

Abstract
Introduction
Sepsis is the most common trigger of acute kidney injury (AKI) in critically ill patients; understanding the structural changes associated with its occurrence is therefore important. Accordingly, we systematically reviewed the literature to assess current knowledge on the histopathology of septic AKI.
Methods
A systematic review of the MEDLINE, EMBASE and CINHAL databases and bibliographies of the retrieved articles was performed for all studies describing kidney histopathology in septic AKI.
Results
We found six studies reporting the histopathology of septic AKI for a total of only 184 patients. Among these patients, only 26 (22%) had features suggestive of acute tubular necrosis (ATN). We found four primate studies. In these, seven out of 19 (37%) cases showed features of ATN. We also found 13 rodent studies of septic AKI. In total, 23% showed evidence of ATN. In two additional studies performed in a dog model and a sheep model there was no evidence of ATN on histopathologic examination. Overall, when ATN was absent, studies reported a wide variety of kidney morphologic changes in septic AKI – ranging from normal (in most cases) to marked cortical tubular necrosis.
Conclusion
There are no consistent renal histopathological changes in human or experimental septic AKI. The majority of studies reported normal histology or only mild, nonspecific changes. ATN was relatively uncommon.
Introduction
Acute kidney injury (AKI) is a common clinical problem in critically ill patients [1,2]. Sepsis is the most important contributing factor for the development of AKI in the critically ill population [3]. Little is known about the pathogenesis of septic AKI. Renal hypoperfusion, and ischemia followed by acute tubular necrosis (ATN), have been repeatedly proposed as central to septic AKI development [4,5]. Recent studies, however, have shown that this paradigm might not be correct in all circumstances [6,7].
A possible strategy aimed at gaining better insight into the pathogenesis of septic AKI could be based on developing a clearer appreciation of the histopathological changes that occur in this condition. For example, if ATN was a consistent histopathological finding, this would strongly suggest that ischemia and tubular cell necrosis are probably an important pathogenetic mechanism. Regrettably, however, no comprehensive review of the histopathological features of septic AKI has yet been performed.
Accordingly, we systematically evaluated all available human and experimental studies describing kidney histopathology in septic AKI.
Methods
Two individuals (CL and SMB) independently identified published articles on the histopathology of septic AKI using both electronic and manual search strategies. An initial screen of identified abstracts was performed followed by a full-text screen of each article identified. Our search was supplemented by scanning the bibliographies of all recovered articles.
The databases MEDLINE (1966 to December 2006), EMBASE (1980 to December 2006) and CINAHL (1982 to December 2006, week 2) were searched. PubMed was also searched. This comprehensive search was updated in July 2007. Only articles written in English were considered.
Three comprehensive search themes were derived. The first search theme was performed using the term 'OR' with the following medical subject headings and text words: 'acute renal failure', 'acute kidney failure', 'acute tubular necrosis', 'kidney dysfunction'. The second search theme was carried out using the term 'OR' with the following medical subject headings and text words: 'sepsis', 'septicemia', 'septic shock', 'bacteremia', 'lipopolysaccharide', 'cecal puncture ligation', 'endotoxin', and 'gram negative'. The final search theme was performed using the term 'OR' with the following medical subject headings and text words: 'pathology', 'histology', 'histopathology', 'microscopy', 'morphology', 'biopsy', 'cytopathology', and 'tubular necrosis'. These three search themes were then combined using the Boolean operator 'AND'.
Study selection
Two individuals independently evaluated all identified articles for eligibility on the basis of four criteria: articles that reported original data from a primary publication, articles that reported on human subjects or experimental models, articles that made specific mention of histopathology in AKI, and articles that included subjects or models with sepsis. Any disagreements on article inclusion were resolved by discussion.
Data extraction and synthesis
The data extracted included the number of patients or animals, the proportion with sepsis, the proportion with AKI, details of sepsis (underlying disease), details of models of sepsis in animals, biopsy/postmortem, the method of assessing samples, histology results, and mortality outcome.
