

Abstract
Glutamic acid decarboxylase (GAD) is the rate-limiting enzyme for γ-aminobutyric acid (GABA) biosynthesis. Previously, we reported the presence of truncated forms of GAD in vivo and in vitro. In addition, an unidentified endogenous protease responsible for proteolytic cleavage of full-length GAD (fGAD) to its truncated form (tGAD) was also observed. In this communication, we report that μ-calpain is a good candidate for conversion of fGAD67 to tGAD67. This conclusion is based on the following observations: 1. Purified recombinant GAD67 is cleaved by μ-calpain at specific sites; 2. In brain synaptosomal preparation, GAD67 is cleaved to its truncated form by an endogenous protease which is inhibited by specific calpain inhibitors; 3. In μ-calpain knockout mice, the level of tGAD in the brain is greatly reduced compared with the wild type; 4. when μ-calpain gene is silenced by siRNA, the level of tGAD is also markedly reduced compared to the control group; 5. μ-calpain is activated by neuronal stimulation and Ca2+-influx. The physiological significance of calpain in regulation of GABA synthesis and GABAergic neurotransmission is also discussed.
Keywords: Calpain, GABA synthesis, GAD, Proteolytic Cleavage
In the central nervous system (CNS), γ-aminobutyric acid (GABA) is synthesized by a single enzymatic reaction catalyzed by L-glutamic decarboxylase (EC 4.1.1.15; GAD) (1, 2). Mammalian species express two isoforms of GAD, namely, GAD65 and GAD67, referring to GAD with a molecular weight of 65 kDa and 67 kDa, respectively. Previously, we reported the presence of the truncated GAD (tGAD) derived from proteolytic cleavage of the full-length (fGAD) in vivo as well as in vitro (3, 4). The presence of smaller forms of GAD was also observed from other laboratories (5-8). However, no information was reported regarding the identity of the endogenous proteases responsible for GAD cleavage and their physiological significance. Recently, we found that recombinant human brain GAD65 is specifically cleaved at arginine 69 from the N-terminal of fGAD65 to produce tGAD65, which is ∼2-3 fold more active than fGAD65 (3). In addition, GAD67 was found to be cleaved at two specific sites, one at arginine 70 and another at arginine 90, to produce two truncated forms of GAD67 (3). Based on these observations, it seems that the formation of tGAD is catalyzed by specific proteases instead of a random degradation of GAD. The presence of truncated GAD prompts us to investigate the identity of the protease and to determine the physiological/pathological significance of the cleavage.
One of the clues to suggest that the endogenous protease responsible for cleavage of GAD is Ca2+-dependent comes from our recent observation that the extent of GAD cleavage is markedly increased when synaptosomes are depolarized in the presence of Ca2+. Hence, we have examined the ability of calpain to cleave GAD. Calpains comprise a family of calcium-dependent, non-lysosomal, neutral, cysteine proteases that are present in most of the mammalian tissues (9). The calpain system is composed of three molecules: two Ca2+-dependent proteases, μ-calpain (Calpain 1) and m-calpain (Calpain 2) and a polypeptide, calpastatin, an endogenous specific calpain inhibitor. Both μ- and m-calpain consist of two subunits: an identical 30 kDa regulatory subunit and an 80 kDa catalytic subunit that shares about 60% homology between the two isoforms. The catalytic subunits of μ-calpain and m-calpain come from different but closely related genes (referred to as Capn 1 and Capn 2, respectively). The two forms differ in their responses to calcium in vitro. The μ-calpain is fully activated by micromolar concentrations of calcium, while the m-calpain requires a millimolar range of calcium. Calpains play an important role in various cellular processes including remodeling cytoskeletal attachments to the plasma membrane during cell fusion and cell motility, regulating signaling pathways, apoptosis and an involvement in long-term potentiation (9). The Capn1-/- mice are viable and fertile, but they have a significant reduction in platelet aggregation and clot retraction (10). In contrast, disruption of the catalytic subunit of calpain 2 caused embryonic death at an early stage (9). Transgenic mice that lack the regulatory subunit die during embryonic development with defects in vascular development (11).
In this communication, evidence of μ-calpain being the protease responsible for the conversion of fGAD67 to tGAD67 in vivo as well as in vitro is presented. The physiological significance of calpain in the brain is also discussed.
Results
Cleavage of GAD67 under neuronal stimulation condition
When the rat brain synaptosomes were stimulated by 55 mM K+ and 2.2 mM Ca2+ for 5 minutes, the extent of the conversion of fGAD67 to the truncated form was increase in comparison with the non-stimulation condition (Fig 1A). The amounts of the truncated GAD67 were significantly increased under stimulation for an additional 5 min (Fig. 1A). When 2 mM EDTA/EGTA was present during the period of stimulation, no significant cleavage was observed (Fig. 1A), suggesting that the cleavage of GAD67 in rat brain is an extracellular Ca2+-dependent process. Similar results were obtained when 10 μM of calpain inhibitor I was included (Fig. 1A), suggesting that calpain might be the endogenous protease responsible for the proteolytic cleavage of GAD67. The truncated form of GAD67 in rat brain synaptosomes has the same gel-mobility on SDS-PAGE as hGAD67(Δ1-70) (Fig. 1A). It is interesting that there was only one truncated form of GAD67 identified. No cleavage product with a similar molecular weight of hGAD67(Δ1-90) was detected.
Figure 1. Effect of neuronal stimulation on cleavage of GAD67 in rat brain synaptosome preparation.
