from Roche, was obtained through the NIH AIDS Research and Reference Reagent Program.
Single-molecule force spectroscopy.
Single-molecule measurements were conducted using a molecular force probe (MFP) (Asylum Research, Santa Barbara, CA). The MFP is similar to an atomic force microscope and utilizes the deflection of a flexible cantilever probe to determine forces between the probe and the sample. The spring constants (in pN/μm) of the individually loaded probes were determined by the nondestructive thermal oscillation method (24). The MFP records the time-dependent position and deflection of the flexible cantilever probe above a sample with microsecond temporal resolution and with subnanometer spatial resolution using laser deflection onto a photodector. Changes in applied force were measured with subpiconewton resolution.
The MFP records outputs from the photodetector (in volts) and the linear variable differential transformer (LVDT). The photodetector output is transformed into force values using the inverse optical lever sensitivity, which is the inverse slope of the sensor output versus the LVDT output while the system is exhibiting constant compliance. The probe-to-sample distance is calculated using the LVDT output by taking the total cantilever movement and subtracting the deformation due to the applied force. Force measurements are computed using Hooke's law, F = kΔx, where F is the applied force, k is the spring constant of the cantilever, and Δx is the measured cantilever deflection. The final output is a time-dependent trace of applied force versus separation distance from the sample.
The largest (and softest) triangular cantilever probe with an average spring constant of 10 pN/nm was used to collect force measurements with the highest possible resolution (<1 pN). For every contact between cell and cantilever, the distance between the cantilever and the cell was adjusted to maintain an impingement force of 100 to 300 pN before retraction (8, 17, 33). Data collection was performed at 1.0 kHz. To extract kinetic parameters using the Bell model, experimental retraction velocities were varied between 5 and 25 μm/s, and the contact time between cantilever and cell surface was kept at a minimum (∼1 μs), which was considered to be no contact time. For measurements of bond maturation, the retraction velocity was kept constant at 10 μm/s, while the contact time was varied between 1 μs and 0.3 s, which was approximately the bond lifetime obtained from Bell model analysis (see text for details).
Data analysis.
Traces of applied force as a function of cell-cantilever separation were analyzed using Igor Pro, version 4.09, software (Wavemetrics, Inc., Lake Oswego, OR). Adhesion forces were determined directly by recording the height of the adhesion peak from the level of zero applied force. Loading rates were calculated for individual adhesion events as the product of the slope of applied force per distance (in pN/μm) prior to rupture and the retraction velocity (in μm/s). Adhesion force measurements were binned according to loading rate at increments of 50 pN s−1 (41, 42). For each set of binned data, a mean adhesion force and loading rate were calculated and used to fit Bell model parameters (41, 42). Specific binding was binned between 20 and 40 pN with a corresponding range of loading rates of ∼200 to 1,000 pN/s. Bell model predicts that:
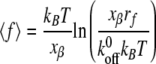 |
where 〈f〉 is the mean adhesion force of a bond, kB is Boltzmann's constant, xβ is reactive compliance, 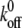 is the unstressed dissociation rate constant, T is the absolute temperature, and rf is the loading rate. We note that for loading rates that were >1,200 pN, nonspecific binding began to occur (i.e., the logarithmic dependence of bond adhesion on loading rate was not observed). To determine if this was characteristic of the virion or an effect of reaching the MFP limit, we performed experiments probing binding from low to high loading rates and again at low loading rates. We observed the logarithmic dependence at low applied forces, but after acquiring data at large applied forces, we could not recover the logarithmic dependence when the applied force was lowered (data not shown).
is the unstressed dissociation rate constant, T is the absolute temperature, and rf is the loading rate. We note that for loading rates that were >1,200 pN, nonspecific binding began to occur (i.e., the logarithmic dependence of bond adhesion on loading rate was not observed). To determine if this was characteristic of the virion or an effect of reaching the MFP limit, we performed experiments probing binding from low to high loading rates and again at low loading rates. We observed the logarithmic dependence at low applied forces, but after acquiring data at large applied forces, we could not recover the logarithmic dependence when the applied force was lowered (data not shown).
For Monte Carlo analysis of Bell model parameters, the theoretical probability of an adhesion event, or bond rupture, Prup, was calculated as
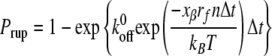 |
where n = 1, 2, 3…, Δt is the time interval, and nΔt is the time step (17, 18). Prup was compared to a random probability, Pran. Theoretical adhesion events and corresponding loading rates were recorded when the calculated Prup was greater than the random Pran.
Distributions of adhesion event probability versus adhesion force were analyzed using MATLAB, version 7.0, software (Mathworks, Inc., Natick, MA). Adhesion forces were obtained as described above and binned to produce probability distributions. For each bin of adhesion forces, a probability was determined as the number of all observed adhesion events within that bin per total observed adhesion events for that condition. The adhesion forces produced for each contact time were averaged and compared using a Student's t test. P values of <0.05 were considered to correspond to distributions of adhesion forces that were statistically different, while P values of >0.5 corresponded to adhesion forces that were statistically similar. The probability of an adhesion event occurring with a particular force f is given by
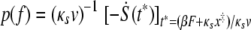 |
where
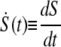 |
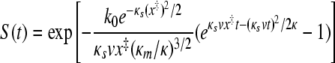 |
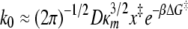 |
and
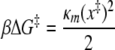 |
as described by Hummer and Szabo (23). Here, κs is the harmonic force constant scaled by kBT = β−1, ν is the retraction velocity of the cantilever, x‡ is the distance along the free energy surface from the well minimum to the energy at bond rupture, κm is the molecular spring constant of the bond, κ is the sum of κs and κm, and D is an effective diffusion coefficient. S is the survival probability or the probability that the rupture has not occurred yet at time t, and t* is the time of rupture. This probability density function was fit to each experimental adhesion force distribution by probing fit parameters using Monte Carlo optimization methods. After independently optimizing using all three fit parameters (κm, x‡, and D), we found that D remained constant at 1,600 ± 3 nm2 s−1; therefore, to obtain an improved fit to the experimental data, we held D constant at 1,600 nm2 s−1.
Error values were obtained by generating synthetic adhesion force probability density distributions and fitting these distributions to the desired model. Each synthetic probability value was randomly chosen from a distribution about the original point obtained when the complete experimental data set was binned. For each of the fitting parameters obtained from the fitting of synthetic data (n = 1,000), separate probability distributions were produced. Error values reported for variables are the standard deviation of these parameter distributions.
RESULTS
Probing interactions between pseudotyped virus and receptor-expressing cells at single-molecule resolution.