Experimental models were classified as having either ATN or no ATN based on the described histopathology. We used the definitions described by Thadhani and colleagues [8]. Binary data were statistically compared using Fisher's exact test with P < 0.05.
Results
Our initial search strategy yielded 378 papers. Only 73 articles, however, were identified as potentially relevant and were reviewed further. In total, 20 papers were included in our study (Figure 1). Of these, six papers were human studies while 14 studies were performed in animals.
Figure 1.
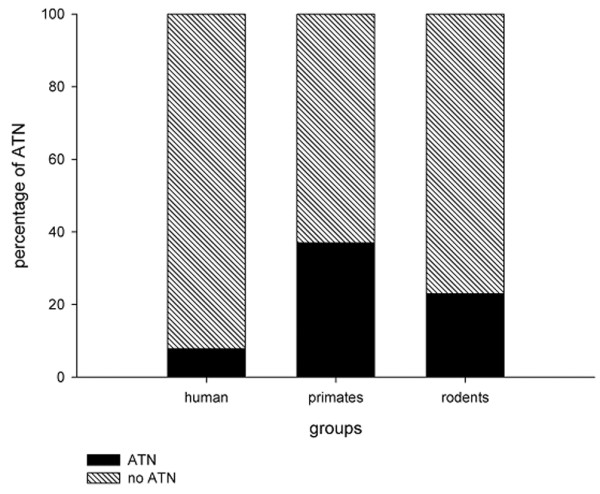
Histogram presenting the percentage of specimens showing ATN from different groups of mammals. Humans appear lest likely to have ATN. ATN, acute tubular necrosis.
We found six human studies examining the renal histopathology of septic AKI (Table 1). These studies were heterogeneous in design, in their definitions for AKI and in their histopathologic findings. While sepsis was attributed as the principal precipitant of AKI in all studies, there was potential for several additional confounding factors. For example, Mustonen and colleagues included only patients with systemic infection, hypovolemia or shock [9], whereas Hotchkiss and colleagues included only patients with septic AKI who were already dead [10]. In the retrospective analysis by Diaz de Leon and colleagues, renal biopsies were performed 6 to 7 days after AKI onset in 40 septic patients (37%, n = 107 with AKI) [11]. The results are therefore potentially biased due to late sampling and confounding from cointerventions (that is, high-dose furosemide). Finally, three studies regrettably only included a small number of septic patients [12-14].
Table 1.
Human studies
| Study | Cause | Acute kidney injury definition | Method | Cases of AKI/number of patients (%) | Acute tubular necrosis (%) |
| Hotchkiss and colleagues [10] | Sepsis/septic shock | Serum creatinine >2 mg/dl and urine output <20 ml/kg/hour × 6 hours | Postmortem | 12/20 (60) | 1 (5) |
| Sato and colleagues [13] | Sepsis | Not available | Postmortem | 6/6 (100) | 1 (17) |
| Mustonen and colleagues [9] | Sepsis/shock/hypovolemia | Not available | Biopsy | 57/57 (100) | 4 (7) |
| Rosenberg and colleagues [12] | Sepsis | Serum creatinine >3.5 mg/dl and urine/plasma osmolality >1 | Biopsy | 1/1 (100) | 0 (0) |
| Zappacosta and Ashby [14] | Sepsis | Not available | Biopsy | 1/1 (100) | 0 (0) |
| Diaz de Leon and colleagues [11] | Severe sepsis | Serum creatinine, urine output, urine/plasma osmolality (not specified) | Biopsy | 107/332 (32) | 20 (50)a |
aRenal biopsy was only performed in 40 patients (37% of the acute kidney injury (AKI) cohort, 12% of the total cohort).
In addition, there were two different methods for acquiring kidney histology specimens across these studies: primary renal biopsy or postmortem examinations.
