

Abstract
APOBEC3 proteins can inhibit human immunodeficiency virus type 1 (HIV-1) replication by inducing G-to-A mutations in newly synthesized viral DNA. However, HIV-1 is able to overcome the antiretroviral activity of some of those enzymes by the viral protein Vif. We investigated the impact of different processivities of HIV-1 reverse transcriptases (RT) on the frequencies of G-to-A mutations introduced by APOBEC3 proteins. Wild-type RT or the M184V, M184I, and K65R+M184V RT variants, which are increasingly impaired in their processivities, were used in the context of a vif-deficient molecular HIV-1 clone to infect H9 cells and peripheral blood mononuclear cells (PBMCs). After two rounds of infection, a part of the HIV-1 env gene was amplified, cloned, and sequenced. The M184V mutation led to G-to-A mutation frequencies that were similar to those of the wild-type RT in H9 cells and PBMCs. The frequencies of G-to-A mutations were increased after infection with the M184I virus variant. This effect was augmented when using the K65R+M184V virus variant (P < 0.001). Overall, the G-to-A mutation frequencies were lower in PBMCs than in H9 cells. Remarkably, 38% ± 18% (mean ± standard deviation) of the env clones derived from PBMCs did not harbor any G-to-A mutation. This was rarely observed in H9 cells (3% ± 3%). Our data imply that the frequency of G-to-A mutations induced by APOBEC3 proteins can be influenced by the processivities of HIV-1 RT variants. The high number of nonmutated clones derived from PBMCs leads to several hypotheses, including that additional antiretroviral mechanisms of APOBEC3 proteins other than their deamination activity might be involved in the inhibition of vif-deficient viruses.
Proteins of the APOBEC3 (apolipoprotein B mRNA- editing enzyme, catalytic polypeptide 3) family, especially APOBEC3G and, to a lesser extent, APOBEC3F, have been identified as host factors which can inhibit the replication of human immunodeficiency virus type 1 (HIV-1) and belong to an intrinsic defense system (6, 37, 57, 68, 72). They are expressed in several cell lines, as well as primary cells like CD4+ T cells and macrophages (12, 57). The HIV-1 protein Vif suppresses the intrinsic antiretroviral effect by binding to APOBEC3G, which results in the degradation of APOBEC3G by the proteasome (38, 43, 58, 60). In the absence of Vif, APOBEC3G and APOBEC3F are packaged into virions through interactions with HIV-1 Gag and/or viral RNA (1, 9, 27, 28, 39, 53, 63, 68, 70).
Several antiretroviral mechanisms of APOBEC3 proteins have been proposed (reviewed in references 11 and 20). One antiretroviral mechanism of those proteins is their ability to deaminate cytosine residues in single-stranded HIV-1 DNA during reverse transcription, leading to G-to-A hypermutation in the viral plus strand (18, 35, 41, 42, 71). Here, APOBEC3F leads preferentially to G-to-A mutation in the context of GA dinucleotides (7, 37, 68), and GG dinucleotides are target sites specific for APOBEC3G, leading to GG-to-AG mutations (6, 18, 37, 68, 69). This process is time dependent, and thus, a 5′→3′ G-to-A mutational gradient is generated in the viral genome in correlation to the exposure time of single-stranded viral DNA to APOBEC3G/F (10, 61, 69). Single-stranded viral DNA is generated during reverse transcription from an RNA-DNA hybrid by the viral RNaseH domain of the HIV-1 reverse transcriptase (RT), and this activity of HIV-1 RNaseH leads to the activation of APOBEC3G (59). APOBEC3G can deaminate cytosine residues independently of the HIV-1 RT in a cell-free deamination assay (62); however, the progression of the synthesis of double-stranded viral DNA terminates the deamination process of APOBEC3 proteins, thereby resulting in the described 5′→3′ G-to-A mutational gradient.
Thus, it is conceivable that the HIV-1 RT is influencing the G-to-A mutation frequency induced by APOBEC3 proteins. Here, we investigated the impact of HIV-1 RT variants differing in their processivities, defined as the number of nucleotides added to a template prior to dissociation of the enzyme from its template, on the G-to-A mutation frequencies induced by APOBEC3 proteins. We hypothesized that reduced processivities lead to an increase of the G-to-A mutation frequencies induced by APOBEC3 proteins, because the time period during which the target viral DNA remains single stranded is prolonged. The rate of polymerization correlates with the processivity (3, 56). Very recently, it has been shown that defects in the rate of polymerization and processivities of M184 variants of HIV-1 RT are both due to defects in deoxynucleoside triphosphate (dNTP) utilization (16). For our approach, we have chosen the RT variants carrying the M184V, M184I, or the K65R+M184V mutations, which confer resistance to certain nucleoside/nucleotide RT inhibitors. Viruses harboring these mutations at codon 184 of the RT are resistant to the RT inhibitors lamivudine and emtricitabine (8, 54, 55). The M184V mutation causes a decrease of the processivity (3, 56), which is even further impaired by RT variants carrying the M184I mutation (3). The additional introduction of the K65R mutation, which confers resistance to tenofovir and abacavir (47), further reduces the processivity of the M184V RT variant (13, 67).
The deaminase activities of APOBEC3G/F and the G-to-A mutation frequencies and pattern have predominantly been studied in T-cell lines, partially using exogenous APOBEC3G/F (18, 35, 41, 42, 68, 71). Only recently has the deaminase activity of endogenous APOBEC3G of several T-cell lines and primary CD4+ T cells been compared with exogenous APOBEC3G derived from APOBEC3G-transfected epithelial cell lines, showing that endogenous APOBEC3G expresses a significantly lower deaminase activity than exogenous APOBEC3G (64). In our study, we determined the G-to-A mutation frequencies of endogenous APOBEC3 proteins in peripheral blood mononuclear cells (PBMCs) in comparison to these mutation frequencies in H9 cells in the context of vif-negative viruses harboring different RT variants in order to gain more insight into the deamination process in physiological target cells of HIV-1.
(This work was presented in part at the Deutsch-Österreichischer AIDS Kongreß, June 27 to 30, 2007, Frankfurt, Germany [30]; Third European Congress of Virology, September 2 to 5, 2007, Nuremberg, Germany [31]; and 15th Conference on Retroviruses and Opportunistic Infections, February 3 to 6, 2008, Boston, MA [32]).
MATERIALS AND METHODS
Plasmids.
The RT mutations M184V and M184I were separately introduced into the molecular HIV-1 clone pNL4-3 (obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases [NIAID], National Institutes of Health [NIH], Bethesda, MD, from Malcolm Martin) as previously described (21). The HIV-1 clone pNL4-3Δvif was kindly provided by Klaus Strebel, NIH, NIAID (25). The M184V and M184I mutations were introduced into this plasmid by restriction with Sph1 and Age1 (New England Biolabs, Frankfurt, Germany), and subsequent ligation using 1 U T4 DNA ligase (Invitrogen, Karlsruhe, Germany). The K65R mutation was introduced into pNL4-3M184V and pNL4-3ΔvifM184V by site-directed mutagenesis (QuikChange XL site-directed mutagenesis kit; Stratagene, La Jolla, CA) using the mutagenesis oligonucleotides sdm K65R (5′-CTCCAGTATTTGCCATAAAGAGAAAAGACAGTACTAAATGGAG-3′) and sdm K65Rrc (5′-CTCCATTTAGTACTGTCTTTTCTCTTTATGGCAAATACTGGAG-3′) (introduced mutation is underlined) following the manufacturer's instructions. The mutations were confirmed by sequence analysis.
Virus stocks.