We used single-molecule force spectroscopy to characterize the initial formation of intermolecular bonds between YU-2 gp120 on the Env of a pseudotyped virion and its primary HIV-1 receptor, CD4, and coreceptor CCR5 or CXCR4 on the surface of living cells (Fig. 1A and B). Virions were tethered to a cantilever and placed in contact with individual cells, which expressed either CD4 (GHOST [3] parental [CD4+ CCR5−]), CCR5 (HOS.CCR5 [CD4− CCR5+]), both CD4 and CCR5 (GHOST [3] Hi-5 [CD4+ CCR5+]), or both CD4 and CXCR4 (GHOST CXCR4 [CD4+ CXCR4+]). The cantilever was repeatedly brought in contact with the host cell surface with a controlled impingement force and then retracted at a controlled velocity (between 5 and 25 μm/s). The value of the force of impingement, ranging between 100 to 300 pN, was selected to promote single-bond formation between host cell surface receptors and Env glycoproteins (8, 17, 33). Adhesion force and separation distance between virion and cell surface were recorded simultaneously with high temporal resolution, resulting in force-deflection traces (Fig. 1C), from which bond adhesion forces were extracted. Histograms of adhesion forces (the force to rupture bonds upon cantilever retraction) were collected and analyzed to extract the average dissociation rate, the reactive compliance (the molecular length over which virion glycoproteins and receptors interact), the adhesion force (tensile strength of the bond), the interaction energy (which measures the stability of the bond), and the lifetime of the initial molecular bonds formed between host cell receptors and Env glycoproteins.
FIG. 1.

Schematic of the instrument used to measure the micromechanics and kinetics properties of single molecular bonds between an infectious HIV-1 virion and individual cell receptors on a live host cell. (A) Schematic of the detection components of the MFP and the flexible cantilever placed just above a host cell. (B) Pseudovirus particles are cross-linked to a triangular cantilever, which is delicately brought into contact with a cell displaying either major receptor CD4, coreceptor CCR5 (or CXCR4), or both on its surface. (C) Typical force deflection traces recorded during the retraction of the cantilever. Ruptures of virion-cell bonds are marked by arrows. (D) Probability of formation of bonds between a virion and a host cell. The distribution displays Poisson characteristics (see text). (Inset graph) Probability of formation of bonds when only force deflection traces displaying at least one bond adhesion are analyzed. The time of contact between cell and virion was ∼1 μs.
Analysis of force deflection traces indicates that the probability of formation of cell-virion contacts that resulted in one or more bond adhesion events adopted a Poisson distribution (Fig. 1D). For the initial characterization of the kinetic and micromechanical properties of individual gp120-receptor bonds, the contact time between pseudotyped virus and host cell was kept as short as possible (<1 μs). These conditions resulted in successful adhesions between CD4+ CCR5− cells and pseudotyped virus in 26% of total contacts, 33% for CD4+ CCR5+ cells, 30% for CD4− CCR5+ cells (in the presence of sCD4), and 26% for CD4+ CXCR4+ cells. According to Poisson distribution statistics (10), when ∼30% of cell-cantilever contacts result in binding events, >80% of these binding events involve a single bond, 15% involve double bonds, and <3% involve triple bonds. Therefore, our experimental setup can detect and characterize the binding of Env glycoproteins to receptors on living cells at single-molecule resolution. Together, these measurements quantify, for the first time at this resolution, the specific binding between a virion and a CD4+ or CCR5+ cell at the earliest stages of molecular recognition prior to fusion.
Probability of binding between virion and host cell and adhesion specificity.
To examine the specificity of our single-molecule force spectroscopy measurements, the probability of successful binding between a virion and a CD4+ CCR5− GHOST parental cell was examined in the presence of either function-blocking antibodies or sCD4. The probability of binding between cell and virion was measured by computing the percentage of force deflection traces that displayed at least one bond de-adhesion event. Binding between cell and virion was deemed successful if the force deflection trace during cantilever retraction displayed at least one discernible peak (i.e., a bond adhesion event) (Fig. 1C). In the absence of virus on the cantilever, in the presence of a monoclonal antibody against CD4 (B4) in the culture medium, or in the presence of saturating amounts of sCD4, the probability of binding between virion and CD4+ CCR5− cells was reduced to <2 to 6% (Fig. 2B). Similarly, for CD4− CCR5+ cells in the absence or presence of sCD4—which promotes a conformational change in gp120 that induces the formation of the glycoprotein's binding site for CCR5—and a monoclonal anti-human CCR5 antibody, successful binding between virus and CD4− CCR5+ cells was reduced to ∼4% (Fig. 2A). In addition, only ∼5% of contacts between this CCR5-tropic virus and CD4− CXCR4+ cells resulted in successful binding. These results indicate that direct binding between gp120 (without sCD4) and CCR5 is not favored and that binding interactions between virions tethered to the cantilever and the cell surface, as detected and measured by our single-molecule force spectroscopy assay, are specific.
FIG. 2.
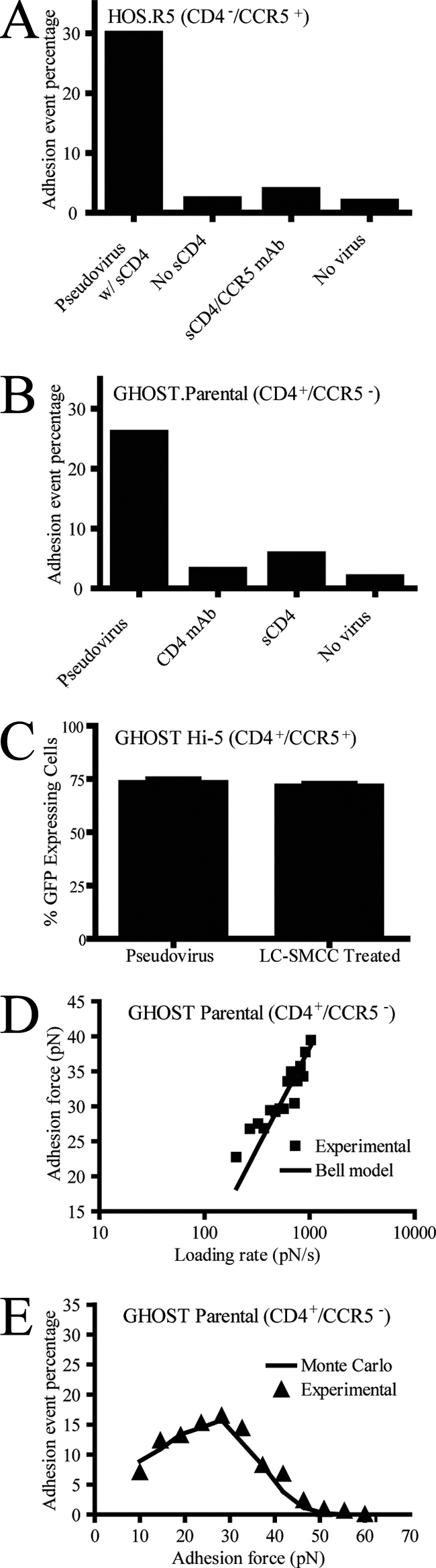
Test of binding specificity and characterization of virion-receptor interactions at single-molecule resolution. (A) Test of specificity of MFP measurements and frequency of binding interactions between Env glycoproteins and CD4− CCR5+ living cells in the presence of sCD4, in the absence of sCD4, in the presence of a function-blocking antibody against CCR5 (CCR5 mAb), or in the absence of virions attached to the cantilever (No virus), respectively. (B) Test of specificity of MFP measurements and frequency of binding interactions between Env glycoproteins and CD4− CCR5+ living cells in the absence of added molecules, in the presence of a function-blocking antibody against CD4 (CD4 mAb; B4), in the presence of sCD4, or in the absence of virions (No virus), respectively. (C) Comparison of CD4+ CCR5+ cells infected to express GFP with pseudotyped virus with and without LC-SMCC treatment. (D) Mean adhesion force of the gp120-CD4 bond as a function of loading rate (pN/s) for CD4+ CCR5− parental cells. Fit of this curve using Bell's model yielded a bond dissociation constant, koff0, of 3.73 s−1 and a bond reactive compliance, xβ, of 0.34 nm. (E) Distribution of adhesion bond forces obtained experimentally (triangles) or computed using a Monte Carlo simulation (line) based on Bell model's kinetic parameters (see text for details). The retraction velocity of the cantilever was maintained at 10 μm s−1.