Overall, of the 417 septic patients included in these six studies, only 44% (n = 184) had evidence of AKI; however, variable definitions were used across studies. Of these 184 patients, only 64% (n = 117) had histopathologic specimens available for evaluation. In total, 26 (22%) patients had features of classic ATN (Table 1). In three studies, renal biopsies were taken to assess histology. Three studies obtained renal histology by means of postmortem examinations following a standardised protocol. The remaining two studies performed standardised postmortem examinations. In studies with postmortem examination, only 11% (n = 2/18) of patients had evidence of ATN; whereas in studies where biopsy was performed, 24% (n = 24/99, P = 0.43) of patients showed evidence of ATN.
Mustonen and colleagues showed that nonspecific tubulointerstitial renal changes were the predominant histopathologic finding [9]. In total, 82% of specimens showed acute tubulointerstitial nephropathy, whereas 7% showed acute glomerulonephritis, 3.5% showed acute pyelonephritis and only four (7%) cases showed classic histopathologic findings consistent with ATN [9]. Similarly, Diaz de Leon and colleagues showed that 11 (27.5%) patients had nonspecific tubular or glomerular damage, whereas nine (22.5%) cases had evidence of vascular involvement [11].
In the postmortem study by Hotchkiss and colleagues, only one patient with septic AKI (8.3%) showed evidence of ATN [10]. In the study by Sato and colleagues, five out of six patients showed evidence of mild nonspecific general cell injury and only one patient had evidence of ATN [13]. Rosenberg and colleagues found mild nonspecific renal changes but no features consistent with ATN [12].
Primate models
We found four studies describing the histopathology of septic AKI in primate models of sepsis (Table 2). In two studies (n = 12), there was evidence of nonspecific tubular damage in 11 animals (92%). Only one specimen revealed ATN [15,16]. In another study, after 48 hours of sepsis, renal histopathology showed evidence of edematous tubular epithelium with the tubules filled with amorphous material; however, no animal had evidence of ATN [17]. Finally, in the study by Welty-Wolf and colleagues all animals (n = 6) showed features suggestive of ATN [18].
Table 2.
Primate studies
| Study | Cause | Cases of acute kidney injury/number of animals (%) | Acute tubular necrosis (%) |
| Carraway and colleagues [17] | Heat-shocked Escherichia coli and live E. coli | 6/6 (100) | 0 (0) |
| Coalson and colleagues [16] | E. coli endotoxin infusion | 4/4 (100) | 1 (25) |
| Coalson and colleagues [15] | Live E. coli infusion | 3/8 (38) | 0 (0) |
| Welty-Wolf and colleagues [18] | Heat-shocked E. coli and live E. coli/gentamicin administration | 6/6 (100) | 6 (100) |
Overall, in experimental primate models of septic AKI, only 37% (n = 7/19) of animals available for analysis showed evidence consistent with of ATN.
Rodent models
We were able to identify only 13 relevant experimental studies in rats (Table 3). The majority did not describe the histopathology of individual specimens in detail. We therefore classified animals into those with ATN and those without ATN. Only three studies (23%) described evidence of ATN. The remaining 10 studies described a variety of histopathologic changes that ranged from normal histology to generalised renal inflammation.
Table 3.
Rodent studies
| Study | Induction of sepsis | Acute tubular necrosis |
| Hurley and colleagues [32] | Salmonella enteritidis endotoxin | No |
| Miyaji and colleagues [33] | Cecal ligation perforation/lipopolysaccharide | Yes |
| Sato and colleagues [34] | Escherichia coli | No |
| Hayashi and colleagues [35] | Lipopolysaccharide-induced sepsis | No |
| Tsao and colleagues [36] | Lipopolysaccharide-induced sepsis | No |
| Kadkhodaee and Qasemi [37] | Lipopolysaccharide-induced sepsis | Yes |
| Zager and Prior [38] | E. coli septicemia | No |
| Wang and colleagues [39] | Lipopolysaccharide-induced sepsis | Yes |
| Tiwari and colleagues [40] | Lipopolysaccharide-induced sepsis | No |
| Gallos and colleagues [41] | Cecal ligation perforation | No |
| Kikeri and colleagues [42] | Lipopolysaccharide-induced sepsis | No |
| Yokota and colleagues [43] | Lipopolysaccharide-induced sepsis | No |
Remaining animal studies
We identified two additional experimental studies, one performed in a dog model and one in a sheep model of septic AKI. In a sheep model of cecal-ligation perforation-induced sepsis, Linton and colleagues found no consistent changes in tubular cells and no evidence of ATN [19]. Hinshaw and colleagues used a dog model of septic AKI and broadly described generalised vascular congestion, often accompanied by hemorrhage, in renal tissue, but found no evidence of ATN [20].