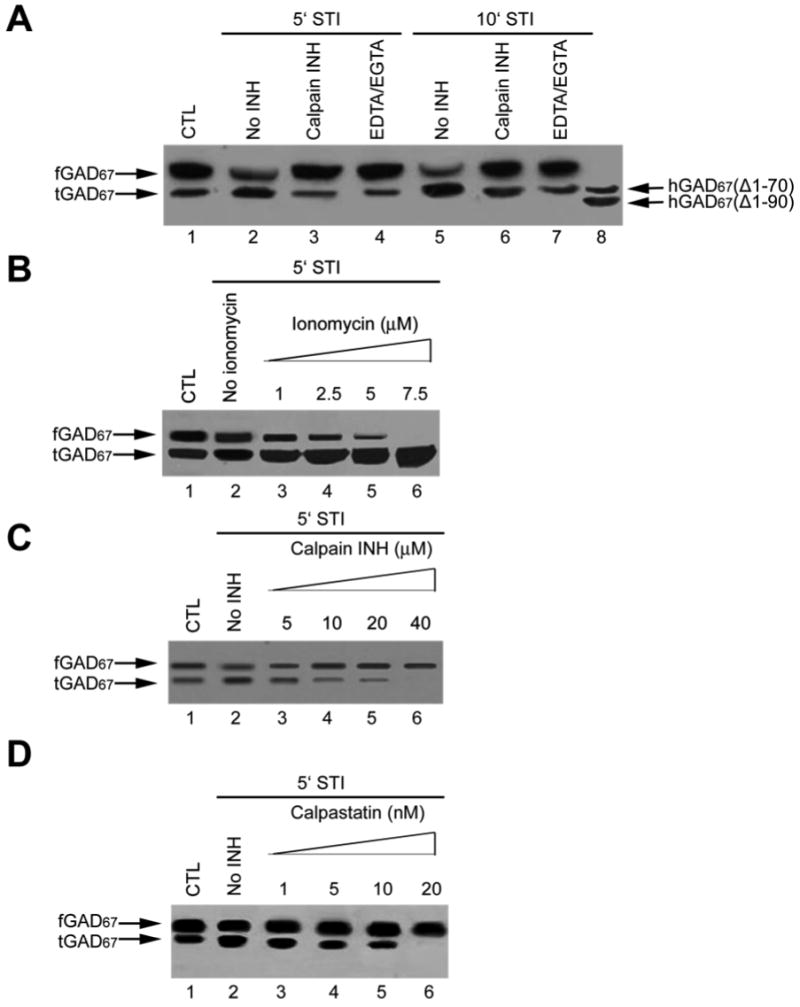
Synaptosome was prepared and stimulated as described. A. Effect of EDTA/EGTA and calpain inhibitor I on stimulation-induced cleavage of GAD67. Lane 1: Synaptosomal soluble fraction under the non-stimulation condition; Lane 2: Synaptosomal soluble fraction under the stimulation condition for 5 min; Lane 3: Same as lane 2 except that the sample was incubated with 2 mM of EDTA /EGTA before the stimulation; Lane 4: Same as lane 2 except that the sample was incubated with 10 μM of calpain inhibitor I before the stimulation; Lane 5: Synaptosomal soluble fraction under the stimulation condition for 10 min; Lane 6: Same as lane 2 except that the sample was incubated with 2 mM of EDTA /EGTA before the stimulation; Lane 7: Same as lane 2 except that the sample was incubated with 10 μM of calpain inhibitor I before the stimulation; Lane 8: Mixture of recombinant hGAD67(Δ1-70) and hGAD67(Δ1-90); B. Effect of ionomycin on cleavage of GAD67 under stimulation condition. Lane 1: Synaptosomal soluble fraction under the non-stimulation condition; Lane 2: Synaptosomal soluble fraction under the stimulation condition for 5 min; Lanes 3-6: same as Lane 2 except that a different concentration of ionomycin is included during the stimulation reaction; C. Effect of calpain inhibitor I on cleavage of GAD67. Lane 1: Synaptosomal soluble fraction under the non-stimulation condition; Lane 2: Synaptosomal soluble fraction under the stimulation condition for 5 min; Lanes 3-6: same as Lane 2 except that synapsome is pre-incubated with a different concentration of calpain inhibitor I; D. Effect of calpastatin on cleavage of GAD67. Lane 1: Synaptosomal soluble fraction under the non-stimulation condition; Lane 2: Synaptosomal soluble fraction under the stimulation condition for 5 min; Lanes 3-6: same as Lane 2 except that the synaptosome is pre-incubated with different concentrations of calpastatin. For each panel, upper arrow indicated the position of fGAD67 and lower arrow indicated the position of tGAD67.
In order to further prove that the cleavage of GAD67 is Ca2+-dependent, effect of ionomycin, a Ca2+ ionophore, on GAD67 cleavage was tested. It was found that the conversion of fGAD67 to tGAD67 was increased with increasing concentrations of ionomycin in a dose-dependent manner (Fig. 1 B).
To further determine if calpain is involved in GAD67 cleavage, the synaptosome was treated with increasing amounts of cell-permeable calpain inhibitors before stimulation. The conversion of fGAD67 to tGAD67 was markedly inhibited by calpain inhibitor I at 5 μM (Fig. 1C) and by calpastatin peptide in a nanomolar range (Fig. 1D), further supporting that calpain might be responsible for the cleavage of GAD67.
Cleavage of hGAD67, hGAD67 (Δ1-70) and hGAD67 (Δ1-90) by μ-calpain in vitro
Recombinant hGAD67 was found to be cleaved by μ-calpain in the presence of 100 μM Ca2+ (Fig. 2A). The conversion resulted in a 50% loss of GAD activity (Fig. 2A), which was compatible with our previous finding that truncated hGAD67 was less active than the full-length form (4). The cleavage and activity change were abolished by 2 mM EDTA/EGTA or 5 μM of calpain inhibitor I (Fig. 2A). In contrast, both hGAD67 (Δ1-70) and hGAD67 (Δ1-90) were not cleaved by μ-calpain along with Ca2+ (Fig. 2B and 2C).
Figure 2. Cleavage of recombinant hGAD67 by μ-calpain in vitro.
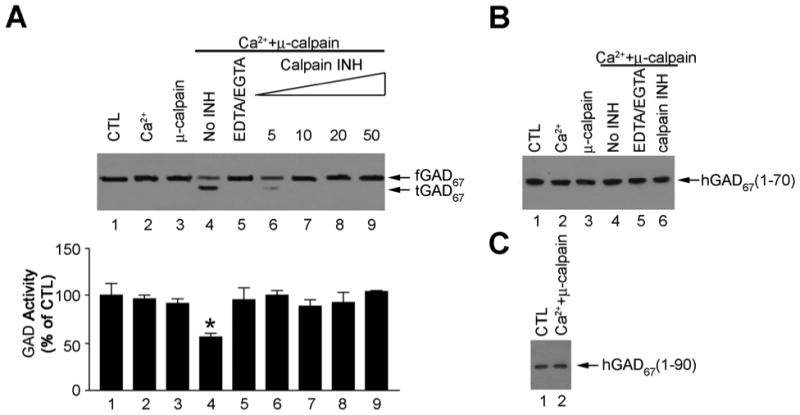
A. Cleavage of recombinant full-length hGAD67. A1 : Immunoblotting test: Lane 1: full-length hGAD67; Lane 2: full-length hGAD67 with 100 μM Ca2+ alone; Lane 3: full-length hGAD67 with μ- calpain (0.15U) alone; Lane 4: full-length hGAD67 with 100 μM Ca2+ and μ-calpain (0.15U); Lane 5: same as lane 4 except that 2 mM EDTA/EGTA was included; Lane 6: same as lane 4 except that 5 μM calpain inhibitor I was included; Lane 7: same as lane 4 except that 10 μM calpain inhibitor I was included; Lane 8: same as lane 4 except that 20 μM calpain inhibitor I was included; Lane 9: same as lane 4 except that 50 μM calpain inhibitor I was included. Upper bands indicated fGAD67 and lower bands indicated tGAD67; A2: GAD activity assay: GAD activity in standard GAD buffer was taken as 100%. Bars indicate the SD with n = 3; B: Effect of μ-calpain treatment on hGAD67 (Δ1-70). Lane 1: hGAD67(Δ1-70) alone; Lane 2: hGAD67 (Δ1-70) with 100 μM Ca2+ alone; Lane 3: hGAD67(Δ1-70) with μ-calpain (0.15U) alone; Lane 4: hGAD67(Δ1-70) with 100 μM Ca2+ and μ-calpain (0.15U); Lane 5: same as lane 4 except that 2 mM EDTA/EGTA was included; Lane 6: same as lane 4 except that 10 μM calpain inhibitor I was included; C: Effect of μ-calpain treatment on hGAD67 (Δ1-90). Lane 1: hGAD67(Δ1-90) alone; Lane 2: hGAD67(Δ1-90) with 100 μM Ca2+ and μ-calpain (0.15U).