Amounts of 3 × 106 293T cells were separately transfected with pNL4-3 or pNL4-3Δvif encoding wild-type RT or the M184V, M184I, or K65R+M184V variants, respectively. After 48 h, supernatants were collected, centrifuged at 2,000 × g at room temperature for 5 min to pellet cell debris, and finally digested with 10 U DNase I/500 μl (Boehringer, Mannheim, Germany) at 25°C for 30 min to avoid possible carryover of plasmids. Virus stocks were stored at −80°C. For characterization, viral RNA of each virus stock was isolated by using a NucleoSpin RNA virus kit (Macherey Nagel, Dueren, Germany) according to the manufacturer's instructions. cDNA synthesis was performed by using random hexamers and Moloney murine leukemia virus RT (Invitrogen) according to the manufacturer's instructions. To evaluate the virus titer, quantitative real-time PCR was performed with the reaction mixture containing 10 μl cDNA, 1× ROX PCR buffer (46), 3.5 mM MgCl2, 0.5 mM dNTPs, 0.4 μM each of the oligonucleotides pol 2981 (5′-TCAGTACAATGTGCTTCCACAGG-3′) and pol 3206 rc (5′-TTTGTCTGGTGTGGTAAATCCCCAC-3′), with Taq DNA polymerase in a final volume of 50 μl. Amplification was performed for 50 cycles (94°C for 30 s, 55°C for 30 s, and 72°C for 30 s) in an Applied Biosystems 7700 Prism spectrofluorometric thermal cycler (Applied Biosystems, Foster City, CA). The oligonucleotides were based on the HIV-1 NL4-3 and HIV-1 HXB2 sequences (GenBank accession numbers AF324493 and K03455, respectively) and synthesized by biomers.net (Ulm, Germany).
Cells and infections.
The T-cell lines H9 and MT4 were maintained under standard conditions. Briefly, cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), 2 mM glutamine, 170 mM penicillin, and 40 mM streptomycin at 37°C and 5% CO2. PBMCs were isolated from buffy coats obtained from healthy donors (Transfusionsmedizin, Suhl, Germany) by using a Ficoll-Hypaque (Sigma, Munich, Germany) gradient. After isolation, cells were washed twice, counted, and transferred into complete RPMI 1640 medium at a density of 3 × 106 cells/ml. PBMCs were stimulated with 3 μg/ml phytohemagglutinin (Sigma). An amount of 10 U/ml interleukin-2 (obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, from Hoffmann-La Roche, Inc.) was added after 24 h to the cell culture and maintained in the medium. After an additional 48 h, the phytohemagglutinin was removed by washing the cells twice. H9 cells (1 × 106) or PBMCs (3 × 106) (72 h after isolation) were infected with each virus stock by spinoculation (48), washed twice, and resuspended in 1 ml medium. At 36 to 48 h postinfection, the supernatants were harvested, centrifuged, and used for a second round of infection, which was carried out as described above. Cells were harvested 6 h later and stored at −20°C until further analyses. To test the replication abilities of the different virus variants, 1 × 106 H9 cells and MT4 cells were infected by spinoculation with the same quantity of all HIV-1 NL4-3 variants as defined by HIV-1 RNA copy numbers, i.e., 1 × 109 HIV-1 RNA copies were used. Cells were washed twice after infection, supernatants were collected daily or every second day for 5 days after infection, and an in-house p24 enzyme-linked immunosorbent assay was performed as previously described (45).
Viral DNA isolation and sequences.
Total DNA of infected cells was isolated by using a NucleoSpin blood kit (Macherey-Nagel) in accordance with the manufacturer's instructions, and DNA was eluted in 130 μl elution buffer (5 mM Tris-HCl, pH 8.5). The DNA was controlled in several ways. First, part of the β-actin gene was amplified to control for the presence of cellular DNA. Briefly, PCR was performed using 1× PCR buffer, 2 mM MgCl2, 0.5 mM dNTPs, 0.4 μM of each of the oligonucleotides bact 532 (5′-CCATCTACGAGGGGTATGC-3′) and bact 728 rc (5′-CGTGGCCATCTCTTGCTC-3′) (β-actin; GenBank accession number X00351), and Taq DNA polymerase in a final volume of 50 μl. DNA was amplified in 35 cycles (94°C for 30 s, 57°C for 30 s, and 72°C for 30 s) with a termination of 5 min at 72°C in a Mastercycler gradient thermocycler (Eppendorf, Hamburg, Germany). Second, PCR for the discrimination between vif-positive and vif-negative DNA was performed using the same conditions described above and the HIV-1 oligonucleotides int 4962 (5′-GGTGAAGGGGCAGTAGTAATAC-3′) and vpr 5685 rc (5′-CCTAAGTTATGGAGCCATATCCTAG-3′). All amplification products were analyzed by gel electrophoresis. Third, M184V, M184I, and K65R allele-specific real-time PCRs were conducted to exclude contaminations between the variants used, as previously described (2, 46). The K65R allele-specific PCR was performed accordingly using the primers pol 2835 rc (5′-GTGGTATTCCTAATTGAACTTCCC-3′) and IN K65 (5′-TCCAGTATTTGCCATAAAGIA-3′) or IN K65R (5′-CCAGTATTTGCCATAAAGIG-3′), respectively (I, inosine). Fourth, contamination by full-length HIV-1 plasmids was ruled out by amplification of the plasmid-specific sequence spanning parts of the HIV-1 5′ long terminal repeat (LTR) and the cloned human genomic DNA. The PCR was carried out according to the conditions described above by using the oligonucleotide LTR 611 (5′-AGTCAGTGTGGAAAATCTCTAGC-3′) plus the pNL4-3-genomic rc (5′-TCTTGTGGTATCAGAGTAGAGG-3′) and an annealing temperature of 55°C. All oligonucleotides were synthesized by biomers.net.
To examine the G-to-A mutation frequencies of viral DNA extracted from infected H9 cells or PBMCs, high-fidelity PCR was performed either by using a Triple Master PCR system (Eppendorf) or an EasyA kit (Stratagene). A part of the HIV-1 env gene was amplified by using the oligonucleotides BamHI/EcoRI env 7045 (5′-GGATCCGAATTCCTGCCAATTTCACAGACAATGC-3′) and ApaI/XhoI env 7552 rc (5′-GGGCCCCTCGAGCACATTTGTCCACTGATGGGAGG-3′) following the manufacturer's instructions. Amplicons were purified by using a NucleoSpin extract II kit (Macherey-Nagel), eluted in 20 μl elution buffer (5 mM Tris-HCl, pH 8.5), and cloned into the TA cloning vector provided in a StrataClone PCR cloning kit (Stratagene) in accordance with the manufacturer's instructions. Plasmids were isolated and sequenced by using the oligonucleotides M13 forward (5′-GTAAAACGACGGCCAG-3′) and M13 reverse (5′-CAGGAAACAGCTATGAC-3′), a BigDye terminator cycle sequencing kit, version 3.1, and an ABI 3100 sequencer (Applied Biosystems). Sequences were analyzed with Edit View 3.7 NT (Applied Biosystems) and Vector NTI suite 7 (Invitrogen). G-to-A mutations were analyzed by using the program Hypermut 2.0 that is freely available at http://www.hiv.lanl.gov/content/sequence/HYPERMUT/hypermut.html (52).
Statistical analysis.
Differences in the G-to-A mutation frequencies were analyzed for statistical significance by using the two-tailed Mann-Whitney test. A P value of <0.05 was considered a significant difference between two groups.
RESULTS
Replication kinetics of HIV-1 NL4-3 and HIV-1 NL4-3Δvif harboring different RT variants in H9 and MT4 cells.