Discussion
We performed a systematic review of the literature using comprehensive search terms to evaluate all human and experimental studies of septic AKI describing renal histopathology. Our principal objective was to determine the nature of the typical histopathological changes seen in septic AKI. In particular, we wanted to evaluate the prevalence of features suggestive of ATN, a widely accepted marker of renal ischemia, in septic AKI to determine whether this was a potential clue to the mechanisms responsible for cell injury in septic AKI. We found very few human or experimental studies, however, which focused on the renal histopathology of septic AKI. We also found that these studies failed to show a consistent or typical renal histopathological pattern. Finally, while the majority of studies showed some general but mild histopathological changes, ATN was relatively uncommon in these human studies and only slightly more common in these experimental investigations. We believe these observations have important clinical and research implications.
The most striking finding of our study is that histopathologic data were evaluated from only 117 patients in total. Considering that an estimated 5% of all intensive care unit patients have severe AKI and that approximately 50% of AKI is primarily due to sepsis [1], one could estimate that more than 100,000 patients will have septic AKI every year in developed countries. Clearly the study sample (117 specimens overall) is inadequate to make robust inferences about the population of intensive care unit patients with septic AKI. The absence of more human data describing the renal histopathology of septic AKI is most probably related to concern about the risk of renal biopsy in acutely ill patients and to the lack of specific treatment options.
Despite such limited human data on the histopathologic changes associated with septic AKI, our review of the available evidence would suggest that ATN might be uncommon in this setting. Indeed, the most striking observation is that there is much heterogeneity of histopathological findings, ranging from totally normal to severe ATN. These observations are consistent with the heterogeneity of sepsis as a clinical condition, and they suggest caution in attributing a particular type of structural injury to this syndrome. Our findings – by failing to confirm the widely held assumption that ATN is the most common or typical histopathological substrate of septic AKI – also challenge the view that ischemia and consequent cell necrosis are most responsible for the loss of the glomerular filtration rate [4]. In fact, only 22% of human renal histopathologic specimens identified in our review showed evidence of ATN. Moreover, in the two studies evaluating postmortem specimens, where one might expect a more significant degree of ATN, only 11% showed evidence of ATN – compared with the 24% found in biopsy specimens. Nonetheless, the paucity of data on the histology of septic AKI in humans naturally led us to evaluate the renal histopathologic findings seen in experimental models of septic AKI.
In primate experimental models of septic AKI, 37% showed evidence of ATN. In particular, Welty-Wolf and colleagues showed a higher incidence of ATN. They present the only study to use vasoactive drugs to maintain blood pressure [18]. Cardiac output was not measured, and therefore we cannot be certain of whether there was a concomitant cardiogenic component to renal injury (hypodynamic sepsis). In addition, the administration of aminoglycosides may further confound the association [21]. Nonetheless, similar to primate studies, 23% of studies performed in rodents showed features consistent with ATN. Again, this would appear to be a considerably higher rate than described in the human data. There are, however, plausible explanations for these differences. Specifically, the methods for sepsis induction, the duration of sepsis prior to tissue sampling and the general supportive conditions of the experimental models (that is, systemic hemodynamics, fluid resuscitation) may contribute to significant heterogeneity across studies.