Sandwich Enzyme-Linked ImmunoSorbent-Assay (SELISA)
The SELISA experiment was used to detect the endogenous protease activity in porcine brains. The principle is that the conversion of GST-fGAD67 to tGAD67 by a protease should cause a decrease in absorbance at 450 nm. The endogenous protease which was responsible for cleavage of recombinant GAD67 was found to be concentrated in combined pools, C2 and C3 as indicated in the decrease of absorbance at 450 nm (Fig. 3B and 3C). It is interesting that μ-calpain detected by an immunoblotting test with anti-μ-calpain was only found in C1-C3, especially in C2 and C3 (Fig. 3D, lanes 2 and 3), the same fractions as the endogenous protease identified by SELISA. These observations are consistent with the idea that calpain may be the endogenous protease for GAD cleavage in the brain. The amount of μ-calpain in C1 and C4 fractions was not comparable with their attenuated absorbance (Fig. 3B and 3D), suggesting the presence of other proteases in the brains that are able to GAD.
Figure 3. Detection of protease activity in proteolytic cleavage of GST-fGAD67 to tGAD67 using sandwich ELISA (SELISA) method.
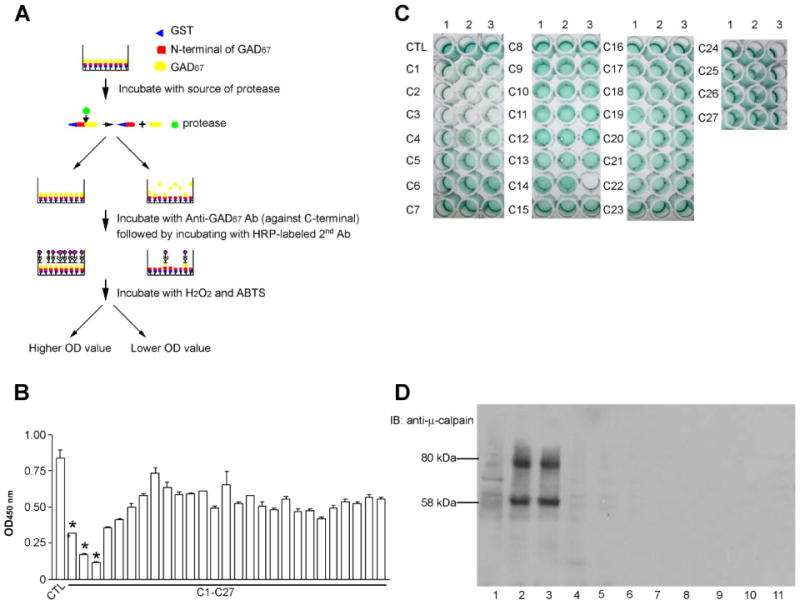
The assay is carried out as described in the text. GST-fGAD67 was used as the substrate and incubated with various combined fractions from the Sephadex G-200 column. Every five fractions from # 110-245 were combined into one combined pool as C1, C2…. C27. Decrease of the substrate, GST-fGAD67, as reflected in decreased of OD at 450 nm is a measurement of protease activity. A. Principle of the methods; B. OD value reading of 96-wells plate: * indicates pools containing high protease activity. Bars indicate the SD with n = 3; C. Picture of 96 –wells plate. Every three columns in each row represent the same group. For instance, a1, a2 and a3 were CTL. Arrow indicates the pool containing high protease activity; D. Immunoblotting test against anti-μ-calpain. Lanes 1-5: C1-C5. Lanes 6-11: C8, C12, C16, C20, C24, C27, respectively.
Effect of μ-calpain KO (Capn 1 -/-) on GAD cleavage
In order to further demonstrate the role of μ-calpain in GAD cleavage, Capn1 (μ-calpain) KO and the wild type (WT) mice were used for the investigations. In the brain soluble fraction, the amount of truncated GAD67 was greatly decreased in Capn 1-/- mice compared to the WT (Fig. 4B). No truncated GAD65 was found in either WT or KO mice (Fig. 4B). In the solubilized membrane extract, both truncated GAD65 and truncated GAD67 were not detected in KO mice (Fig. 4D). Equal loading of the protein was demonstrated by immunoblotting analysis of soluble and pellet fractions using anti-actin antibody (Fig 4E). The positive staining of the sample from Capn 1-/- mice by polyclonal anti-μ-calpain antibodies (Fig. 4A and 4C) was due to the cross activity between m-calpain and anti-μ-calpain antibodies. However, the RT-PCR data clearly showed the complete deletion of mRNA of μ-calpain in brain tissue (Fig. 4F). RT-PCR analysis carried out with specific primers targeted to different regions of calpain-1 confirmed the loss of calpain-1 RNA message in calpain-1 null mice. Primer pairs Cal 1F (boundary exon 2-3) and Cal 2R (exon 9), Cal 1F and Cal 3R (exon 5), and Cal 4F (exon 4) and Cal 5R (exon 5) yielded products of expected sizes corresponding to 615 bp (lane 1), 248 bp (lane 3), and 171 bp (lane 6) in the wild type mice whereas the corresponding product was not detected in the calpain-1 null mice (Fig. 4F). A similar analysis for calpain-2 and GAPDH using specific primers revealed no differences in their expression between wild type and calpain-1 null mice (Fig. 4F). Real-time quantitative PCR analysis confirmed the loss of calpain-1 message in the calpain-1 null mouse brain tissue whereas no difference in the expression of calpain-2 was observed between wild type and calpain-1 null mice. Since the primer pair used for calpain-1 resulted in some primer-dimer formation (Fig. 4F), the residual signal as seen in the calpain-1 null mice (Fig. 4F) presumably originates from the primer-dimer formation, which was later confirmed using same primers when used in the absence of any cDNA (data not shown).