sCD4 can induce the dissociation of gp120 from the membrane-anchored gp41 (20, 36). To determine the potential effect of this dissociation on our micromechanical results, we examined the binding of pseudovirus carrying gp160, in which gp120 is covalently linked to gp41. The pseudovirus was produced by cells transfected to inhibit gp160 cleavage and normalized to virus carrying wild-type HIV-1 Env based on p24 enzyme-linked immunosorbent assay. Pseudovirus carrying gp160 units was unable to productively infect CD4+ CCR5+ cells (see Fig. S1 in the supplemental material) but produced MFP binding frequencies similar to normal pseudovirus (data not shown). Experiments performed using the noninfectious pseudovirus and CD4+ CCR5+ or CD4− CCR5+ cells resulted in thermodynamic and micromechanical properties displaying comparable trends and values similar to or lower than those of normally produced virions (see Fig. S2 in the supplemental material). This suggests that bond rupture events in force displacement traces (Fig. 1C) correspond mostly to the breakages of CD4-gp120 or CCR5-gp120 bonds, not the cleavage of gp120 from gp41.
Interactions between pseudovirus and host-cell receptors at single-molecule resolution.
The Bell model (4), which has been successfully applied to analyze the binding kinetics of many molecular pairs (8, 17, 18, 41, 42), has become the standard model to analyze single-molecule force spectroscopy data (48). Here, it was exploited to characterize the micromechanical and kinetic properties of a single gp120-CD4 bond formed between cell and virion during their initial interaction. The Bell model relates the force of adhesion (also called tensile strength) of a single molecular bond to the rate at which a force of retraction, or loading rate, is applied to that bond. Bell model parameters, the unstressed dissociation rate, 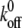 , and the reactive compliance, xβ, were obtained by a nonlinear least-squares fit of binned adhesion forces as a function of the logarithm of the loading rate. As predicted by the Bell model (4), the force of adhesion of a single gp120-CD4 bond increased logarithmically with loading rate (Fig. 2D). The Bell model fit yielded an unstressed dissociation rate of the bond between viral gp120 and CD4 on live CD4+ CCR5− cells of 3.73 ± 0.45 s−1, corresponding to an equilibrium bond lifetime, 1/
, and the reactive compliance, xβ, were obtained by a nonlinear least-squares fit of binned adhesion forces as a function of the logarithm of the loading rate. As predicted by the Bell model (4), the force of adhesion of a single gp120-CD4 bond increased logarithmically with loading rate (Fig. 2D). The Bell model fit yielded an unstressed dissociation rate of the bond between viral gp120 and CD4 on live CD4+ CCR5− cells of 3.73 ± 0.45 s−1, corresponding to an equilibrium bond lifetime, 1/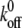 , of 0.27 ± 0.03 s (Table 1). The fit also yielded a reactive compliance of 3.4 ± 0.6 Å for the gp120-CD4 bond.
, of 0.27 ± 0.03 s (Table 1). The fit also yielded a reactive compliance of 3.4 ± 0.6 Å for the gp120-CD4 bond.
TABLE 1.
Summary of the biochemical and biomechanical properties of pseudotyped virus Env gp120-CD4 bond and purified gp120-CD4 bond using the Bell modela
| Molecular pair | Dissociation rate, koff0 (s−1) | Adhesion strength, F (pN), at loading rate of:
|
Relative compliance xβ (nm) | Source or reference | |
|---|---|---|---|---|---|
| 200 pN/s | 500 pN/s | ||||
| gp120-CD4 (pseudotyped) | 3.73 ± 0.45 | 22 ± 1 | 29 ± 1 | 0.34 ± 0.06 | This work |
| gp120-CD4 (purified) | 4.10 ± 0.20 | 26 ± 1 | 34 ± 1 | 0.14 ± 0.01 | 8 |
| P-selectin/PSGL-1 | 0.22 ± 0.05 | 82 | 130 | 0.14 ± 0.01 | 18 |
Values are means ± standard errors. The values for P-selectin/PSGL-1 are used for comparison.
Importantly, Monte Carlo simulations of gp120-CD4 bond ruptures under constant loading rate, which assumed a single dissociation rate and a single reactive compliance, showed good agreement with the experimental data (Fig. 2E). This agreement indicates that the early interactions between the pseudotyped virus and a CD4+ parental cell depend on CD4 and gp120, not other receptors on the surfaces of the cell and molecules on the virus.
The Bell model is an excellent tool for the characterization of molecular bonds, and our system offers another example of its value. In an attempt to monitor subtle time-dependent changes in the kinetic properties of cell-virion molecular bonds under different binding conditions, we employed a recently developed method to analyze adhesion force distributions.
Monitoring single gp120-receptor bond maturation.
So far, we have analyzed the interactions between host cell and virion at extremely short contact times (<1 μs). Here, we asked how individual bonds between gp120 and CD4 in live CD4+ CCR5− and CD4+ CCR5+ cells, as well as gp120 and CCR5 in the presence of sCD4 in live CD4− CCR5+ cells developed over time, or “matured.” We monitored the kinetics and micromechanical properties of these bonds by recording force deflection traces for increasing contact times between host cell and virion. Cell-virion contact times were increased from 1 μs to up to 0.3 s, an upper limit chosen to be longer than gp120-CD4 bond lifetime. The percentage of traces resulting in at least one bond rupture event did not increase >33% for all tested conditions and contact times, which ensures that all results corresponded to single-molecule observations. Statistical analysis of adhesion force distributions was performed using equations derived by Hummer and Szabo (23) (see Materials and Methods). Fits of each adhesion force distribution were independently optimized using two fitting parameters, including the distance from the free energy minimum to bond rupture, x‡, and the molecular stiffness, κm.