Unfortunately, in most of the studies, there were limited data provided on systemic hemodynamics such as cardiac output. Several of these models may therefore have been characterised by hypodynamic shock with decreased cardiac output, which would combine the effect of cardiogenic shock with septic shock. This hemodynamic pattern is not representative of the classical hemodynamic pattern found in human septic shock, where the circulation is generally hyperdynamic, characterised by an elevated cardiac output [22-28]. Recent evidence suggests that cardiac output may be the most important determinant of renal perfusion and that a hypodynamic circulation is likely to be a significant confounder in experimental models of septic AKI [6,7]. In contrast, experimental models of hyperdynamic sepsis (preserved or elevated cardiac output) have shown significant increases in global renal blood flow, decreases in renal vascular resistance and maintenance of renal ATP levels [29-31].
The present review has strengths and limitations. To our knowledge, this is the first study to comprehensively appraise the available English literature on the renal histopathologic changes associated with septic AKI. While our study is strengthened by performing a systematic and reproducible search and by using predefined study inclusion criteria, we only evaluated studies published in the English language. We recognise this may have contributed to omission of additional small investigations reported in other languages.
In addition, we used criteria for describing classic ATN as proposed by Thadhani and colleagues [8]; we recognise that if we used a broader definition for ATN, by incorporating more subtle renal histopathologic changes (that is, endothelial injury, evidence of apoptosis), the sensitivity of our search would probably have been increased. Our study was primarily focused, however, on describing the occurrence of classic ATN in septic AKI.
Finally, we acknowledge that many of the studies included (both experimental and human) were observational, were small, were limited in design (that is, no controls), were published several decades ago, and showed findings with considerable heterogeneity. Therefore, while these studies may present a biased perspective and global inferences may be limited, we cautiously question the strength of association of evidence of classic ATN in septic AKI and draw attention to the urgent need for a broader understanding to the renal histopathologic correlation in septic AKI.
Conclusion
The available experimental and human evidence does not, at present, support the notion that ATN is the typical histopathological lesion associated with septic AKI. Experimental findings further support the notion that ATN might be relatively uncommon in sepsis. Moreover, the reviewed studies also suggest no specific or characteristic histological features exist that are reliably associated with septic AKI. In fact, if a typical histopathological pattern exists, it is one of great heterogeneity.
A complete understanding of the histopathology of any disorder represents a fundamental step in comprehending its pathogenesis and is needed long before the development of potential therapeutic interventions. Evidence of histopathologic correlation between ATN and septic AKI, from the data available, would appear weak and lacking in robustness. We contend that further investigations of validated experimental models of septic AKI along with autopsies studies in human septic shock are clearly needed to better evaluate the true renal histopathologic appearance (along with temporal trends) associated with septic AKI.
Key messages
• Only a very small number of renal biopsies or renal postmortem assessments have been reported in humans with septic AKI.
• In these human studies, ATN was a relatively uncommon (<25%) finding.
• A limited number of experimental studies have reported the histopathological findings of septic AKI.
• In experimental studies, ATN was also a relatively uncommon histopathological finding.
• Across these experimental and human studies, there appears to be no single typical renal histopathological finding associated with septic AKI. The heterogeneity of histopathology in this condition (from normal to severe ATN) is striking.
Abbreviations
AKI = acute kidney injury; ATN = acute tubular necrosis.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
CL and SMB designed the study protocol, performed the literature search, evaluated studies, extracted data, analysed data and wrote the manuscript. RB and CNM aided in the study design, and provided critical review of successive drafts of the manuscript.
Acknowledgments
Acknowledgements
The authors thank Ms Marie Cousinery for her assistance with retrieving relevant articles.
Contributor Information
Christoph Langenberg, Email: christoph.langenberg@gmail.com.
Sean M Bagshaw, Email: bagshaw@ualberta.ca.
Clive N May, Email: c.may@hfi.unimelb.edu.au.