To determine the impact of differences in the processivities of HIV-1 RT on the G-to-A mutation frequencies induced by APOBEC3 proteins, we introduced several mutations into the RT gene of the HIV-1 clones pNL4-3 and pNL4-3Δvif. Virus stocks of all variants were generated and tested with regard to their replication abilities. First, we infected nonpermissive H9 cells, which express APOBEC3G/F and, therefore, inhibit the replication of HIV-1 NL4-3Δvif variants. None of the HIV-1 NL4-3Δvif viruses replicated in H9 cells, whereas HIV-1 NL4-3, HIV-1 NL4-3M184V, and HIV-1 NL4-3M184I showed similar replication abilities (Fig. 1A). Next, we infected permissive MT4 cells, which do not express APOBEC3G/F, and thus, both vif-positive and vif-negative HIV-1 variants could replicate in those cells (17). The replication abilities of all eight HIV-1 NL4-3 and Δvif variants were similar in MT4 cells (Fig. 1B). As expected, HIV-1 NL4-3 variants were able to replicate in both cell lines, whereas HIV-1 NL4-3Δvif variants could only replicate in MT4 cells.
FIG. 1.

Replication kinetics of HIV-1 NL4-3 and HIV-1 NL4-3Δvif harboring different RT variants in H9 and MT4 cells. Shown are the results of a representative example of three independent experiments. Supernatants from uninfected cells served as negative controls and are indicated by crosses. (A) H9 cells were infected with HIV-1 NL4-3 (black squares), HIV-1 NL4-3M184V (black circles), HIV-1 NL4-3M184I (black diamonds), HIV-1 NL4-3Δvif (open squares), HIV-1 NL4-3ΔvifM184V (open circles), and HIV-1 NL4-3ΔvifM184I (open diamonds). (B) MT4 cells were infected with all six HIV-1 variants listed above, HIV-1 NL4-3K65R+M184V (black triangles), and HIV-1 NL4-3ΔvifK65R+M184V (open triangles). Supernatants were collected daily or every second day and assayed for viral p24 antigen by enzyme-linked immunosorbent assay.
Lack of differences in total mutation frequencies of HIV-1 NL4-3 carrying different RT variants in infected H9 cells and PBMCs.
It has been reported that the processivities of HIV-1 RT variants are inversely correlated to their fidelities, i.e., slower enzymes introduce fewer mutations (reviewed in reference 51). To account for those differences in mutation frequencies in the following experiments with the HIV-1 NL4-3Δvif viruses, we investigated the frequencies of mutations of the HIV-1 NL4-3 variants containing vif. Thus, the impact of APOBEC3 proteins on the mutation frequencies is precluded. Two independent experiments were performed using H9 cells, and another two independent experiments with PBMCs from donors A and B, and the results are summarized in Fig. 2 and Table 1. In H9 cells infected with HIV-1 NL4-3, HIV-1 NL4-3M184V, and HIV-1 NL4-3M184I, no mutations in the env sequences were observed in approximately 50% (48 to 57%) of clones. The majority of mutated clones harbored only one mutation (36 to 39%), clones carrying two or three mutations were infrequently observed, and the maximum of four mutations was only detected in one of 80 clones (Fig. 2A). G-to-A mutations were observed in seven clones, each carrying one G-to-A mutation. There were no significant differences observed between the mutation frequencies regarding the three different viral variants. In total, 53 mutations in 80 clones were observed; 7 of 53 mutations were G-to-A changes (Fig. 2B). Remarkably, A-to-G mutations were the most-frequently detected (24/53), followed by T-to-C mutations (12/53) (Fig. 2B).
FIG. 2.

Total mutation frequencies of viral DNA do not differ in H9 cells and PBMCs infected with HIV-1 NL4-3 harboring different RT variants. (A) H9 cells and PBMCs were infected with HIV-1 NL4-3 containing vif and the different RT variants, wild-type RT, M184V, M184I, and K65R+M184V. After the second round of infection, viral DNA was isolated and 466 nucleotides of the env gene were amplified, cloned, and sequenced. The percentages of HIV-1 env clones carrying the indicated numbers of mutations are shown. The numbers of clones analyzed are given. Two independent experiments using H9 cells were performed, and PBMCs from two different donors, A and B, were used. The infection with HIV-1 NL4-3K65R+M184V was only performed in PBMCs from donor B. (B) Preferences of nucleotide substitutions in HIV-1 env clones in H9 cells and PBMCs infected with HIV-1 NL4-3 harboring the different RT variants. The total numbers of mutations are indicated.
TABLE 1.
Summary of total and G-to-A mutation frequencies of analyzed HIV-1 env clonesa
| Item analyzed | Cell type infected | No. (%) of clones, base pairs, or mutations for:
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| HIV-1 NL4-3
|
HIV-1 NL4-3Δvif
|
||||||||
| WT RT | M184V | M184I | K65R+ M184V | WT RT | M184V | M184I | K65R+M184V | ||
| Total no. of clones sequenced | H9 | 29 | 28 | 23 | − | 31 | 34 | 33 | − |
| PBMC | 11 | 13 | 13 | 6 | 78 | 105 | 90 | 68 | |
| Total no. of base pairs sequenced | H9 | 13,514 | 13,048 | 10,718 | − | 14,446 | 15,844 | 15,378 | − |
| PBMC | 5,126 | 6,058 | 6,058 | 2,796 | 36,348 | 48,930 | 41,940 | 31,688 | |
| Total no. of mutations | H9 | 21 | 15 | 17 | − | 257 | 232 | 354 | − |
| PBMC | 5 | 19 | 11 | 1 | 221 | 312 | 416 | 287 | |
| Total no. of G-to-A mutations | H9 | 4 | 1 | 2 | − | 246 | 210 | 334 | − |
| PBMC | 3 | 12 | 4 | 0 | 190 | 270 | 371 | 254 | |
| No. of clones without G-to-A mutations | H9 | 25 (86.2) | 27 (96.4) | 21 (91.3) | − | 0 | 2 (5.9) | 1 (3.0) | − |
| PBMC | 9 (81.8) | 11 (84.7) | 11 (84.7) | 6 (100) | 29 (37.2) | 40 (38.1) | 33 (36.7) | 36 (52.9) | |
| Avg no. of mutations/100 bp | H9 | 0.16 | 0.11 | 0.16 | − | 1.78 | 1.47 | 2.30 | − |
| PBMC | 0.10 | 0.31 | 0.18 | 0.04 | 0.61 | 0.64 | 0.99 | 0.91 | |
| Avg no. of G-to-A mutations/ 100 bp | H9 | 0.03 | 0.01 | 0.02 | − | 1.70 | 1.33 | 2.17 | − |
| PBMC | 0.06 | 0.20 | 0.07 | 0.00 | 0.52 | 0.55 | 0.88 | 0.80 | |
| Avg no. of G-to-A mutations/ 100 bp (clones without G-to-A mutations excluded) | H9 | NA | NA | NA | − | 1.70 | 1.41 | 2.24 | − |
| PBMC | NA | NA | NA | NA | 0.83 | 0.89 | 1.40 | 1.70 | |
WT, wild-type; −, not performed; NA, not applicable.
Similar results were obtained from the infection of PBMCs from donors A and B with those viruses. Forty-six to 83% of clones did not show any mutation, 16 to 27% harbored one mutation, and 9 to 21% carried two or three mutations (Fig. 2A). In comparing the mutation frequencies of the four viral variants HIV-1 NL4-3, HIV-1 NL4-3M184V, HIV-1 NL4-3M184I, and HIV-1 NL4-3K65R+M184V, no significant differences were observed. In addition, the mutation frequencies in H9 cells (0.13% ± 0.04% [mean ± standard deviation]) and PBMCs (0.20% ± 0.11%) revealed no significant differences (Table 1). In total, 43 env clones derived from infected PBMCs were analyzed and 36 mutations were detected. As seen in env sequences derived from infected H9 cells, A-to-G transitions were frequently detected (Fig. 2B). More than 50% of all mutations were G-to-A changes (19/36) (Fig. 2B). However, this includes one single clone obtained from PBMCs infected with HIV-1 NL4-3M184V, which, in contrast to the other 35 clones, exhibited G-to-A hypermutation. In this clone, we detected 11 G-to-A mutations; 7 of those occurred within GG dinucleotides (GG to AG), which is the target site specific for APOBEC3G (6, 18, 37, 68, 69) (Fig. 2A). As this result is very unusual in the presence of Vif, all DNA samples isolated after the second round of infection were controlled by real-time PCRs specific for RT mutant alleles and PCRs specific for wild-type vif and vif-deficient DNA, as well as pNL4-3 vector-specific PCRs. No contamination was observed (data not shown). This suggests that in rare events, APOBEC3-induced G-to-A hypermutation can occur despite the presence of Vif.