This analysis revealed that the initial gp120-CD4 bond between a CD4+ CCR5− cell and the pseudotyped virus had an average adhesion force of 35 ± 1.4 pN, which decreased to 26 ± 1.8 pN within 0.3 s (Fig. 3B). Initially, the adhesion force of the gp120-CD4 bond in the presence of a CCR5 (gp120-CD4/CCR5) bond between a CD4+ CCR5+ cell and the pseudotyped virus was 32 ± 0.7 pN, statistically similar to that obtained in the absence of CCR5. However, the adhesion force decreased to 16 ± 0.5 pN within 0.3 s (Fig. 3D). These results suggest that gp120-CD4 bonds weakened over time and also argue for a possible synergistic effect of CCR5, resulting in the rapid progression toward a weaker gp120-CD4 bond than with CD4 binding alone.
FIG. 3.

Histograms of adhesion forces of single intermolecular bonds for increasing contact time between pseudovirus and host cell. Time-dependent histograms of adhesion forces and mean adhesion forces of intermolecular bonds between Env glycoproteins and receptors on CD4+ CCR5− GHOST parental cells (A and B), CD4+ CCR5+ GHOST Hi-5 cells (C and D), and CD4− CCR5+ HOS.R5 cells (E and F). Mean adhesion forces at times >0 s were considered statistically significant different from the values at no contact time. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data were collected with a retraction velocity of 10 μm/s.
Correspondingly, the minimum free energy, ΔG‡ = (1/2)κmx‡2, decreased steadily for increasing cell-virion contact time. ΔG‡ describes the interaction energy barrier between gp120 and receptors CD4 and CCR5. The interaction energy of the gp120-CD4 bond between virus and CD4+ CCR5− cells decreased from 6.6 ± 0.15 kBT to 5.7 ± 0.2 kBT, while the gp120-CD4+/CCR5+ bond decreased from 6.7 ± 0.2 kBT to 3.9 ± 0.1 kBT (Fig. 4A). Moreover, the dissociation rate of the gp120-CD4 bond increased only 2.5-fold in the absence of CCR5 and >11-fold in the presence of CCR5 (Fig. 5A). The molecular stiffness of the bonds remained constant and did not approach values obtained with CD4− CCR5+ cells, which is consistent with the assay probing only gp120-CD4 interactions in both cases. This is especially apparent when κm values for CD4+ cells are compared to those obtained for gp120-CCR5 bonds with CD4− CCR5+ cells (Fig. 4C). These results suggest that there is a rapid destabilization of the gp120-CD4 bond and that CCR5 mediates an enhancement of that destabilization.
FIG. 4.

Energy barriers, lifetimes, and molecular elastic constant of bonds between Env glycoprotein and cellular receptors. (A and B) Time-dependent minimum value of the free energy (ΔG‡) describing the binding adhesion interactions between Env glycoproteins and receptors on CD4+ CCR5−, CD4+ CCR5+, CD4− CCR5+, and CD4+ CXCR4+ cells in the absence (A) and the presence (B) of small-molecule inhibitors. (C and D) Time-dependent molecular spring constants, κm, of gp120-CD4 and gp120(sCD4)-CCR5 bonds in the absence (C) and the presence (D) of small-molecule inhibitors. (E and F) Time-dependent distance from the free energy minimum to the point of bond rupture, x‡, of gp120-CD4 and gp120(sCD4)-CCR5 bonds in the absence (E) and presence (F) of small-molecule inhibitors. Data were collected with a retraction velocity of 10 μm/s.
FIG. 5.
Dissociation rates calculated from adhesion distributions and normalized by the initial (i) dissociation rate of the gp120-CD4 bond. (A) Dissociation rates k0 for virion gp120 adhesion with CD4+ CCR5−, CD4+ CCR5+, CD4− CCR5+, and CD4+ CXCR4+ cell lines normalized by the no-contact time k0 for CD4+ CCR5− cells. (B) k0 values (normalized by the initial control CD4+ CCR5− value as in A) for CD4+ CCR5+ and CD4− CCR5+ cell lines in the presence of small-molecule inhibitors. (C) Example fit of the theoretical probabilistic equation to experimental data obtained from CD4+ CCR5+ cells with no contact time. (D) Effect of small-molecule inhibitors (w/inhibitor) on k0 from CD4+ CCR5+ and CD4− CCR5+ cell lines normalized by the corresponding values without (w/o) small-molecule inhibitors. Data were collected with a retraction velocity of 10 μm/s.
The adhesion force of the bond between gp120 and CCR5 on HOS.CCR5 cells in the presence of sCD4 had a mean value of 45 ± 2 pN (Fig. 3F). Unlike CD4+ cells, the mean adhesion force did not change significantly when the contact time was increased to 0.3 s (Fig. 3F). The difference between time-dependent bond strength distributions (Fig. 3D and F) suggests that deadhesion events occur more slowly post-CCR5 binding than immediately following CD4 binding. Moreover, the large forces of adhesion produced ΔG‡ values larger (∼7.5 kBT) than obtained with CD4+ cells, indicating a more stabile bond.
To determine if the destabilization of the gp120-CD4 bond can be induced by the presence of any coreceptor or CCR5 specifically, we examined gp120-CD4 bond progression on CD4+ CXCR4+ cells. Initially, the change in free energy suggests a more dynamic effect than with coreceptor-negative cells up to 0.2 s. However, ΔG‡ then approaches the coreceptor-negative value at the final 0.3-s point (Fig. 4A). Also, the change in k0 compares much better to the CD4+ CCR5− binding than that observed with CD4+ CCR5+ cells (Fig. 5A). These results suggest that the bond instability observed in CD4+ CCR5+ cells for the CCR5-tropic HIV-1 strain is due to the presence of CCR5 specifically, not simply the presence of a coreceptor.
Effect of small-molecule viral entry inhibitors on bond progression.
To determine whether the rapid destabilization of gp120-CD4/CCR5 bonds was caused by a conformational change in gp120 initiated by CD4, we utilized the BMS-806 viral entry inhibitor. We focused on CD4+ CCR5+ cells because they produced a more dramatic change in the kinetic properties of the bonds than CD4+ parental cells Fig. 3B and D). BMS-806 promotes a nonproductive conformational change in gp120 that prevents viral fusion but does not inhibit binding to CD4 or CCR5 (53). In the presence of BMS-806, the mean adhesion force of the bond remained constant at a value of 22 ± 1 pN (Fig. 6B). BMS-806 had a complex effect on the minimum free energy of gp120-CD4 binding in the presence of CCR5: while originally ΔG‡ decreased by 3 kBT over time, ΔG‡ remained constant at 5.1 ± 0.2 kBT. Interestingly, this value was both lower than the initial ΔG‡ and larger than that observed after 0.3 s of contact (Fig. 4B). In addition, BMS-806 completely abrogated the increase in the koff rate of the gp120-CD4 bond with time of contact (Fig. 5B). The effect of BMS-806 on sCD4-induced gp120 binding to CCR5 was also explored using CD4− CCR5+ cells. Initially, gp120 bound CCR5 in the presence of sCD4 with a constant strength of 45 ± 2 pN (P > 0.05) (Fig. 3F). The addition of BMS-806 to the system decreased the mean adhesion force to a statistically different value of 30 ± 2 pN (Fig. 6F). BMS-806 effectively decreased the ΔG‡ of gp120 to CCR5 at all time points. Both in the presence and the absence of BMS-806, ΔG‡ remained relatively constant; however, the mean ΔG‡ obtained with BMS-806 was 6.1 kBT, slightly lower than normal. It is tempting to infer from this that the destabilization of the gp120-CD4/CCR5 bond may be a measure of the extent of conformation change in gp120.