Rinaldo Bellomo, Email: rinaldo.bellomo@med.monash.edu.au.
References
- Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C, Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294:813–818. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- Bagshaw S, George C, Bellomo R, ANZICS Database Management Committee Changes in the incidence and outcome for early acute kidney injury in a cohort of Australian intensive care units. Crit Care. 2007;11:R68. doi: 10.1186/cc5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagshaw SM, George C, Bellomo R. Early acute kidney injury and sepsis: a multicentre evaluation. Crit Care. 2008;12:R47. doi: 10.1186/cc6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351:159–169. doi: 10.1056/NEJMra032401. [DOI] [PubMed] [Google Scholar]
- Lameire N, Van Biesen W, Vanholder R. Acute renal failure. Lancet. 2005;365:417–430. doi: 10.1016/S0140-6736(05)17831-3. [DOI] [PubMed] [Google Scholar]
- Langenberg C, Bellomo R, May CN, Egi M, Wan L, Morgera S. Renal vascular resistance in sepsis. Nephron Physiol. 2006;104:p1–p11. doi: 10.1159/000093275. [DOI] [PubMed] [Google Scholar]
- Langenberg C, Bellomo R, May C, Wan L, Egi M, Morgera S. Renal blood flow in sepsis. Crit Care. 2005;9:R363–R374. doi: 10.1186/cc3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334:1448–1460. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- Mustonen J, Pasternack A, Helin H, Pystynen S, Tuominen T. Renal biopsy in acute renal failure. Am J Nephrol. 1984;4:27–31. doi: 10.1159/000166768. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- Diaz de Leon M, Moreno SA, Gonzalez Diaz DJ, Briones GJ. Severe sepsis as a cause of acute renal failure. Nefrologia. 2006;26:439–444. [PubMed] [Google Scholar]
- Rosenberg IK, Gupta SL, Lucas CE, Khan AA, Rosenberg BF. Renal insufficiency after trauma and sepsis. A prospective functional and ultrastructural analysis. Arch Surg. 1971;103:175–183. doi: 10.1001/archsurg.1971.01350080091013. [DOI] [PubMed] [Google Scholar]
- Sato T, Kamiyama Y, Jones RT, Cowley RA, Trump BF. Ultrastructural study on kidney cell injury following various types of shock in 26 immediate autopsy patients. Adv Shock Res. 1978;1:55–69. [PubMed] [Google Scholar]
- Zappacosta AR, Ashby BL. Gram-negative sepsis with acute renal failure. Occurrence from acute glomerulonephritis. JAMA. 1977;238:1389–1390. doi: 10.1001/jama.238.13.1389. [DOI] [PubMed] [Google Scholar]
- Coalson JJ, Archer LT, Benjamin BA, Beller-Todd BK, Hinshaw LB. A morphologic study of live Escherichia coli organism shock in baboons. Exp Mol Pathol. 1979;31:10–22. doi: 10.1016/0014-4800(79)90003-0. [DOI] [PubMed] [Google Scholar]
- Coalson JJ, Benjamin B, Archer LT, Beller B, Gilliam CL, Taylor FB, Hinshaw LB. Prolonged shock in the baboon subjected to infusion of E. coli endotoxin. Circ Shock. 1978;5:423–437. [PubMed] [Google Scholar]
- Carraway MS, Welty-Wolf KE, Miller DL, Ortel TL, Idell S, Ghio AJ, Petersen LC, Piantadosi CA. Blockade of tissue factor: treatment for organ injury in established sepsis. Am J Respir Crit Care Med. 2003;167:1200–1209. doi: 10.1164/rccm.200204-287OC. [DOI] [PubMed] [Google Scholar]