Increase of G-to-A mutation frequency of viral DNA in H9 cells infected with HIV-1 NL4-3ΔvifM184I.
To determine the impact of each RT variant on the G-to-A mutation frequency induced by APOBEC3G/F, we infected H9 cells with HIV-1 NL4-3Δvif and its variants HIV-1 NL4-3ΔvifM184V and HIV-1 NL4-3ΔvifM184I in two independent experiments. A total of 99 clones were analyzed (Fig. 3; Table 1). Three clones harbored no G-to-A mutations. Two clones from the remaining 96 clones harboring G-to-A mutations were identical. Thus, one of those two clones was excluded from the final analysis to avoid any unwanted bias due to possible clonal amplification.
FIG. 3.

Impact of impaired processivities of HIV-1 RT variants and the cell type on the G-to-A mutation frequencies induced by APOBEC3 proteins after infection with HIV-1 NL4-3Δvif variants. Shown are summarized results of two independent experiments using H9 cells and of independent experiments using PBMCs from four different donors, A to D. Cells were infected with HIV-1 NL4-3Δvif, HIV-1 NL4-3ΔvifM184V, and HIV-1 NL4-3ΔvifM184I. PBMCs from donors B to D were also infected with HIV-1 NL4-3ΔvifK65R+M184V. Viral DNA was isolated after the second round of infection, and 466 nucleotides of the env gene were cloned, sequenced, and analyzed. (A) The percentages of HIV-1 env clones carrying the indicated numbers of G-to-A mutations are shown. The numbers of clones analyzed are given. (B) Preferences of nucleotide substitutions in HIV-1 env clones in H9 cells and PBMCs infected with HIV-1 NL4-3Δvif harboring different RT variants. The total numbers of mutations are indicated. (C) Comparison of G-to-A mutation frequencies in HIV-1 env sequences harboring G-to-A mutations and generated from H9 cells and PBMCs after infection with HIV-1 NL4-3Δvif, HIV-1 NL4-3ΔvifM184V, HIV-1 NL4-3ΔvifM184I, and HIV-1 NL4-3ΔvifK65R+M184V. Error bars show standard deviations. WT, wild type. Asterisks represent statistical significance within both groups, i.e., H9 cell- and PBMC-derived viral env sequences. *, P < 0.05; ***, P < 0.001.
All 31 individual env sequences derived from HIV-1 NL4-3Δvif infected H9 cells harbored G-to-A mutations (Fig. 3A). Eighteen of 31 clones (58.1%) carried one to eight and 13 of 31 clones (31.9%) nine and more G-to-A mutations. A maximum of 18 G-to-A mutations was found in one clone, i.e., 19.8% of all guanine residues were mutated in this sequence. A similar pattern was observed in the 34 env sequences derived from H9 cells infected with HIV-1 NL4-3ΔvifM184V. Eight of 34 clones (23.5%) harbored nine and more G-to-A mutations, with a maximum of 19 G-to-A mutations in one clone; 24 of 34 clones (70.6%) carried one to eight G-to-A mutations, and no G-to-A changes were observed in two clones (Fig. 3A). Taken together, we observed no significant differences between those two viruses (Fig. 3C).
In contrast, the infection of H9 cells with HIV-1 NL4-3ΔvifM184I led to an increased frequency of G-to-A mutations. In one clone, no G-to-A mutations were observed, and 15 of 33 clones (45.5%) harbored one to eight G-to-A mutations. Seventeen of 33 env sequences (51.5%) carried nine and more G-to-A mutations (Fig. 3A). In four of those clones, 22, 23, 24, and 40 of 91 guanine residues (24.2 to 44.0%), respectively, were mutated to adenine. These results differ significantly from the G-to-A mutation frequency observed in H9 cells infected with HIV-1 NL4-3ΔvifM184V (P = 0.04) (Fig. 3C). Although the infection of H9 cells with HIV-1 NL4-3ΔvifM184I led to more G-to-A mutations than infection with HIV-1 NL4-3Δvif, those differences were not significant (P = 0.24) (Fig. 3C).
As already seen in the results for H9 cells infected with vif-positive HIV-1 NL4-3 variants, the most-frequently observed non-G-to-A mutations were A-to-G and T-to-C mutations (Fig. 3B). No differences in the non-G-to-A mutation frequencies or patterns in regard to the different HIV-1 NL4-3 variants were observed.
Inverse correlation between processivities of RTs and G-to-A mutation frequencies of viral DNA in PBMCs infected with HIV-1 NL4-3Δvif variants.
It has been shown that the replication capacities of HIV-1 variants differing in the 184th amino acid position of the RT are very similar in T-cell lines, suggesting that the differences in the processivities of those RT variants may not delay substantially the process of reverse transcription in HIV-1-infected T-cell lines (3). However, a correlation between the processivities of those RT variants and their replication abilities has been observed in PBMCs (3). This underlines the role of this effect in vivo, and thus, we expanded our experiments and investigated the impact of different RT variants on the G-to-A mutation frequencies in PBMCs (the physiological target cells of HIV-1) from four different HIV-1-negative donors. In addition, we included HIV-1 NL4-3ΔvifK65R+M184V, which exhibits a lower processivity than the M184V RT variant (13, 67), to broaden the spectrum of RT variants with reduced processivities.
In total, 363 HIV-1 env clones were sequenced and analyzed. Two hundred three clones showed at least one G-to-A mutation. In order to avoid any unwanted bias due to possible clonal amplification, 22 of those 203 clones were excluded from the final analysis, because identical clones derived from the same cell cultures were detected. Thus, 341 clones were included in the final analysis (Fig. 3; Table 1). Forty-nine of 78 clones derived from PBMCs infected with HIV-1 NL4-3Δvif showed G-to-A mutations. Nearly all of those HIV-1 env sequences harbored 1 to 7 G-to-A mutations, only one clone showed 9 mutations, and two clones showed 11 G-to-A mutations (Fig. 3A). A similar pattern was observed in HIV-1 env clones derived from PBMCs infected with HIV-1 NL4-3ΔvifM184V. Sixty-five of 105 HIV-1 env sequences carried G-to-A mutations. The majority of those clones showed one to seven G-to-A changes (81.8 to 100%). Eight, 9, 12, and 13 G-to-A mutations were observed once, and 10 or 11 G-to-A mutations twice. HIV-1 env sequences derived from PBMCs infected with HIV-1 NL4-3ΔvifM184I showed higher G-to-A mutation frequencies than those infected with clones of HIV-1 NL4-3Δvif and HIV-1 NL4-3ΔvifM184V; however, this increase was not significant (P = 0.06 and P = 0.09, respectively). Here, 57 of 90 clones revealed G-to-A mutations, with 33 of 57 clones (57.9%) harboring one to seven and 20 clones ≥nine G-to-A mutations. In contrast to the other two previously described HIV-1 RT variants, we detected eight clones with 14 to 18 G-to-A mutations; one clone even carried 38 G-to-A mutations, i.e., 41.8% of all guanine residues were mutated to adenine. Such a highly mutated sequence was also observed once in H9 cells infected with the same HIV-1 variant, NL4-3ΔvifM184I. The HIV-1 variant NL4-3ΔvifK65R+M184V was used to infect PBMCs from donors B, C, and D (Fig. 3A). In 68 clones analyzed, we detected 32 HIV-1 env sequences with G-to-A mutations. Here, only 15 of 32 clones (46.9%) carried one to seven G-to-A mutations. Seventeen of 32 clones (53.1%) harbored 9 to 16 G-to-A mutations (Fig. 3; Table 1). Thus, HIV-1 NL4-3ΔvifK65R+M184V showed the highest G-to-A mutation frequency, which differed significantly from the mutation frequencies of HIV-1 NL4-3Δvif and HIV-1 NL4-3ΔvifM184V (P < 0.001) (Fig. 3C).