FIG. 6.

Histograms of adhesion forces of single intermolecular bonds between pseudovirus and host cell in the presence of small-molecule inhibitors. Time-dependent histograms of adhesion forces and mean adhesion forces of single intermolecular bonds formed between HIV-1 and CD4+ CCR5+ GHOST Hi-5 cells in the presence and absence (control) of BMS-806 (A and B), CD4− CCR5+ HOS.R5 cells in the presence of sCD4 and in the presence and absence of BMS-806 (C and D), and CD4− CCR5+ HOS.R5 cells in the presence of sCD4 and in the presence and absence of T20 (E and F). Data were collected with a retraction velocity of 10 μm/s.
T20 is a small-molecule inhibitor of HIV-1 that mimics the coiled-coil complex formed by the HR regions of gp41, subsequently locking the complex in a conformation that does not support viral fusion (27). For short contact times, T20 had no effect on the mean adhesion force of the sCD4-induced gp120-CCR5 [gp120(sCD4)-CCR5] bond (Fig. 6F). Though the initial adhesion forces (no increased contact time) were indistinguishable, they changed significantly at later times (Fig. 6F). While gp120(sCD4)-CCR5 bonds without T20 were not statistically different at later time points (P > 0.05), each subsequent time point with T20 was statistically similar to the no-contact time mean. Also, while T20 does not directly target gp120, it slightly decreased the interaction energy of the gp120-CCR5 bond. Originally the ΔG‡ was stable at ∼7.5 kBT; however, the presence of T20 lowered the ΔG‡ of gp120(sCD4)-CCR5 bonds to ∼6.7 kBT (Fig. 4B). Additionally, T20 only slightly increased the koff rate, k0 (Fig. 5B). Neither BMS-806 nor T20 had an effect on x‡ (Fig. 4F). As T20 binds to a protein that acts downstream of gp120 binding to CCR5, observing an effect of T20 on the gp120(sCD4)-CCR5 bond was unexpected. The indirect effect that T20 has on gp120(sCD4)-CCR5 bond kinetics is evidence of the effect of gp41 on the dynamics of gp120 binding to its receptors.
DISCUSSION
Using single-molecule force spectroscopy, we have determined the mechanical and kinetic properties as well as energy barriers of intermolecular bonds between HIV-1 pseudotyped virus and receptors CD4 and CCR5 as a function of time of interaction, at single-molecule resolution, and in living cells.
Rapid destabilization of the gp120-CD4 bond aided by coreceptor CCR5.
Our analysis demonstrates a significant difference between the growing instability of gp120-CD4 bonds with and without the coexpression of CCR5 on the host cell surface (Fig. 4A). Within a time of contact of 300 ms with cells expressing both CD4 and CCR5, we did not observe a transition from gp120-CD4 bonds to gp120-CCR5 bonds. First, distributions of adhesion forces contained only one quantized peak. A transition from the initial gp120-CD4 bond distributions to those measured from only CCR5-expressing cells would ultimately contain two distinct peaks of adhesion force probability. Second, if we measured a transition from gp120-CD4-dominated binding to gp120-CCR5-dominated binding, we would note an increasing similarity between κm and other micromechanical properties to values obtained from CD4− CCR5+ cells (Fig. 4A). Finally, dissociation rates measured with CD4+ CXCR4+ cells did not approach the level of instability produced by CCR5+ binding (Fig. 5A), suggesting that it is the expression of CCR5 that is significant for this virus, not simply an underlying protein organization due to unspecific coreceptor expression.
The rapid instability of gp120-CD4 bonds is unexpected. Typically, bond strength has been observed to increase in strength over time, as with cadherin-cadherin bonds (43). Increased cadherin binding is thought to be due to an increase in intramolecular interactions between cadherin pairs. Here, HIV-1 fusion events begin with the formation of gp120-CD4 bonds, which progress toward other protein associations for effective fusion. Therefore, we speculate that the destabilization of the gp120-CD4 bond over time is a beneficial step for effective infection.
The drastic change in bond stability mediated by coreceptor CCR5 suggests a synergistic effect of CCR5 on gp120-CD4 bond progression. The colocalization of CD4 and fusion coreceptors at the surface of live cells has been suggested, and colocalization enhancement through capsianoside G treatment has been observed to increase infection (25, 54). Also, in an attempt to explain the inhibitory effects of tetraspanin EC2 proteins for HIV-1 (22), it has been suggested that membrane protein reorganization with a disruption of CD4-coreceptor complexes would result in decreased fusion. However, a synergistic effect on gp120 interaction had not been directly observed.
Contribution of gp120 conformational change to virion-receptor bond mechanics.
It is tempting to infer that gp120-CD4 bond instability stems directly from the conformational change that gp120 undergoes upon binding with CD4. The association of CD4 with gp120 once gp120 begins to bind with CCR5 is not well understood. Our result showing that the gp120-CD4 bond becomes unstable with time (i.e., as indicated by a decrease of the bond free energy and an increase of its dissociation rate) implies that the bond progresses rapidly toward dissociation.
To test this hypothesis, we used the small-molecule inhibitor BMS-806 specific to this portion of early fusion events. BMS-806 produces a nonproductive conformational change in the glycoprotein while simultaneously allowing for the binding of gp120 to both CD4 and CCR5 (52). In the presence of saturating amounts of BMS-806, the early fusion dynamics of gp120 binding to a CD4+ CCR5+ cell is drastically altered. Interestingly, while the free energy of the initial binding with no incremented contact time decreases, the increasing instability of the complex is abrogated, which leads to a more stable complex with the drug at later time points. By demonstrating that a drug that has been shown to decrease infection prevents destabilization, our results suggest that the growing instability of the gp120-CD4 bond is preferential for viral fusion. Previous knowledge of gp120 expression on an individual virion, the spherical geometry of the virus (56), and this destabilizing effect could result in a highly effective probing mechanism of the cell surface by the virus. The virion could employ these kinetic features either to remain associated with a CD4+ cell while not remaining statically bound to one small area of the cell or even to relocate to a neighboring cell to search its surface for coreceptor binding.
Viral bond progression and dynamic organization of the virion-cell adhesion complex before fusion.