- Welty-Wolf KE, Carraway MS, Miller DL, Ortel TL, Ezban M, Ghio AJ, Idell S, Piantadosi CA. Coagulation blockade prevents sepsis-induced respiratory and renal failure in baboons. Am J Respir Crit Care Med. 2001;164:1988–1996. doi: 10.1164/ajrccm.164.10.2105027. [DOI] [PubMed] [Google Scholar]
- Linton AL, Walker JF, Lindsay RM, Sibbald WJ. Acute renal failure and tubular damage due to sepsis in an animal model. Proc Eur Dial Transplant Assoc Eur Ren Assoc. 1985;21:837–842. [PubMed] [Google Scholar]
- Hinshaw LB, Taylor FB, Jr, Chang AC, Pryor RW, Lee PA, Straughn F, Murray CK, Flournoy DJ, Peer GT, Kosanke SD. Staphylococcus aureus-induced shock: a pathophysiologic study. Circ Shock. 1988;26:257–265. [PubMed] [Google Scholar]
- Beauchamp D, Labrecque G. Aminoglycoside nephrotoxicity: do time and frequency of administration matter? Curr Opin Crit Care. 2001;7:401–408. doi: 10.1097/00075198-200112000-00006. [DOI] [PubMed] [Google Scholar]
- Parker MM, Shelhamer JH, Natanson C, Alling DW, Parrillo JE. Serial cardiovascular variables in survivors and nonsurvivors of human septic shock: heart rate as an early predictor of prognosis. Crit Care Med. 1987;15:923–929. doi: 10.1097/00003246-198710000-00006. [DOI] [PubMed] [Google Scholar]
- Thijs A, Thijs LG. Pathogenesis of renal failure in sepsis. Kidney Int Suppl. 1998;66:S34–S37. [PubMed] [Google Scholar]
- Winslow EJ, Loeb HS, Rahimtoola SH, Kamath S, Gunnar RM. Hemodynamic studies and results of therapy in 50 patients with bacteremic shock. Am J Med. 1973;54:421–432. doi: 10.1016/0002-9343(73)90038-7. [DOI] [PubMed] [Google Scholar]
- Villazon SA, Sierra UA, Lopez SF, Rolando MA. Hemodynamic patterns in shock and critically ill patients. Crit Care Med. 1975;3:215–221. doi: 10.1097/00003246-197511000-00002. [DOI] [PubMed] [Google Scholar]
- Mathiak G, Szewczyk D, Abdullah F, Ovadia P, Feuerstein G, Rabinovici R. An improved clinically relevant sepsis model in the conscious rat. Crit Care Med. 2000;28:1947–1952. doi: 10.1097/00003246-200006000-00043. [DOI] [PubMed] [Google Scholar]
- Koo DJ, Zhou M, Chaudry IH, Wang P. The role of adrenomedullin in producing differential hemodynamic responses during sepsis. J Surg Res. 2001;95:207–218. doi: 10.1006/jsre.2000.6013. [DOI] [PubMed] [Google Scholar]
- Wang P, Chaudry IH. Mechanism of hepatocellular dysfunction during hyperdynamic sepsis. Am J Physiol. 1996;270:R927–R938. doi: 10.1152/ajpregu.1996.270.5.R927. [DOI] [PubMed] [Google Scholar]
- Langenberg C, Wan L, Bagshaw SM, Egi M, May CN, Bellomo R. Urinary biochemistry in experimental septic acute renal failure. Nephrol Dial Transplant. 2006;21:3389–3397. doi: 10.1093/ndt/gfl541. [DOI] [PubMed] [Google Scholar]
- Langenberg C, Wan L, Egi M, May CN, Bellomo R. Renal blood flow in experimental septic acute renal failure. Kidney Int. 2006;69:1996–2002. doi: 10.1038/sj.ki.5000440. [DOI] [PubMed] [Google Scholar]
- Langenberg C, Wan L, Egi M, May CN, Bellomo R. Renal blood flow and function during recovery from experimental septic acute kidney injury. Intensive Care Med. 2007;33:1614–8. doi: 10.1007/s00134-007-0734-8. [DOI] [PubMed] [Google Scholar]