In regard to non-G-to-A mutations, we observed frequencies and distributions that were similar to those of HIV-1 env sequences derived from H9 cells infected with vif-negative viruses (Fig. 3B), as well as to HIV-1 env clones obtained after infection with vif-positive HIV-1 variants (Fig. 2B). Of the non-G-to-A mutations, 41.7% were A-to-G changes, followed by T-to-C mutations (27.2%) and C-to-T mutations (19.2%) (Fig. 3B).
High number of HIV-1 env clones without G-to-A mutations in PBMCs infected with HIV-1 NL4-3Δvif variants.
Only 3 of 98 HIV-1 env sequences (3.1%) derived from H9 cells did not show any G-to-A mutation, and one of those three clones carried an A-to-G mutation. In contrast, no G-to-A mutations were found in 138 of 341 HIV-1 env clones (40.5%) derived from PBMCs. This effect was slightly donor dependent. PBMCs from donor B harbored the lowest number of HIV-1 env clones without G-to-A mutations (9.1 to 28.6%) (Fig. 3A), and PBMCs from donor C the highest quantity (39.3 to 73.1%) (Fig. 3A). Overall, 51 of those 138 clones harbored different non-G-to-A mutations, the number of identical clones harboring G-to-A mutations was low, and the results of all our control experiments did not reveal evidence for clonal amplification or contamination.
Finally, we compared the G-to-A mutation frequencies of each HIV-1 NL4-3Δvif variant in H9 cells and PBMCs. We observed significantly lower frequencies of G-to-A mutations for all HIV-1 NL4-3Δvif variants in PBMCs than in H9 cells (HIV-1 NL4-3Δvif, P < 0.001, and HIV-1 NL4-3ΔvifM184V and HIV-1 NL4-3ΔvifM184I, P = <0.01) (Fig. 3C).
G-to-A mutations predominantly occur within the GG dinucleotide context in viral DNA in H9 cells and PBMCs infected with HIV-1 NL4-3Δvif variants.
The analyzed HIV-1 env fragment contains 466 nucleotides, of which 91 are guanines. We analyzed all 290 clones derived from H9 cells and PBMCs infected with HIV-1 NL4-3Δvif variants which contained G-to-A mutations in regard to their GN dinucleotide context. Only 6 of 13 GC and 11 of 22 GT dinucleotides were mutated to AC and AT, respectively, at low frequencies of 0.3 to 1.0%. In total, less than 1.5% of all G-to-A mutations occurred within those two GN dinucleotides. GA dinucleotides, the preferred target site of APOBEC3F (7, 37, 68), were more-frequently mutated to AA, accounting for 13.5% of all G-to-A mutations. Only 1 of 31 GA dinucleotides was not mutated; 27 of 31 GA-to-AA mutations were observed with frequencies of 0.7 to 4.1%. Three GA nucleotides showed mutation frequencies of 6.9, 10.3, and 12.8% (Fig. 4). However, 85.1% of all G-to-A mutations occurred within GG dinucleotides, which is the target site specific for APOBEC3G (6, 18, 37, 68, 69), suggesting that the deamination of viral cDNA occurs predominantly due to APOBEC3G. Twenty-two of the 25 GG dinucleotides were mutated in at least 5% of the HIV-1 env clones (Fig. 4). Only three GG dinucleotides were less-frequently mutated, and one of those GG dinucleotides was not mutated to AG in any of the 290 clones. Nine of 25 GG dinucleotides were mutated frequently, showing GG-to-GA mutation frequencies of 26.9 to 71.7% (Fig. 4). No clear patterns in regard to the 5′ and 3′ neighboring nucleotides of GG dinucleotides appeared; however, this might be due to the limited number of GG dinucleotides in our HIV-1 env sequence. The G-to-A mutation frequencies, which were significantly higher in viral DNA derived from H9 cells than in PBMCs, were reflected in the GG-to-AG mutation frequencies, which were 1.7 ± 0.6 times higher in viral DNA derived from H9 cells. However, the pattern of frequently versus rarely mutated GG dinucleotides was similar in both cell populations.
FIG. 4.

G-to-A mutation spectrum of viral DNA in H9 cells and PBMCs. Shown are nucleotides 7057 to 7522 of the 466-bp env fragment of HIV-1 NL4-3, which was cloned and sequenced. The V3 and V4 loops are underlined, and all first guanine residues of the GG motifs, which are preferentially targeted by APOBEC3G, are depicted in bold. Given in red are the percentages of guanine residues mutated to adenine in a total of 290 HIV-1 env sequences obtained by the infection of H9 cells (95 clones) and PBMCs from four different donors (195 clones) harboring at least one G-to-A mutation. Percentages of G-to-A mutations within GA motifs, which are the preferred motifs of APOBEC3F, are depicted in black when more than 5% of those motifs have been mutated.
DISCUSSION
In this study, we have shown that the G-to-A mutation frequencies in the viral genome that are induced mainly by APOBEC3G/F are increased in HIV-1 variants expressing RTs which exhibit low processivities. Furthermore, we discovered that the G-to-A mutation frequencies are generally lower in PBMCs than in the T-cell line H9.
Our HIV-1 NL4-3-derived viruses carrying different RT variants do not exhibit different overall mutation frequencies in the context of vif-positive viruses, either in the T-cell line H9 or in PBMCs from various donors. This is in contrast to results obtained with purified RT enzymes carrying the M184I, M184V, or M814V+K65R mutations in cell-free nucleotide misincorporation, mismatch extension, and M13-based forward-mutation assays (13, 49, 50, 66). In these biochemical assays, the fidelity of those RT variants was enhanced. In the virus replication assays, the results were contradictory. Lower fidelities of M184I and M184V variants have been observed than for wild-type HIV-1 (14). In contrast, no differences have been shown in regard to the fidelities of those variants and wild-type viruses in cell cultures (24, 26). This suggests that additional viral or cellular factors might also influence the diversity of HIV-1.
In the context of Vif expression, G-to-A, A-to-G, and T-to-C mutations were the most-frequent substitutions observed in H9 cells and PBMCs. This is in accordance with the results of a large survey based on the Los Alamos HIV-1 drug resistance database which has shown that 47% of the resistance-conferring mutations are due to those three substitutions (5). In PBMCs, we found one clone with G-to-A hypermutation, which occurred predominantly in the context of GG dinucleotides and, therefore, is probably caused by APOBEC3G deaminase activity. G-to-A hypermutation has occasionally been detected in cell cultures infected with vif-positive HIV-1 (65). Hypermutated proviral DNA has also been detected in patients in up to ∼20% of sequences, predominantly in the GG and GA dinucleotide context, without evidence of infection with vif-deficient HIV-1 (15, 22, 29). This suggests that APOBEC3G/F can deaminate the viral genome even in the presence of Vif. It is also possible that Vif occasionally became defective due to mutation(s) during reverse transcription in the first round of infection, leading to the incorporation of APOBEC3G/F into virions and subsequently causing G-to-A hypermutation in newly infected cells.