T20 (also called enfurvatide) is a small-molecule inhibitor that has recently begun to be used clinically (26). Its ability to produce a nonfunctional coiled-coil complex within gp41 is sufficiently effective to produce an impressive log 2 difference in viral loads. T20 binds the gp41 HR regions specifically (27). The gp41 binding site for T20 is available after gp120 binds CD4, and by interfering with a post-CCR5-binding mechanism, T20 prevents membrane fusion (28, 53). However, using sCD4-induced pseudotyped virus and CD4− CCR5+ cells, we were able to detect that T20 slightly affected the free energy of gp120 binding to CCR5.
Heparin and heparan sulfate (HS) may play a role in the attachment of virions to various cell types (19, 35, 49, 50), although their effect on R5-tropic strains is somewhat controversial (38, 58). Our control experiments suggest that heparin and HS binding do not play a significant role in R5-specific virion attachment with our cells. However, cell surface concentrations of heparin and HS vary greatly from one cell line to another (46). Our assay could prove very useful in heparin interactions by utilizing multiple viral strains and cell lines with well-characterized heparin and HS surface profiles.
We observed a slight decrease in ΔG‡ and increase in k0 for gp120(sCD4)-CCR5. The sensitivity of these proteins to changes in conformation allows force spectroscopy to analyze subtle differences in binding. Therefore, this may indeed be a new method for analyzing the effect of entry-inhibiting drugs on their targets. Here, we summarize how the bond parameters change in an uninhibited system (Fig. 7). How these bond parameters differ from strain to strain and how effects of individual drugs relate to the ease with which HIV-1 may mutate toward resistance are important questions for future work. We note that our measurements of bond lifetime and energetics of the gp120-CD4 bond differ significantly from those obtained by Myszka et al. (39), who used surface plasmon resonance spectroscopy (Bioacore). Surface plasmon resonance requires the use of purified viral proteins instead of whole virions and purified ligands instead of full-length receptors expressed on the surface of living cells. Hence, these differences may be due to the drastic differences in experimental approaches. Our use of live cells and whole virions ensures the proper orientations of proteins on the viral surface and receptors on the cell surface, preserves their posttranslational modifications, and allows for intracellular signaling pathways to be functional. Therefore, our assay allows us to monitor early fusion dynamics in a more physiological environment than surface plasmon resonance.
FIG. 7.

Schematic of the kinetic and mechanical parameters describing early fusion dynamics of HIV-1. Mean adhesion force (f), dissociation rate (k0), change in free energy (ΔG‡), and distance from the free energy minimum (x‡) of the bonds involved in early HIV-1 fusion dynamics. The values above and below the arrows compare the mean adhesion forces and dissociation rates of the bonds corresponding to the binding and conformational states linked by the arrows. The initial binding of CD4 to gp120, the conformation change of gp120, and finally gp120 binding to CCR5 are illustrated here above their corresponding energy potentials. Data were collected with a retraction velocity of 10 μm/s.
Virion-cell attachment is governed by complexes composed of reversibly bound CD4 and CCR5 receptors. The number of receptors required to form these functional complexes is dependent on coreceptor affinity (45). Detailed electron microscopy studies of what have been termed “entry claw” structures reveal a dense packing of viral Env spikes between the virion and cell membranes (55). The assembly of such complexes would likely explain discrepancies between the time scales of typical fusion assays and those examined here. While previous kinetic studies describing the stoichiometric requirements for fusion-competent complexes also describe the dynamic relationship of the coreceptor population (45), our results highlight how these dynamic interactions are driven by dynamic binding between individual CD4 and CCR5 receptors with gp120 (56). Our findings together with those presented by Subramaniam and coworkers suggest that the adhesion interface between cell and virion could eventually self organize into a “synapse” before fusion. This synapse-like complex would be initially dominated by short-lived gp120-CD4 interactions, which individually undergo rapid destabilization. If CCR5 is absent from the first point of contact between the virion and host cell, the virion can exploit these short-lived interactions to efficiently scan the cell surface to seek CCR5. When the virion successfully forms a first CCR5 bond, we hypothesize that the other (short-lived) gp120-CD4 bonds become organized about this central stable CCR5 bond, which acts as an anchor for the virion. This reorientation of the gp120-CD4 bonds about a gp120-coreceptor bond might then lead to the previously hypothesized (55) formation of the fusion pore at the center of the cell-virus adhesion complex.
Supplementary Material
Acknowledgments
We thank Ernesto Freire for his generous gift of BMS-806, Andres Klein-Szanto and Gary Thomas for their generous gift of cPDX DNA, Christina Alves for her help with flow cytometry, and Brian Daniels for fruitful discussions.
This work was partially supported by NIH grants GM075305-01 (S.X.S. and D.W.) and EB006890-02 (D.W.). T.M.D. was partially supported by the HHMI graduate training program in nanotechnology for biology and medicine (NBMed) in the Johns Hopkins Institute for NanoBioTechnology.
Footnotes
Published ahead of print on 14 May 2008.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Alkhatib, G., C. C. Broder, and E. A. Berger. 1996. Cell type-specific fusion cofactors determine human immunodeficiency virus type 1 tropism for T-cell lines versus primary macrophages. J. Virol. 705487-5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arthos, J., K. C. Deen, M. A. Chaikin, J. A. Fornwald, G. Sathe, Q. J. Sattentau, P. R. Clapham, R. A. Weiss, J. S. McDougal, C. Pietropaolo, and et al. 1989. Identification of the residues in human CD4 critical for the binding of HIV. Cell 57469-481. [DOI] [PubMed] [Google Scholar]
- 3.Bassi, D. E., R. Lopez De Cicco, H. Mahloogi, S. Zucker, G. Thomas, and A. J. Klein-Szanto. 2001. Furin inhibition results in absent or decreased invasiveness and tumorigenicity of human cancer cells. Proc. Natl. Acad. Sci. USA 9810326-10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell, G. I. 1978. Models for the specific adhesion of cells to cells. Science 200618-627. [DOI] [PubMed] [Google Scholar]
- 5.Berger, E. A., R. W. Doms, E. M. Fenyo, B. T. M. Korber, D. R. Littman, J. P. Moore, Q. J. Sattentau, H. Schuitemaker, J. Sodroski, and R. A. Weiss. 1998. A new classification for HIV-1. Nature 391240. [DOI] [PubMed] [Google Scholar]