- Hurley RM, Nayyar RP, Goto M, Zeller WP. Renal lesions in young rats induced by Salmonella enteritidis endotoxin. Pediatr Nephrol. 1989;3:156–161. doi: 10.1007/BF00852898. [DOI] [PubMed] [Google Scholar]
- Miyaji T, Hu X, Yuen PS, Muramatsu Y, Iyer S, Hewitt SM, Star RA. Ethyl pyruvate decreases sepsis-induced acute renal failure and multiple organ damage in aged mice. Kidney Int. 2003;64:1620–1631. doi: 10.1046/j.1523-1755.2003.00268.x. [DOI] [PubMed] [Google Scholar]
- Sato T, Kono Y, Shimahara Y, Tanaka J, Jones RT, Cowley RA, Trump BF. The pathophysiology of septic shock: acute renal failure in rats following live E. coli injection. A histochemical study of the proximal tubules. Adv Shock Res. 1982;7:61–70. [PubMed] [Google Scholar]
- Hayashi H, Imanishi N, Ohnishi M, Tojo SJ. Sialyl Lewis X and anti-P-selectin antibody attenuate lipopolysaccharide-induced acute renal failure in rabbits. Nephron. 2001;87:352–360. doi: 10.1159/000045942. [DOI] [PubMed] [Google Scholar]
- Tsao CM, Ho ST, Chen A, Wang JJ, Li CY, Tsai SK, Wu CC. Low-dose dexamethasone ameliorates circulatory failure and renal dysfunction in conscious rats with endotoxemia. Shock. 2004;21:484–491. doi: 10.1097/00024382-200405000-00014. [DOI] [PubMed] [Google Scholar]
- Kadkhodaee M, Qasemi A. Inhibition of inducible nitric oxide synthase reduces lipopolysaccharide-induced renal injury in the rat. Clin Exp Pharmacol Physiol. 2004;31:842–846. doi: 10.1111/j.1440-1681.2004.04096.x. [DOI] [PubMed] [Google Scholar]
- Zager RA, Prior RB. Gentamicin and gram-negative bacteremia. A synergism for the development of experimental nephrotoxic acute renal failure. J Clin Invest. 1986;78:196–204. doi: 10.1172/JCI112552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Faubel S, Ljubanovic D, Mitra A, Falk SA, Kim J, Tao Y, Soloviev A, Reznikov LL, Dinarello CA, Schrier RW, Edelstein CL. Endotoxemic acute renal failure is attenuated in caspase-1-deficient mice. Am J Physiol Renal Physiol. 2005;288:F997–F1004. doi: 10.1152/ajprenal.00130.2004. [DOI] [PubMed] [Google Scholar]
- Tiwari MM, Brock RW, Megyesi JK, Kaushal GP, Mayeux PR. Disruption of renal peritubular blood flow in lipopolysaccharide-induced renal failure: role of nitric oxide and caspases. Am J Physiol Renal Physiol. 2005;289:F1324–F1332. doi: 10.1152/ajprenal.00124.2005. [DOI] [PubMed] [Google Scholar]
- Gallos G, Jones DR, Nasr SH, Emala CW, Lee HT. Local anesthetics reduce mortality and protect against renal and hepatic dysfunction in murine septic peritonitis. Anesthesiology. 2004;101:902–911. doi: 10.1097/00000542-200410000-00015. [DOI] [PubMed] [Google Scholar]
- Kikeri D, Pennell JP, Hwang KH, Jacob AI, Richman AV, Bourgoignie JJ. Endotoxemic acute renal failure in awake rats. Am J Physiol. 1986;250:F1098–F1106. doi: 10.1152/ajprenal.1986.250.6.F1098. [DOI] [PubMed] [Google Scholar]
- Yokota M, Kambayashi J, Tahara H, Kawasaki T, Shiba E, Sakon M, Mori T. Renal insufficiency induced by locally administered endotoxin in rabbits. Methods Find Exp Clin Pharmacol. 1990;12:487–491. [PubMed] [Google Scholar]