In H9 cells and PBMCs infected with vif-deficient viruses, we observed an increase in G-to-A mutation frequencies in the viral DNA derived from HIV-1 NL4-3ΔvifM184I compared to the frequencies in DNA from HIV-1 NL4-3Δvif and HIV-1 NL4-3ΔvifM184V. The G-to-A mutation frequencies were further and significantly enhanced when PBMCs were infected with HIV-1 NL4-3ΔvifK65R+M184V. These observations indicate that the G-to-A mutation frequencies induced by APOBEC3G/F are inversely correlated to the processivity of the HIV-1 RT. It is unlikely that this is due to direct interactions of the HIV-1 RT with APOBEC3G/F, because it has been reported that the deaminase capacity of APOBEC3G does not require the presence of the HIV-1 RT (62). Thus, it can be assumed that RT enzymes with impaired processivities increase the time period when viral DNA remains single stranded and, therefore, is accessible for deaminases, like APOBEC3G/F. In two elaborative studies, a 5′→3′ G-to-A mutational gradient has been observed (61, 69). Yu and colleagues have found the highest frequencies of G-to-A mutations in the 5′ region of nef and the lowest in the 5′ LTR (69). A twin gradient was observed in the other study, with two maxima of G-to-A mutation frequencies, one at the 5′ region of nef and the second one just 5′ of the central polypurine tract (61). Those results fit nicely to the model of lentiviral reverse transcription and demonstrate that the deamination process is time dependent.
Our analysis of G-to-A mutation sites in regard to the GN dinucleotide context revealed that approximately 85% of all G-to-A mutations occurred at the first guanine of GG dinucleotides, although GG dinucleotides represented only 27.5% of all four possible GN dinucleotides within the chosen 466-bp fragment of HIV-1 NL4-3 env. GT and GC dinucleotides were only rarely mutated to AT and AC, respectively. No differences were observed between H9 cells and PBMCs from four different donors. GG dinucleotides are the specific target for APOBEC3G (6, 18, 37, 68, 69), suggesting that APOBEC3G is mainly responsible for the G-to-A hypermutation seen in the viral DNA of vif-deficient viruses in H9 cells and PBMCs.
Some GG dinucleotides within the analyzed part of env were frequently mutated, whereas others were only rarely mutated. Moreover, one GG dinucleotide was not mutated in any clone. Remarkably, the corresponding amino acid, W427, of gp120 is important for the binding of the viral envelope to the CD4 receptor and highly conserved among primate immunodeficiency viruses (33). Here, the GG-to-AG mutation would result in a stop codon. A cytosine is positioned immediately downstream of this GG dinucleotide. It has been shown that G-to-A mutations do not or only rarely occur in the context of GGC (4, 18, 34, 69). This suggests that some protective mechanisms against APOBEC3 protein-mediated deamination might exist.
In all of our experiments performed with different vif-deficient viruses, we have observed that the G-to-A mutation frequencies were significantly higher in H9 cells than in PBMCs. This was contrary to the expected results. It has been shown that the replication abilities of HIV-1 variants harboring the M184V or M184I mutation are impaired in PBMCs, which correlates with the processivity of each RT variant. This effect has not been observed in a T-cell line. The authors argued that the dNTP concentrations, which are lower in primary cells than in T-cell lines, caused the impaired replication abilities of those HIV-1 variants in PBMCs (3). It has recently been confirmed that a dNTP utilization defect is one reason for decreased processivities and slower polymerization rates of HIV-1 RT variants carrying M184I or M184A mutations, which is exacerbated by limiting dNTP concentrations (16). Thus, dNTP concentrations being lower in PBMCs than in H9 cells should lead to slower polymerization rates and subsequently to higher frequencies of the G-to-A mutations induced by APOBEC3 proteins. In fact, we have obtained opposite results. Furthermore, on average 40% of viral env clones derived from PBMCs did not carry any G-to-A mutation. This effect was not observed to the same extent in H9 cells. Several other factors might explain those different observations in H9 cells and PBMCs. Recently, it has been shown that the deaminase activity of endogenous APOBEC3G in T-cell lines and PBMCs is lower than the deaminase activity of exogenous APOBEC3G derived from APOBEC3G-transfected epithelial cells (64). The authors showed that the deaminase activity of APOBEC3G is suppressed by a yet-unidentified cellular factor(s) which is present in T-cell lines and primary CD4+ cells, but not in epithelial cells. This inhibitory activity might be lower in H9 cells than in PBMCs, and the expression levels of this factor(s) could vary in PBMCs from different donors. In addition, the level of APOBEC3G/F expression might be lower in PBMCs than in H9 cells. Variations in the level of APOBEC3G mRNA expression have been observed in PBMCs from different donors (23).
In regard to the high number of sequences without G-to-A mutations in PBMCs, it is also conceivable that other antiretroviral mechanisms besides the deaminase activity of APOBEC3G/F are responsible for this observation and that those mechanisms are not similarly present and active in T-cell lines and PBMCs from different donors. Several groups have shown that APOBEC3G exhibits additional antiretroviral activities; for instance, postentry restriction of HIV-1 in resting CD4+ T cells (12), inhibition of the accumulation of reverse transcripts (17, 19, 44), and inhibition of DNA strand transfer during reverse transcription and the integration of proviral DNA (36, 40, 44). Another possibility might be the existence of a population of cells within PBMCs which are susceptible to HIV-1 infection and support virus replication but do not express APOBEC3G/F.
In summary, we have shown that the frequencies of G-to-A mutations induced by APOBEC3G/F are increased when viruses are expressing RT enzymes with impaired processivities. Moreover, the deaminase activity of endogenous APOBEC3G/F is substantially lower, if not partly absent, in PBMCs than in H9 cells. Further experiments are required to explore whether this is caused by antiretroviral mechanisms of APOBEC3G/F other than their deaminase activity, by suppressive cellular factors inhibiting APOBEC3G/F, by primary cell populations not expressing a sufficient quantity of APOBEC3G/F, and/or for other reasons.
Acknowledgments
We thank Bernhard Fleckenstein, Brigitte Biesinger-Zwosta, Robert Slany, and Michael Mach for their constant support and Kristina Allers for technical assistance. The pNL4-3 plasmid was kindly provided by the AIDS Research and Reference Reagent Program, division of AIDS, NIAID, NIH (M. Martin). We are grateful to Klaus Strebel for kindly providing pNL4-3Δvif.
This work was supported by the Deutsche Forschungsgemeinschaft (graduate program 1071, Viruses of the Immune System; SFB 466), Wilhelm Sander Stiftung (grant no. 2002.079.1), and the ELAN Foundation (grant no 07.07.09.1).
Footnotes
Published ahead of print on 30 April 2008.