- 6.Bourinbaiar, A. S. 1994. The ratio of defective HIV-1 particles to replication-competent infectious virions. Acta Virol. 3859-61. [PubMed] [Google Scholar]
- 7.Chan, D. C., D. Fass, J. M. Berger, and P. S. Kim. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89263-273. [DOI] [PubMed] [Google Scholar]
- 8.Chang, M. I., P. Panorchan, T. M. Dobrowsky, Y. Tseng, and D. Wirtz. 2005. Single-molecule analysis of human immunodeficiency virus type 1 gp120-receptor interactions in living cells. J. Virol. 7914748-14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chertova, E., J. W. Bess, Jr., B. J. Crise, I. R. Sowder, T. M. Schaden, J. M. Hilburn, J. A. Hoxie, R. E. Benveniste, J. D. Lifson, L. E. Henderson, and L. O. Arthur. 2002. Envelope glycoprotein incorporation, not shedding of surface envelope glycoprotein (gp120/SU), is the primary determinant of SU content of purified human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 765315-5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chesla, S. E., P. Selvaraj, and C. Zhu. 1998. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys. J. 751553-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deen, K. C., J. S. Mcdougal, R. Inacker, G. Folenawasserman, J. Arthos, J. Rosenberg, P. J. Maddon, R. Axel, and R. W. Sweet. 1988. A soluble form of Cd4 (T4) protein inhibits AIDS virus infection. Nature 33182-84. [DOI] [PubMed] [Google Scholar]
- 12.Doms, R. W., and J. P. Moore. 2000. HIV-1 membrane fusion: targets of opportunity. J. Cell Biol. 151F9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Earl, P. L., R. W. Doms, and B. Moss. 1990. Oligomeric structure of the human immunodeficiency virus type 1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA 87648-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallo, S. A., J. D. Reeves, H. Garg, B. Foley, R. W. Doms, and R. Blumenthal. 2006. Kinetic studies of HIV-1 and HIV-2 envelope glycoprotein-mediated fusion. Retrovirology 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haim, H., I. Steiner, and A. Panet. 2007. Time frames for neutralization during the human immunodeficiency virus type 1 entry phase, as monitored in synchronously infected cell cultures. J. Virol. 813525-3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hallenberger, S., V. Bosch, H. Angliker, E. Shaw, H. D. Klenk, and W. Garten. 1992. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 360358-361. [DOI] [PubMed] [Google Scholar]
- 17.Hanley, W., O. McCarty, S. Jadhav, Y. Tseng, D. Wirtz, and K. Konstantopoulos. 2003. Single molecule characterization of P-selectin/ligand binding. J. Biol. Chem. 27810556-10561. [DOI] [PubMed] [Google Scholar]
- 18.Hanley, W. D., D. Wirtz, and K. Konstantopoulos. 2004. Distinct kinetic and mechanical properties govern selectin-leukocyte interactions. J. Cell Sci. 1172503-2511. [DOI] [PubMed] [Google Scholar]
- 19.Harrop, H. A., and C. C. Rider. 1998. Heparin and its derivatives bind to HIV-1 recombinant envelope glycoproteins, rather than to recombinant HIV-1 receptor, CD4. Glycobiology 8131-137. [DOI] [PubMed] [Google Scholar]
- 20.Hart, T. K., R. Kirsh, H. Ellens, R. W. Sweet, D. M. Lambert, S. R. Petteway, Jr., J. Leary, and P. J. Bugelski. 1991. Binding of soluble CD4 proteins to human immunodeficiency virus type 1 and infected cells induces release of envelope glycoprotein gp120. Proc. Natl. Acad. Sci. USA 882189-2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He, Y., R. Vassell, M. Zaitseva, N. Nguyen, Z. Yang, Y. Weng, and C. D. Weiss. 2003. Peptides trap the human immunodeficiency virus type 1 envelope glycoprotein fusion intermediate at two sites. J. Virol. 771666-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho, S. H., F. Martin, A. Higginbottom, L. J. Partridge, V. Parthasarathy, G. W. Moseley, P. Lopez, C. Cheng-Mayer, and P. N. Monk. 2006. Recombinant extracellular domains of tetraspanin proteins are potent inhibitors of the infection of macrophages by human immunodeficiency virus type 1. J. Virol. 806487-6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hummer, G., and A. Szabo. 2001. Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc. Natl. Acad. Sci. USA 983658-3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutter, J. L., and J. Bechhoefer. 1993. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 641868-1873. [Google Scholar]
- 25.Iyengar, S., J. E. Hildreth, and D. H. Schwartz. 1998. Actin-dependent receptor colocalization required for human immunodeficiency virus entry into host cells. J. Virol. 725251-5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jablonowski, H. 2003. Antiretroviral therapy 2003. The current status. MMW Fortschr. Med. 1454-8. [In German.] [PubMed] [Google Scholar]
- 27.Kilby, J. M., S. Hopkins, T. M. Venetta, B. DiMassimo, G. A. Cloud, J. Y. Lee, L. Alldredge, E. Hunter, D. Lambert, D. Bolognesi, T. Matthews, M. R. Johnson, M. A. Nowak, G. M. Shaw, and M. S. Saag. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 41302-1307. [DOI] [PubMed] [Google Scholar]
- 28.Kliger, Y., S. A. Gallo, S. G. Peisajovich, I. Munoz-Barroso, S. Avkin, R. Blumenthal, and Y. Shai. 2001. Mode of action of an antiviral peptide from HIV-1. Inhibition at a post-lipid mixing stage. J. Biol. Chem. 2761391-1397. [DOI] [PubMed] [Google Scholar]
- 29.Kozak, S. L., E. J. Platt, N. Madani, F. E. Ferro, Jr., K. Peden, and D. Kabat. 1997. CD4, CXCR-4, and CCR-5 dependencies for infections by primary patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J. Virol. 71873-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393648-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landau, N. R., M. Warton, and D. R. Littman. 1988. The envelope glycoprotein of the human immunodeficiency virus binds to the immunoglobulin-like domain of CD4. Nature 334159-162. [DOI] [PubMed] [Google Scholar]
- 32.Layne, S. P., M. J. Merges, M. Dembo, J. L. Spouge, S. R. Conley, J. P. Moore, J. L. Raina, H. Renz, H. R. Gelderblom, and P. L. Nara. 1992. Factors underlying spontaneous inactivation and susceptibility to neutralization of human immunodeficiency virus. Virology 189695-714. [DOI] [PubMed] [Google Scholar]
- 33.Li, F. Y., S. D. Redick, H. P. Erickson, and V. T. Moy. 2003. Force measurements of the alpha(5)beta(1) integrin-fibronectin interaction. Biophys. J. 841252-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melikyan, G. B., R. M. Markosyan, H. Hemmati, M. K. Delmedico, D. M. Lambert, and F. S. Cohen. 2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151413-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mondor, I., S. Ugolini, and Q. J. Sattentau. 1998. Human immunodeficiency virus type 1 attachment to HeLa CD4 cells is CD4 independent and gp120 dependent and requires cell surface heparans. J. Virol. 723623-3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore, J. P., J. A. McKeating, R. A. Weiss, and Q. J. Sattentau. 1990. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 2501139-1142. [DOI] [PubMed] [Google Scholar]