Figure 4. Effect of μ-calpain knockout (KO) on GAD cleavage in mice.

μ-calpain knockout (KO) mice brains were processed as described. A. Immunoblotting analysis of brain soluble extract using anti-μ-calpain. B. Immunoblotting analysis of brain soluble extract using anti-GAD antibodies. C and D were same as A and B except that solubilized membrane extract was used; E. RT-PCR analysis. E1: RT-PCR analysis of wild type and calpain-1 null mice. Control lanes show results of reactions in which no RNA was added. These lanes provide evidence for primer-dimer formation in the absence of RNA (lanes 5, 8) both in wild type and calpain-1 null reactions (lanes 3, 4, 6, 7); E2: RT-PCR analysis of wild type and calpain-1 null mice using calpain-2 specific primers. The GAPDH specific primers served as internal controls to normalize the amount of RNA; E3: Bar graph represents real-time quantitative PCR analysis of calpain-1 and calpain-2 message in wild type and calpain-1 null mouse brain RNA. Data represent mean ± SEM (N=3).
Effect of silencing μ-calpain on GAD cleavage in neuronal cultures
In order to further rule out the possibility that in addition to μ-calpain, m-calpain may also be involved in GAD cleavage in vivo, an RNAi experiment was designed and carried out in primary neuronal cultures. First, three pairs of siRNA duplexes were selected for either Capn1 or Capn2 and obtained commercially (Invitrogen). Among the siRNAs for μ-calpain (Capn1-1, 1-2, 1-3, respectively), Capn 1-1 gave the best silencing results, showing no truncated GAD67 in the Capn 1-1 siRNA treatment group (Fig. 5). The other two siRNA duplexes also caused silencing of μ-calpain, but to a lesser extent as Capn1-1 based on the formation of tGAD67 (Fig.). It is interesting that three pairs of siRNA duplexes for Capn 2 completely silenced the m-calpain expression observed from the immunoblotting test (Fig. 5). The Capn 2 siRNAs treated group showed no difference in the amount of tGAD67 produced (Fig. 5). Based on these observations, Capn 1-1 and Capn 2-3 were selected for further investigation.
Figure 5. Selection of siRNA duplex for silencing calpain in primary neuronal cultures.

A. Sequence of siRNA duplexes for Capn 1 (μ-calpain) and Capn 2 (m-calpain); B. Detection of calpain silencing by immunoblotting. 8 DIV neuronal cultures were transfected by siRNA duplex as indicated using lipofection method. 48 hr after transfection, cells were lysed and extract was applied to immunoblotting against anti-μ-calpain and m-calpain antibodies. C. same as B except that anti-GAD67 was used. Upper bands indicated fGAD67 and lower bands indicated tGAD67.
Transfection of Capn 1-1 and Capn 2-3 inhibited the expression of capn 1 and capn 2 at both protein and mRNA levels (Fig. 6B and 6C). Silencing of capn 1 attenuated the amount of tGAD67 and tGAD65 in the neuronal cultures (Fig. 6A). When the cell cultures were stimulated by high K+, tGAD was significantly enhanced, which was antagonized by 10 nM calpastatin (Fig. 6A). However, silencing capn1 abolished the conversion of GAD to truncated form caused by stimulation conditions, whereas silencing capn 2 didn't have the same effect (Fig. 6A). In addition, silencing either capn 1 or capn 2 had no effect on the mRNA level of both GAD65 and GAD67 (Fig. 6C). These observations suggest that μ-calpain, but not m-calpain, is the protease responsible for conversion of GAD to the truncated form in neuronal cultures.
Figure 6. Effect of silencing Capn 1 and Capn 2 on GAD cleavage in neuronal cultures.

8 DIV neuronal cultures were transfected by siRNA duplex and using the lipofection method. Capn 1-1 and Capn 2-3 siRNA control were included. 48 hr after transfection, cells were treated as indicated and lysed. Cell extract was applied to immunoblotting analysis. A: Immunoblotting test using anti-GAD67 and anti-GAD65.; B: Immunoblotting test using anti-μ-calpain and anti-m-calpain. Lane1: Mock; Lane 2: Capn 1-1 CTL; Lane 3: Capn 1-1; Lane 4: Capn 2-3 CTL; Lane 5: Capn 2-3; C: RT-PCR results. For individual primers, Lane 1: mock; Lane 2: Capn 1-1 control; Lane 3: Capn 1-1; Lane 4: Capn 2-3 control; Lane 5: Capn 2-3. Glyceraldehyde 3-phosphate dehydrogenase (G3PDH) was included as a control to normalize the samples.
Effect of GAD phosphorylation on calpain cleavage
Previously we have reported that recombinant hGAD67 was phosphorylated by PKA and recombinant hGAD65 was phosphorylated by PKCε (18). In order to determine role of GAD phosphorylation on calpain cleave, we performed calpain cleavage assay following phosphorylation reaction. Phosphorylation of recombinant hGAD67 by PKA and recombinant hGAD65 by PKCε were confirmed by immunoblotting probed with anti-phospho-ser/thr (Fig. 7A and 7B). After phosphorylation by PKA, hGAD67 cleavage by μ-calpain was strongly inhibited (Fig. 7A). In contrast, phosphorylation of hGAD65 by PKCε facilitates the μ-calpain cleavage (Fig. 7B). In addition, the truncated hGAD65 contained phospho-ser/thr, indicating that the cleavage site must be before the phosphorylation site for PKCε (Fig.7B).
Figure 7. Effect of GAD phosphorylation on calpain cleavage.