REFERENCES
- 1.Alce, T. M., and W. Popik. 2004. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J. Biol. Chem. 27934083-34086. [DOI] [PubMed] [Google Scholar]
- 2.Allers, K., S. A. Knoepfel, P. Rauch, H. Walter, M. Opravil, M. Fischer, H. F. Gunthard, and K. J. Metzner. 2007. Persistence of lamivudine-sensitive HIV-1 quasispecies in the presence of lamivudine in vitro and in vivo. J. Acquir. Immune Defic. Syndr. 44377-385. [DOI] [PubMed] [Google Scholar]
- 3.Back, N. K., M. Nijhuis, W. Keulen, C. A. Boucher, B. O. Oude Essink, A. B. van Kuilenburg, A. H. van Gennip, and B. Berkhout. 1996. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a processivity defect of the reverse transcriptase enzyme. EMBO J. 154040-4049. [PMC free article] [PubMed] [Google Scholar]
- 4.Beale, R. C., S. K. Petersen-Mahrt, I. N. Watt, R. S. Harris, C. Rada, and M. S. Neuberger. 2004. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J. Mol. Biol. 337585-596. [DOI] [PubMed] [Google Scholar]
- 5.Berkhout, B., and A. de Ronde. 2004. APOBEC3G versus reverse transcriptase in the generation of HIV-1 drug-resistance mutations. AIDS 181861-1863. [DOI] [PubMed] [Google Scholar]
- 6.Bishop, K. N., R. K. Holmes, A. M. Sheehy, N. O. Davidson, S. J. Cho, and M. H. Malim. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 141392-1396. [DOI] [PubMed] [Google Scholar]
- 7.Bishop, K. N., R. K. Holmes, A. M. Sheehy, and M. H. Malim. 2004. APOBEC-mediated editing of viral RNA. Science 305645. [DOI] [PubMed] [Google Scholar]
- 8.Boucher, C. A., N. Cammack, P. Schipper, R. Schuurman, P. Rouse, M. A. Wainberg, and J. M. Cameron. 1993. High-level resistance to (−) enantiomeric 2′-deoxy-3′-thiacytidine in vitro is due to one amino acid substitution in the catalytic site of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 372231-2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cen, S., F. Guo, M. Niu, J. Saadatmand, J. Deflassieux, and L. Kleiman. 2004. The interaction between HIV-1 Gag and APOBEC3G. J. Biol. Chem. 27933177-33184. [DOI] [PubMed] [Google Scholar]
- 10.Chelico, L., P. Pham, P. Calabrese, and M. F. Goodman. 2006. APOBEC3G DNA deaminase acts processively 3′ → 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 13392-399. [DOI] [PubMed] [Google Scholar]
- 11.Chiu, Y. L., and W. C. Greene. 2006. Multifaceted antiviral actions of APOBEC3 cytidine deaminases. Trends Immunol. 27291-297. [DOI] [PubMed] [Google Scholar]
- 12.Chiu, Y. L., V. B. Soros, J. F. Kreisberg, K. Stopak, W. Yonemoto, and W. C. Greene. 2005. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature 435108-114. [DOI] [PubMed] [Google Scholar]
- 13.Deval, J., K. L. White, M. D. Miller, N. T. Parkin, J. Courcambeck, P. Halfon, B. Selmi, J. Boretto, and B. Canard. 2004. Mechanistic basis for reduced viral and enzymatic fitness of HIV-1 reverse transcriptase containing both K65R and M184V mutations. J. Biol. Chem. 279509-516. [DOI] [PubMed] [Google Scholar]
- 14.Diallo, K., B. Brenner, M. Oliveira, D. Moisi, M. Detorio, M. Gotte, and M. A. Wainberg. 2003. The M184V substitution in human immunodeficiency virus type 1 reverse transcriptase delays the development of resistance to amprenavir and efavirenz in subtype B and C clinical isolates. Antimicrob. Agents Chemother. 472376-2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gandhi, S. K., J. D. Siliciano, J. R. Bailey, R. F. Siliciano, and J. N. Blankson. 2008. Role of APOBEC3G/F-mediated hypermutation in the control of human immunodeficiency virus type 1 in elite suppressors. J. Virol. 823125-3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao, L., M. N. Hanson, M. Balakrishnan, P. L. Boyer, B. P. Roques, S. H. Hughes, B. Kim, and R. A. Bambara. 2008. Apparent defects in processive DNA synthesis, strand transfer, and primer elongation of Met-184 mutants of HIV-1 reverse transcriptase derive solely from a dNTP utilization defect. J. Biol. Chem. 2839196-9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
17.Guo, F., S. Cen, M. Niu, J. Saadatmand, and L. Kleiman. 2006. Inhibition of
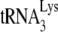 -primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 8011710-11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 8011710-11722. [DOI] [PMC free article] [PubMed] [Google Scholar] - 18.Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113803-809. [DOI] [PubMed] [Google Scholar]
- 19.Holmes, R. K., F. A. Koning, K. N. Bishop, and M. H. Malim. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2822587-2595. [DOI] [PubMed] [Google Scholar]
- 20.Holmes, R. K., M. H. Malim, and K. N. Bishop. 2007. APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 32118-128. [DOI] [PubMed] [Google Scholar]
- 21.Huelsmann, P. M., P. Rauch, K. Allers, M. J. John, and K. J. Metzner. 2006. Inhibition of drug-resistant HIV-1 by RNA interference. Antivir. Res. 691-8. [DOI] [PubMed] [Google Scholar]
- 22.Janini, M., M. Rogers, D. R. Birx, and F. E. McCutchan. 2001. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4+ T cells. J. Virol. 757973-7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin, X., A. Brooks, H. Chen, R. Bennett, R. Reichman, and H. Smith. 2005. APOBEC3G/CEM15 (hA3G) mRNA levels associate inversely with human immunodeficiency virus viremia. J. Virol. 7911513-11516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonckheere, H., M. Witvrouw, E. De Clercq, and J. Anne. 1998. Lamivudine resistance of HIV type 1 does not delay development of resistance to nonnucleoside HIV type 1-specific reverse transcriptase inhibitors as compared with wild-type HIV type 1. AIDS Res. Hum. Retrovir. 14249-253. [DOI] [PubMed] [Google Scholar]
- 25.Karczewski, M. K., and K. Strebel. 1996. Cytoskeleton association and virion incorporation of the human immunodeficiency virus type 1 Vif protein. J. Virol. 70494-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keulen, W., W. A. van, R. Schuurman, B. Berkhout, and C. A. Boucher. 1999. Increased polymerase fidelity of lamivudine-resistant HIV-1 variants does not limit their evolutionary potential. AIDS 131343-1349. [DOI] [PubMed] [Google Scholar]
- 27.Khan, M. A., R. Goila-Gaur, S. Opi, E. Miyagi, H. Takeuchi, S. Kao, and K. Strebel. 2007. Analysis of the contribution of cellular and viral RNA to the packaging of APOBEC3G into HIV-1 virions. Retrovirology 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan, M. A., S. Kao, E. Miyagi, H. Takeuchi, R. Goila-Gaur, S. Opi, C. L. Gipson, T. G. Parslow, H. Ly, and K. Strebel. 2005. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 795870-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kieffer, T. L., P. Kwon, R. E. Nettles, Y. Han, S. C. Ray, and R. F. Siliciano. 2005. G→A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J. Virol. 791975-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knoepfel, S. A., P. Rauch, N. Salisch, K. Allers, P. M. Huelsmann, and K. J. Metzner. 2007. Impact of impaired processivity of HIV-1 reverse transcriptase on the rate of G to A mutations induced by APOBEC-3F/-3G, abstr. F2. Deutsch-Österreich. AIDS Kongr., 27 to 30 June 2007, Frankfurt, Germany.
- 31.Knoepfel, S. A., P. Rauch, N. Salisch, K. Allers, P. M. Huelsmann, and K. J. Metzner. 2007. Impact of HIV-1 RT on the G to A mutation rate induced by APOBEC-3G/F, abstr. INI004. Third Eur. Congr. Virol., 2 to 5 September 2007, Nuremberg, Germany.
- 32.Knoepfel, S. A., P. Rauch, N. Salisch, K. Allers, P. M. Huelsmann, and K. J. Metzner. 2008. Impact of impaired processivity of HIV-1 reverse transcriptase on the G-to-A mutation rate induced by APOBEC-3G/-3F, abstr. C122. 15th Conf. Retrovir. Opportun. Infect., 3 to 6 February 2008, Boston, MA.