- 37.Morner, A., A. Bjorndal, J. Albert, V. N. Kewalramani, D. R. Littman, R. Inoue, R. Thorstensson, E. M. Fenyo, and E. Bjorling. 1999. Primary human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1 isolates, frequently use CCR5 but show promiscuity in coreceptor usage. J. Virol. 732343-2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moulard, M., H. Lortat-Jacob, I. Mondor, G. Roca, R. Wyatt, J. Sodroski, L. Zhao, W. Olson, P. D. Kwong, and Q. J. Sattentau. 2000. Selective interactions of polyanions with basic surfaces on human immunodeficiency virus type 1 gp120. J. Virol. 741948-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Myszka, D. G., R. W. Sweet, P. Hensley, M. Brigham-Burke, P. D. Kwong, W. A. Hendrickson, R. Wyatt, J. Sodroski, and M. L. Doyle. 2000. Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. USA 979026-9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neumann, T., I. Hagmann, S. Lohrengel, M. L. Heil, C. A. Derdeyn, H. G. Krausslich, and M. T. Dittmar. 2005. T20-insensitive HIV-1 from naive patients exhibits high viral fitness in a novel dual-color competition assay on primary cells. Virology 333251-262. [DOI] [PubMed] [Google Scholar]
- 41.Panorchan, P., J. P. George, and D. Wirtz. 2006. Probing intercellular interactions between vascular endothelial cadherin pairs at single-molecule resolution and in living cells. J. Mol. Biol. 358665-674. [DOI] [PubMed] [Google Scholar]
- 42.Panorchan, P., M. S. Thompson, K. J. Davis, Y. Tseng, K. Konstantopoulos, and D. Wirtz. 2006. Single-molecule analysis of cadherin-mediated cell-cell adhesion. J. Cell Sci. 11966-74. [DOI] [PubMed] [Google Scholar]
- 43.Perret, E., A. Leung, H. Feracci, and E. Evans. 2004. Trans-bonded pairs of E-cadherin exhibit a remarkable hierarchy of mechanical strengths. Proc. Natl. Acad. Sci. USA 10116472-16477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pierson, T. C., Y. Zhou, T. L. Kieffer, C. T. Ruff, C. Buck, and R. F. Siliciano. 2002. Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J. Virol. 768518-8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Platt, E. J., J. P. Durnin, and D. Kabat. 2005. Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus. J. Virol. 794347-4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rabenstein, D. L. 2002. Heparin and heparan sulfate: structure and function. Nat. Prod. Rep. 19312-331. [DOI] [PubMed] [Google Scholar]
- 47.Rizzuto, C., and J. Sodroski. 2000. Fine definition of a conserved CCR5-binding region on the human immunodeficiency virus type 1 glycoprotein 120. AIDS Res. Hum. Retroviruses. 16741-749. [DOI] [PubMed] [Google Scholar]
- 48.Robert, P., A. M. Benoliel, A. Pierres, and P. Bongrand. 2007. What is the biological relevance of the specific bond properties revealed by single-molecule studies? J. Mol. Recognit. 20432-447. [DOI] [PubMed] [Google Scholar]
- 49.Roderiquez, G., T. Oravecz, M. Yanagishita, D. C. Bou-Habib, H. Mostowski, and M. A. Norcross. 1995. Mediation of human immunodeficiency virus type 1 binding by interaction of cell surface heparan sulfate proteoglycans with the V3 region of envelope gp120-gp41. J. Virol. 692233-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saphire, A. C., M. D. Bobardt, Z. Zhang, G. David, and P. A. Gallay. 2001. Syndecans serve as attachment receptors for human immunodeficiency virus type 1 on macrophages. J. Virol. 759187-9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sattentau, Q. J., and J. P. Moore. 1991. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble Cd4 binding. J. Exp. Med. 174407-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sattentau, Q. J., J. P. Moore, F. Vignaux, F. Traincard, and P. Poignard. 1993. Conformational changes induced in the envelope glycoproteins of the human and simian immunodeficiency viruses by soluble receptor binding. J. Virol. 677383-7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Si, Z., N. Madani, J. M. Cox, J. J. Chruma, J. C. Klein, A. Schon, N. Phan, L. Wang, A. C. Biorn, S. Cocklin, I. Chaiken, E. Freire, A. B. Smith, 3rd, and J. G. Sodroski. 2004. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. USA 1015036-5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song, W., S. Yahara, Y. Maeda, K. Yusa, Y. Tanaka, and S. Harada. 2001. Enhanced infection of an X4 strain of HIV-1 due to capping and colocalization of CD4 and CXCR4 induced by capsianoside G, a diterpene glycoside. Biochem. Biophys. Res. Commun. 283423-429. [DOI] [PubMed] [Google Scholar]
- 55.Sougrat, R., A. Bartesaghi, J. D. Lifson, A. E. Bennett, J. W. Bess, D. J. Zabransky, and S. Subramaniam. 2007. Electron tomography of the contact between T cells and SIV/HIV-1: implications for viral entry. PLoS Pathog. 3e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun, S. X., and D. Wirtz. 2006. Mechanics of enveloped virus entry into host cells. Biophys. J. 90L10-L12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomas, D. J., J. S. Wall, J. F. Hainfeld, M. Kaczorek, F. P. Booy, B. L. Trus, F. A. Eiserling, and A. C. Steven. 1991. gp160, the envelope glycoprotein of human immunodeficiency virus type 1, is a dimer of 125-kilodalton subunits stabilized through interactions between their gp41 domains. J. Virol. 653797-3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ugolini, S., I. Mondor, and Q. J. Sattentau. 1999. HIV-1 attachment: another look. Trends Microbiol. 7144-149. [DOI] [PubMed] [Google Scholar]
- 59.Wang, C. Y., L. S. Sawyer, K. K. Murthy, X. Fang, A. M. Walfield, J. Ye, J. J. Wang, P. D. Chen, M. L. Li, M. T. Salas, M. Shen, M. C. Gauduin, R. W. Boyle, R. A. Koup, D. C. Montefiori, J. R. Mascola, W. C. Koff, and C. V. Hanson. 1999. Postexposure immunoprophylaxis of primary isolates by an antibody to HIV receptor complex. Proc. Natl. Acad. Sci. USA 9610367-10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387426-430. [DOI] [PubMed] [Google Scholar]
- 61.Wyatt, R., and J. Sodroski. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 2801884-1888. [DOI] [PubMed] [Google Scholar]
- 62.Yang, X., S. Kurteva, X. Ren, S. Lee, and J. Sodroski. 2005. Stoichiometry of envelope glycoprotein trimers in the entry of human immunodeficiency virus type 1. J. Virol. 7912132-12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang, X., S. Kurteva, X. Ren, S. Lee, and J. Sodroski. 2006. Subunit stoichiometry of human immunodeficiency virus type 1 envelope glycoprotein trimers during virus entry into host cells. J. Virol. 804388-4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang, W., G. Canziani, C. Plugariu, R. Wyatt, J. Sodroski, R. Sweet, P. Kwong, W. Hendrickson, and I. Chaiken. 1999. Conformational changes of gp120 in epitopes near the CCR5 binding site are induced by CD4 and a CD4 miniprotein mimetic. Biochemistry 389405-9416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.