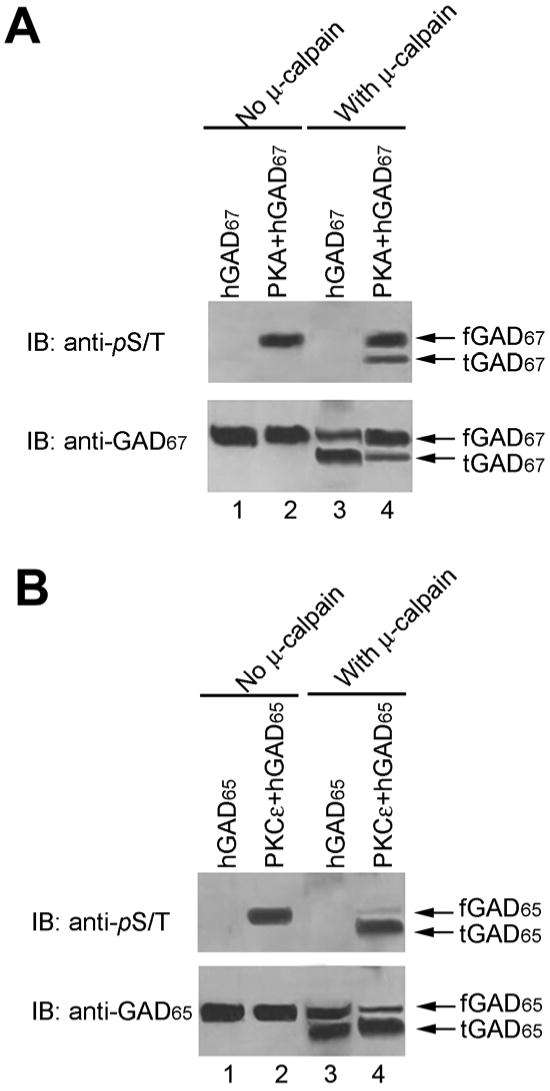
Cleavage assay was carried out after recombinant hGAD67 and hGAD65 were phosphorylated by PKA and PKCε, respectively, as described. A. Effect of PKA phosphorylation on hGAD67 cleavage. Lane 1: hGAD67 CTL; Lane 2: phosphorylated hGAD67 by PKA; Lane 3: hGAD67 CTL cleaved by calpain; Lane 4: phosphorylated hGAD67 cleaved by calpain. B. Effect of PKCε phosphorylation on hGAD65 cleavage. Lane 1: hGAD65 CTL; Lane 2: phosphorylated hGAD65 by PKCε; Lane 3: hGAD65 CTL cleaved by calpain; Lane 4: phosphorylated hGAD65 cleaved by calpain;
Discussion
An important mechanism involved in the regulation of enzyme activity is through proteolytic cleavage of enzyme proteins. For instance, protein tyrosine phosphatases (PTPs) is regulated by a proteolytic mechanism and the cleaved proteins show higher activity (20). In striatum neurons, striatal tyrosine phosphatase (STEP) is proteolytically cleaved by glutamate-induced calcium influx and calpain activation. The cleaved STEP can reach its targets and perform its function. In this communication, we demonstrated that another neuronal protein, the GABA synthesizing enzyme, GAD, is also regulated by a similar mechanism involving μ-calpain. Calpain, as a mediator in Ca2+ signal transduction, plays an important role in the basic cell life processes such as cell differentiation, cell division and growth (See review, 9). In the central nervous system, the activation of calpain has been implicated in the process of neuronal damage due to global or focal cerebral ischemia (21, 22). The NMDA receptor function is down-regulated through calpain-cleavage of its NR2 subunits induced by prolonged glutamate stimulation (23). Recently, it was also reported that α-processing of Alzheimer's β-amyloid precursor protein (APP) requires μ-calpain activation (24), suggesting the possibility of involvement of μ-calpain in the pathological process of the nervous system (see review, 25).
The presence of tGAD derived from proteolytic cleavage of fGAD has been observed during the course of purification of recombinant GAD from a bacterial expression system (3, 7, 8) or from mammalian brain e.g., porcine, mouse or rat brain (5, 6). However, no detailed information was reported regarding the site of proteolytic cleavage, the properties of the truncated GAD and their physiological significance.
In this communication, we reported that calpain, particularly μ-calpain, is responsible for proteolytic regulation of GAD. This notion is supported by the following findings: Firstly, upon stimulation by high K+ and Ca2+, a portion of full-length GAD67 is converted to truncated GAD67. This reaction is Ca2+-dependent since EDTA/EGTA inhibits the cleavage and Ca2+ ionophore and ionomycin facilitates the cleavage. Secondly, the conversion is calpain-related since calpain inhibitor significantly inhibits the cleavage of recombinant human brain GAD67 by μ-calpain (Fig. 2A). Truncation of GAD67 by μ-calpain results in a marked activity loss, which is compatible with our previous findings (4). Thirdly, brain fractions responsible for cleavage of GAD are also enriched with calpain (Fig. 3 B, C and D); Fourthly, in μ-calpain KO mice brain, the amount of both truncated GAD67 and GAD65 is significantly decreased (Fig. 4B and D); Fifthly, silencing of m-calpain gene has no effect on the level of truncated GAD in the cultures, whereas silencing μ-calpain greatly decreases the conversion of full-length GAD65 and GAD67 to their respective truncated forms (Fig. 5 and 6). It is interesting that PKA-mediated GAD67 phosphorylation inhibits its cleavage by μ-calpain, while PKCε-mediated GAD65 phosphorylation facilitates the process. The physiological significance of proteolytic cleavage of GAD by μ-calpain remains unclear. It is possible that cleavage of GAD could be pathological since cleavage of GAD67 to its truncated forms reduces GAD activity. It has been shown that GAD67 is important for proper neuronal development since knock out of GAD67 is lethal (26). Hence it is reasonable to propose that cleavage of GAD67 may result in neuronal injury, especially during brain development. On the other hand, the following observations favor a physiological role of proteolytic cleavage of GAD. Firstly, since neurotransmitter GABA is synthesized primarily by SV-associated GAD65, but not cytosolic GAD67, the conversion of fGAD65 to tGAD65 may enhance GABA transmission because of increase of GABA synthesis by tGAD65; Secondly, the proteolytic cleavage of GAD is dependent upon neuronal activity. It is likely that increase in GABA synthesis by tGAD65 would replenish GABA released during neuronal activity.
Based on these observations together with our previous observation of regulation of GAD by protein phosphorylation (18), a model leading from neuronal stimulation to regulation of GABA neurotransmission is proposed: When GABAergic neurons are stimulated, there is an increase in calcium influx (step 1). Elevation of calcium activates calpain (step 2). Activated calpain then converts fGAD65 to tGAD65 (step 3). The activated tGAD65 could increase the GABA biosynthesis to meet the high demand of GABA release upon stimulation (step 4). In addition, increased intracellular Ca2+ level will also facilitate translocation of both PKCε and GAD65 to SV (step 5), resulting in phosphorylation and activation of GAD65. Phosphorylated GAD65 is then cleaved more efficiently to tGAD65 (step 6) resulting in further facilitation of GABA synthesis and GABA transmission (step 7). The cleavage of GAD67 by calpain to tGAD67 will decrease GAD67 activity, which will conserve L-glutamate as substrate for synthesis of neurotransmitter GABA by GAD65 (step 8). A similar regulation mechanism has been found for striatal tyrosine phosphatase (STEP) which is enriched within neurons of the striatum (27). A number of other protein tyrosine phosphatases (PTPs) have a similar regulatory mechanism (20, 28, 29), and some of the PTPs show enhanced activity by proteolysis (20, 29) which is similar to what is described here for tGAD65.