- 33.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393648-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langlois, M. A., R. C. Beale, S. G. Conticello, and M. S. Neuberger. 2005. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 331913-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lecossier, D., F. Bouchonnet, F. Clavel, and A. J. Hance. 2003. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 3001112. [DOI] [PubMed] [Google Scholar]
- 36.Li, X. Y., F. Guo, L. Zhang, L. Kleiman, and S. Cen. 2007. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J. Biol. Chem. 28232065-32074. [DOI] [PubMed] [Google Scholar]
- 37.Liddament, M. T., W. L. Brown, A. J. Schumacher, and R. S. Harris. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 141385-1391. [DOI] [PubMed] [Google Scholar]
- 38.Liu, B., X. Yu, K. Luo, Y. Yu, and X.-F. Yu. 2004. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J. Virol. 782072-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo, K., B. Liu, Z. Xiao, Y. Yu, X. Yu, R. Gorelick, and X. F. Yu. 2004. Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J. Virol. 7811841-11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo, K., T. Wang, B. Liu, C. Tian, Z. Xiao, J. Kappes, and X. F. Yu. 2007. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 817238-7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mangeat, B., P. Turelli, G. Caron, M. Friedli, L. Perrin, and D. Trono. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 42499-103. [DOI] [PubMed] [Google Scholar]
- 42.Mariani, R., D. Chen, B. Schrofelbauer, F. Navarro, R. Konig, B. Bollman, C. Munk, H. Nymark-McMahon, and N. R. Landau. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 11421-31. [DOI] [PubMed] [Google Scholar]
- 43.Marin, M., K. M. Rose, S. L. Kozak, and D. Kabat. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 91398-1403. [DOI] [PubMed] [Google Scholar]
- 44.Mbisa, J. L., R. Barr, J. A. Thomas, N. Vandegraaff, I. J. Dorweiler, E. S. Svarovskaia, W. L. Brown, L. M. Mansky, R. J. Gorelick, R. S. Harris, A. Engelman, and V. K. Pathak. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 817099-7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKeating, J. A., A. McKnight, and J. P. Moore. 1991. Differential loss of envelope glycoprotein gp120 from virions of human immunodeficiency virus type 1 isolates: effects on infectivity and neutralization. J. Virol. 65852-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metzner, K. J., S. Bonhoeffer, M. Fischer, R. Karanicolas, K. Allers, B. Joos, R. Weber, B. Hirschel, G. Kostrikis, and H. F. Gunthard. 2003. Emergence of minor populations of human immunodeficiency virus type 1 carrying the M184V and L90M mutations in subjects undergoing structured treatment interruptions. J. Infect. Dis. 1881433-1443. [DOI] [PubMed] [Google Scholar]
- 47.Miller, M. D. 2004. K65R, TAMs and tenofovir. AIDS Rev. 622-33. [PubMed] [Google Scholar]
- 48.O'Doherty, U., W. J. Swiggard, and M. H. Malim. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 7410074-10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oude Essink, B. B., N. K. Back, and B. Berkhout. 1997. Increased polymerase fidelity of the 3TC-resistant variants of HIV-1 reverse transcriptase. Nucleic Acids Res. 253212-3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rezende, L. F., W. C. Drosopoulos, and V. R. Prasad. 1998. The influence of 3TC resistance mutation M184I on the fidelity and error specificity of human immunodeficiency virus type 1 reverse transcriptase. Nucleic Acids Res. 263066-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rezende, L. F., and V. R. Prasad. 2004. Nucleoside-analog resistance mutations in HIV-1 reverse transcriptase and their influence on polymerase fidelity and viral mutation rates. Int. J. Biochem. Cell. Biol. 361716-1734. [DOI] [PubMed] [Google Scholar]
- 52.Rose, P. P., and B. T. Korber. 2000. Detecting hypermutations in viral sequences with an emphasis on G → A hypermutation. Bioinformatics 16400-401. [DOI] [PubMed] [Google Scholar]
- 53.Schafer, A., H. P. Bogerd, and B. R. Cullen. 2004. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology 328163-168. [DOI] [PubMed] [Google Scholar]
- 54.Schinazi, R. F., R. M. Lloyd, Jr., M. H. Nguyen, D. L. Cannon, A. McMillan, N. Ilksoy, C. K. Chu, D. C. Liotta, H. Z. Bazmi, and J. W. Mellors. 1993. Characterization of human immunodeficiency viruses resistant to oxathiolane-cytosine nucleosides. Antimicrob. Agents Chemother. 37875-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schuurman, R., M. Nijhuis, R. van Leeuwen, P. Schipper, D. de Jong, P. Collis, S. A. Danner, J. Mulder, C. Loveday, and C. Christopherson. 1995. Rapid changes in human immunodeficiency virus type 1 RNA load and appearance of drug-resistant virus populations in persons treated with lamivudine (3TC). J. Infect. Dis. 1711411-1419. [DOI] [PubMed] [Google Scholar]
- 56.Sharma, P. L., and C. S. Crumpacker. 1999. Decreased processivity of human immunodeficiency virus type 1 reverse transcriptase (RT) containing didanosine-selected mutation Leu74Val: a comparative analysis of RT variants Leu74Val and lamivudine-selected Met184Val. J. Virol. 738448-8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418646-650. [DOI] [PubMed] [Google Scholar]
- 58.Sheehy, A. M., N. C. Gaddis, and M. H. Malim. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 91404-1407. [DOI] [PubMed] [Google Scholar]
- 59.Soros, V. B., W. Yonemoto, and W. C. Greene. 2007. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 3e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stopak, K., C. de Noronha, W. Yonemoto, and W. C. Greene. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12591-601. [DOI] [PubMed] [Google Scholar]
- 61.Suspène, R., C. Rusniok, J. P. Vartanian, and S. Wain-Hobson. 2006. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 344677-4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suspène, R., P. Sommer, M. Henry, S. Ferris, D. Guetard, S. Pochet, A. Chester, N. Navaratnam, S. Wain-Hobson, and J. P. Vartanian. 2004. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 322421-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Svarovskaia, E. S., H. Xu, J. L. Mbisa, R. Barr, R. J. Gorelick, A. Ono, E. O. Freed, W. S. Hu, and V. K. Pathak. 2004. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J. Biol. Chem. 27935822-35828. [DOI] [PubMed] [Google Scholar]
- 64.Thielen, B. K., K. C. Klein, L. W. Walker, M. Rieck, J. H. Buckner, G. W. Tomblingson, and J. R. Lingappa. 2007. T cells contain an RNase-insensitive inhibitor of APOBEC3G deaminase activity. PLoS Pathog. 3e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vartanian, J. P., A. Meyerhans, M. Sala, and S. Wain-Hobson. 1994. G→A hypermutation of the human immunodeficiency virus type 1 genome: evidence for dCTP pool imbalance during reverse transcription. Proc. Natl. Acad. Sci. USA 913092-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wainberg, M. A., W. C. Drosopoulos, H. Salomon, M. Hsu, G. Borkow, M. Parniak, Z. Gu, Q. Song, J. Manne, S. Islam, G. Castriota, and V. R. Prasad. 1996. Enhanced fidelity of 3TC-selected mutant HIV-1 reverse transcriptase. Science 2711282-1285. [DOI] [PubMed] [Google Scholar]
- 67.White, K. L., N. A. Margot, T. Wrin, C. J. Petropoulos, M. D. Miller, and L. K. Naeger. 2002. Molecular mechanisms of resistance to human immunodeficiency virus type 1 with reverse transcriptase mutations K65R and K65R+M184V and their effects on enzyme function and viral replication capacity. Antimicrob. Agents Chemother. 463437-3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wiegand, H. L., B. P. Doehle, H. P. Bogerd, and B. R. Cullen. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 232451-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu, Q., R. Konig, S. Pillai, K. Chiles, M. Kearney, S. Palmer, D. Richman, J. M. Coffin, and N. R. Landau. 2004. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11435-442. [DOI] [PubMed] [Google Scholar]
- 70.Zennou, V., D. Perez-Caballero, H. Gottlinger, and P. D. Bieniasz. 2004. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 7812058-12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang, H., B. Yang, R. J. Pomerantz, C. Zhang, S. C. Arunachalam, and L. Gao. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 42494-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zheng, Y.-H., D. Irwin, T. Kurosu, K. Tokunaga, T. Sata, and B. M. Peterlin. 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 786073-6076. [DOI] [PMC free article] [PubMed] [Google Scholar]