In summary, GAD is regulated by μ-calpain and Ca2+-dependent proteolytic cleavage in the brain as well as in neuronal cultures. In addition, regulation of GAD by protein phosphorylation alters its reaction to μ-calpain cleavage. There is an interaction between these two regulation mechanisms to provide GABA synthesized by SV-associated GAD65 for neurotransmission and GABA synthesized by GAD67 for other functions. We propose that proteolytic cleavage of GAD could be either physiology or pathological. Further studies using μ-calpain knockout mice may provide more definitive conclusion regarding the physiological significance of proteolytic cleavage of GAD. If μ-calpain plays an important role in the regulation of GABA transmission by converting GAD65 to a much more active tGAD65 resulting in increased GABA synthesis and packaging into SV, the deficiency of GABA transmission should be detected in μ-calpain knockout mice. In addition, if cleavage of GAD by μ-calpain results in enhancement of GABA neurotransmission, one would expect that μ-calpain-/- mice would exhibit some behavioral signs of deficiency in GABA transmission such as susceptibility to seizure or anxiety as the GAD65-/- mice (30, 31). These aspects of functional and behavioral studies are currently under investigation.
Experimental Procedures
Materials
Protein inhibitor cocktail, ionomycin, μ-calpain, calpain inhibitor I, polyclonal anti-μ-calpain, polyclonal anti-m-calpain, goat anti-rabbit IgG conjugated with horseradish peroxidase (HRP), ABTS [2,2′-azino-bis (3-ethylbenziazoline-6-sulfonic acid)], and Igepal CA 630 were purchased from Sigma (St. Louise, MO). Calpastatin peptide was purchased from Calbiochem (San Diego, CA). Lipofetamine, Opti-MEM medium, TRIZOL and the cDNA synthesis kit were from Invitrogen (Calsbad, CA). pfu polymerase was purchased from Stratagen (La Jolla, CA). Goat anti-glutathoine-S-transferase (GST), L-[14C] glu-tamic acid and the ECL™ western blotting analysis system were purchased from Amersham Biosciences (Piscataway, NJ). Pregnant Sprague-Dawley rats were purchased from Harland Sprague-Dawley (Indianapolis, IN). The protein kinase A (PKA) assay kit, protein kinase C (PKC) assay kit, recombinant PKA catalytic subunit, PKCε isoforms and anti-phosphpo-ser/thr antibodies were purchased from Upstate Biotechnology (Lake Placid, NY). Rabbit polyclonal anti-GAD67 and anti-GAD65 were produced in our lab. Fresh porcine brains were purchased from a local slaughter house. Mouse brains of μ-calpain knockout (KO) (Capn1-/-) mice were produced and characterized as described (Azam et al., 2001).
Methods
Preparation of crude synaptosome
Preparation of crude synaptosome was performed as described previously (12, 13). Unless otherwise specified, all procedures were carried out at 4°C. Briefly, fresh rat brains were homogenized in 0.32M sucrose, and the homogenate was centrifuged at 1,000 × g for 10 minutes. The supernatant solution was collected and centrifuged at 12,000 × g for 30 minutes. The resulting supernatant liquid was discarded and the pellet was gently suspended in Krebs-Ringer phosphate buffer (123mM NaCl, 3mM KCl, 0.4mM MgCl2, 0.5mM NaH2PO4, 0.25mM Na2HPO4, and 1mg/ml glucose, pH 7.4) to serve as the crude synaptosome preparation.
Stimulation of synaptosome
Aliquots of freshly prepared synaptosomes were stimulated by the addition of KCl and CaCl2 with final concentrations of 55 mM and 2.2 mM, respectively, at 37°C. For drug treatment, synaptosomes were first incubated with EGTA/EDTA, calpain inhibitor I, or calpastatin at 37°C for 15 minutes prior to the stimulation. Ionomycin was added to give the concentrations as indicated to stimulate the synaptosomes. After stimulation, the suspension was sonicated and centrifuged at 100,000 × g for 1 hour. The supernatant was collected and used as the source of cytosolic GAD, which is mostly GAD67.
Purification of recombinant hGAD65, hGAD67, hGAD67 (Δ1-70) and hGAD67 (Δ1-90)
Cloning, expression and purification of full-length hGAD65 and hGAD67 were conducted as described previously (3, 14). Briefly, recombinant full-length hGAD65 or hGAD67 was expressed as a glutathione-S-transferase (GST) fusion protein in E coli DH5α using a pGEX-3X vector, and GST fusion proteins were purified by glutathione affinity column. After the GST tag was removed by Factor Xa, free GAD protein was further purified by a second glutathione affinity column. hGAD67 (Δ1-70) and hGAD67 (Δ1-90) were subcloned in a frame into a pGEX-6P-1 vector and expressed as a GST fusion protein in E.coli BL21. The recombinant proteins were purified as described above except that the GST tag was removed by PreScission protease cleavage as described (4).
Calpain cleavage assay in vitro
Recombinant hGAD67, hGAD67 (Δ1-70) and hGAD67 (Δ1-90) were incubated with μ-calpain at 30°C for 30 minutes. The reaction was stopped by the addition of SDS sample buffer. Samples were analyzed by SDS-PAGE and immunoblotting as described (4).
Sandwich Enzyme-Linked ImmunoSorbent-Assay (SELISA)
We have developed a SELISA method to detect endogenous protease activity in porcine brains. Briefly, the ELISA 96 well plate was first coated with goat anti-GST antibodies (1:5000) in coating solution (100mM Na2CO3, pH 9.2) at 4°C overnight. The wells were further blocked in blocking buffer consisting of phosphate buffered saline (PBS) with 0.1% Tween 20 (PBS-T buffer) containing 5% milk, pH 7.4 at 22°C for 2 hours. The plate was then washed in PBS-T buffer three times. GST-fGAD67 was added to each well and incubated with the coated anti-GST for 2 hours at 22°C. After extensive washing, an aliquot containing endogenous protease was added to the wells and incubated at 4° overnight, followed by extensive washing with PBS-T buffer. The first antibody, anti-GAD67, diluted in blocking buffer (1:3000) was added and incubated at 22°C for 1 hour followed by washing. Although anti-GAD67 would recognize both of the substrates, the uncleaved GST-fGAD67 and the cleaved product, tGAD67, only the uncleaved GST-fGAD67 would bind to the coated anti-GST on the ELISA plate. Hence, GST-fGAD67 and anti-GAD67 complex could be detected by using the secondary antibody, goat anti-rabbit IgG conjugated with horseradish peroxidase (HRP), followed by a colorimetric determination of peroxidase reaction products. Briefly, the secondary antibody, goat anti-rabbit IgG conjugated with HRP, diluted in blocking buffer (1:4000) was added and incubated at 22°C for an additional 1 hour, followed by washing. The reaction was determined by adding a mixture of HRP substrate which is composed of ABTS [2,2′-azino-bis (3-ethylbenziazoline-6-sulfonic acid)] and H2O2 according to the ratio (1000:1). The color was developed in 5-20 minutes, and the absorbance was read with an ELISA plate reader at 450 nm. Since the SELISA method used recognized the uncleaved GST-fGAD67 protein, the decrease in absorbance indicated the conversion of fGAD67 to tGAD67, an indication of protease activity.
Sephadex G-200 gel filtration was carried out as described (3) and every five fractions from # 110- # 244 were combined. These combined fractions were referred to as C1, C2…C27, as the source of endogenous protease.
Preparation of brain tissue extractions from μ-calpain KO mice
Frozen brains of μ-calpain KO mice were first homogenized in standard GAD buffer (50 mM potassium phosphate, 1mM AET, 0.2 mM PLP, pH 7.2) with protease inhibitor cocktail, followed by sonication. The suspension was centrifuged at 100,000 × g for 1 hour. The supernatant was collected as a source of soluble fraction. The pellet was resuspended in GAD buffer and solubilized by 1% Triton X-100 overnight at 4°C. The Triton X-100 was further removed by SM-2 adsorbent beads (Bio-Rad) as described (15).
RT-PCR from μ-calpain KO brain total RNA
Total RNA was isolated from 200 mg of brain tissue from the wild type and μ-calpain mutant mice (N=3). Frozen brain tissue was homogenized in the Dounce homogenizer, mixed with 4.0 ml TRIzol® reagent (GIBCOBRL), and processed according to the manufacturers' protocol for the isolation of total RNA. Total brain RNA (2.0 μg) from wild type and μ-calpain mutant animal was analyzed by one-step RT-PCR kit (Qiagen). The μ-calpain gene specific primers targeted to different regions are designated as Cal 1F (exon 2-3 boundary), Cal 2R (exon 9), Cal 3R (exon 5), Cal 4F (exon4) and Cal 5R (exon 5). The m-calpain primers were targeted to exon 12 (Calp 2F) and exon 13 (Calp 2R). Internal control primers, GAPDH-F and GAPDH-R, for the housekeeping gene GAPDH were used under the same experimental conditions.
Real-time quantitative PCR
Total RNA (1.0 μg) was isolated individually from the brain tissue of 3 wild type and 3 μ-calpain null mice, and reverse transcribed in a 20 μl reaction using the ImProm-II™ Reverse Transcription System Kit from Promega Corporation, USA. All samples including the negative RT controls were prepared using a common reaction mixture. Real-time quantitative PCR was performed using a cDNA equivalent of 50 ng of total RNA from wild type and calpain-1 null samples with primers specific for mouse μ-calpain (Cal 4F and Cal 5R), m-calpain (Calp 2F and Calp 2R), and a housekeeping gene GAPDH (GAPDH-F and GAPDH-R). The reaction was carried out in 25 μl using SYBR® GREEN PCR Master Mix (Applied Biosystems) according to the manufacturers' instructions. PCR was monitored using the ABI PRISM 7000 Sequence Detection System (Applied Biosystems).
Preparation of primary neuronal culture
Whole-brain primary neuronal cultures were prepared from fetal rat brains obtained from female Sprague-Dawley rats as previously described (16, 17). Briefly, brains dissected from fetal rats were mechanically dissociated in basal medium Eagle (BME) supplemented with 7.6 mM sodium bicar-bonate, 26.8 mM glucose, 2 mM glutamine, and 20% heat-inactivated fetal bovine serum. This medium was referred to as GME. Cell suspensions were centrifuged at 200 × g for 3 minutes, and the pellet was resuspended in GME (4 ml GME/brain) and plated in 24-well clusters (1 ml/well). The medium was then replaced with serum-free BME after incubation in a humidified incubator (37°C, 5% CO2) for 1–2 hours. The cultures were then maintained in the incubator for different time intervals. Cultures prepared under these conditions usually contain about 80–85% neurons as estimated by immunohistochemical staining using antibodies against neurofilament protein (16).
siRNA transfection
Three sets of siRNA duplexes specific to rat Capn 1 (μ-calpain) and Capn 2 (m-calpain) mRNA were designed by BLOCK-it™ RNAi Designer (Invitrogen, Fig 5A). Seven to eight days of primary neuronal cultures were transfected with the siRNA duplexes using the protocol supplied with the Lipofectamine™ 2000 CD Transfection Reagent (Invitrogen). Briefly, the Lipofectamine was first diluted (1:50) into Opti-MEM medium for about 5 minutes and then siRNA duplex was added to the medium to form a lipid-siRNA complex. Following additional 20-minute incubation, transfection was initiated by the addition of the lipid-siRNA complex to 24 well plates. 20 pmol of siRNA duplex was used for each well according to the manufacturer's instruction. The cell cultures after transfection were incubated in a humidified incubator (37°C, 5% CO2) for 48 hours.
Stimulation of cultured neurons
Culture medium was removed and the culture was washed and incubated with 1ml EBSS (116.4 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 1 mM NaH2PO4, 26.2 mM NaHCO3, 1.8 mM CaCl2, 5.6 mM D-glucose, pH 7.4). For the calpastatin treated group, calpastatin was added 15 minutes prior to the stimulation to give a final concentration of 10 nM. The cell cultures were stimulated by a high K+ EBSS (20.6 mM NaCl, 100.7 mM KCl, 0.8 mM MgSO4, 1 mM NaH2PO4, 26.2 mM NaHCO3, 1.8 mM CaCl2, 5.6 mM D-glucose, pH 7.4), which was used instead of normal EBSS buffer. After a 15-minute incubation, the media was removed and the wells were washed twice with normal EBSS buffer.
RT-PCR
After siRNA transfection for 48 hours, cells were lysed in TRIZOL reagent and total RNA was first isolated by chloroform extraction, followed by precipitation by ethanol. The first strand cDNA was synthesized by using the SuperScript II first-strand synthesis system (Invitrogen) according to the manufacturer's instruction. PCR was performed with respective primers in a DNA thermal cycler as indicated in Fig. 6F. The amplified products were visualized on 1% agarose gel.
Preparation of neuronal culture extract
Briefly, the cells were washed with ice-cold PBS and lysed for 10 minutes with 100 μl lysis buffer/4 wells (50 mM Tris, 150 mM NaCl, 0.1% BSA, 1% Igepal CA630, 1:100 protease inhibitor cocktail, pH 7.5). Cell lysates were sonicated, followed by centrifu-gation at 10,000 × g for 10 minutes. The supernatant was collected and referred to as cell culture extract.
Phosphorylation of recombinant hGAD65 and hGAD67
Previously we have shown that recombinant hGAD65 could be phosphorylated by PKCε, and hGAD67 could be phos-phorylated by PKA in vitro (18). In order to test whether GAD phosphorylation may have any effect on cleavage by μ-calpain, phos-phorylated GAD was prepared as described (18). Briefly, highly purified hGAD67 and hGAD65 in standard GAD buffer (50 mM KP, 1mM AET, 0.2 mM PLP, pH 7.2) were first extensively dialyzed against dialysis buffer (20 mM MOPS, 1mM AET, 0.2 mM PLP, pH 7.2). Phosphorylation reactions were conducted using PKA and PKC assay kits as described below.
PKA treatment of hGAD67
10 μg of hGAD67 was incubated at 30°C in a total volume of 200μl containing 20 mM MOPS, pH 7.2, 25 mM β-phosphoglycerol, 1mM sodium orthovanadate, 1 mM dithio-threitiol, 1mM AET, 0.2 mM PLP, 1 μg PKA catalytic subunit, 3 μM cAMP, 0.3 μM PKC inhibitor, 3 μM CaMKII inhibitor, 5 mM MgCl2 and 200 μM ATP.
PKC ε treatment of hGAD65
10 μg of hGAD65 was added to a 200μl reaction buffer containing 20 mM MOPS, pH 7.2, 25 mM β-phosphoglycerol, 1mM sodium orthovanadate, 1 mM dithiothreitiol, 1mM AET, 0.2 mM PLP, 5 mM MgCl2, 200 μM ATP, 25 ng of PKC ε, 75 μg/ml phosphatidyl serine, 75 μg/ml diacylglycerol, 0.3 μM PKA inhibitor and 3 μM CaMKII inhibitor.
Both PKA and PKC reactions were carried out at 30°C for 45 minutes. The extent of phosphorylation of GAD was tested by immunoblotting test with anti-phospho-ser/thr as previously described (18).
Enzyme assay
GAD was assayed by a radiometric method measuring the formation of 14CO2 from [1-14C]glutamic acid as described previously (4, 19).
Acknowledgments
This work was supported in part by National Institutes of Health Grant NS37851 (to J.-Y.W.), National Science Foundation grant IBN-9723079 (to J.-Y.W), the Schmidt Family Foundation and the Center of Excellence for Biomedical and Marine Biotechnology at Florida Atlantic University.
The abbreviations used are
- GAD
Glutamic acid decarboxylase GAD
- GABA
γ-Aminobutyric acid
- fGAD
full-length GAD
- tGAD
truncated GAD
- SELISA
Sandwich Enzyme-Linked ImmunoSorbent-Assay
- HRP
horseradish peroxidase
- ABTS
2,2′-azino-bis (3-ethylbenziazoline-6-sulfonic acid)
- PLP
pyridoxal phosphate
- AET
2-aminoethylisothiuronium bromide
- GAPDH
hGAD, human GAD
- GST
glutathione-S-transferase
- PKA
cAMP-dependent protein kinase A
- PKCε
protein kinase Cε
- Capn
calpain
- CTL
control group
- STI
stimulation group
- KO
knockout
- WT
wild type
- anti-pS/T
anti-phospho-serine and threonine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wu JY. GABA in Nervous System Function. 1st. Raven Press; New York: 1976. [Google Scholar]
- 2.Watanabe M, Maemura K, Kanbara K, Tamayama T, Hayasaki H. Int Rev Cytol. 2002;213:1–47. doi: 10.1016/s0074-7696(02)13011-7. [DOI] [PubMed] [Google Scholar]
- 3.Wei J, Jin Y, Wu H, Sha D, Wu JY. J Biomed Sci. 2003;10:617–624. doi: 10.1159/000073527. [DOI] [PubMed] [Google Scholar]
- 4.Sha D, Wei JN, Wu H, Jin Y, Wu JY. Brain Res Mol Brain Res. 2005;136:255–261. doi: 10.1016/j.molbrainres.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Legay F, Henry S, Tappaz M. J Neurochem. 1987;48:1022–1026. doi: 10.1111/j.1471-4159.1987.tb05620.x. [DOI] [PubMed] [Google Scholar]
- 6.Chang YC, Gottlieb DI. J Neurosci. 1988;8:2123–2130. doi: 10.1523/JNEUROSCI.08-06-02123.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu WC, Metzler DE. Arch Biochem Biophys. 1994;313:287–295. doi: 10.1006/abbi.1994.1390. [DOI] [PubMed] [Google Scholar]
- 8.Buss K, Drewke C, Lohmann S, Piwonska A, Leistner E. J Med Chem. 2001;44:3166–3174. doi: 10.1021/jm010868f. [DOI] [PubMed] [Google Scholar]
- 9.Goll DE, Thompson VF, Li H, Wei W, Cong J. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 10.Azam M, Andrabi SS, Sahr KE, Kamath L, Kuliopulos A, Chishti AH. Mol Cell Biol. 2001;21:2213–2220. doi: 10.1128/MCB.21.6.2213-2220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arthur JS, Elce JS, Hegadorn C, Williams K, Greer PA. Mol Cell Biol. 2000;20:4474–4481. doi: 10.1128/mcb.20.12.4474-4481.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu CC, Thomas C, Chen W, Davis KM, Foos T, Chen JL, Wu E, Floor E, Schloss JV, Wu JY. J Biol Chem. 1999;274:24366–24671. doi: 10.1074/jbc.274.34.24366. [DOI] [PubMed] [Google Scholar]
- 13.Sha D, Jin H, Kopke RD, Wu JY. Neurochem Res. 2004;29:199–207. doi: 10.1023/b:nere.0000010449.05927.f9. [DOI] [PubMed] [Google Scholar]
- 14.Davis KM, Foos T, Bates CS, Tucker E, Hsu CC, Chen WQ, Jin H, Tyburski JB, Schloss JV, Tobin AJ, Wu JY. Biochem Biophys Res Commun. 2000;267:777–782. doi: 10.1006/bbrc.1999.2038. [DOI] [PubMed] [Google Scholar]
- 15.Jin H, Wu H, Osterhaus G, Wei J, Davis KM, Sha D, Floor E, Hsu CC, Kopke RD, Wu JY. Proc Natl Acad Sci USA. 2003;100:4293–4298. doi: 10.1073/pnas.0730698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee YH, Deupree DL, Chen SC, Kao LS, Wu JY. J Neurochem. 1994;62:2325–2332. doi: 10.1046/j.1471-4159.1994.62062325.x. [DOI] [PubMed] [Google Scholar]
- 17.Wu H, Jin Y, Wei J, Jin H, Sha D, Wu JY. Brain Res. 2005a;1038:123–131. doi: 10.1016/j.brainres.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 18.Wei J, Davis KM, Wu H, Wu JY. Biochemistry. 2004;43:6182–6189. doi: 10.1021/bi0496992. [DOI] [PubMed] [Google Scholar]
- 19.Wu JY, Denner LA, Wei SC, Lin CT, Song GX, Xu YF, Liu JW, Lin HS. Brain Res. 1986;373:1–14. doi: 10.1016/0006-8993(86)90309-4. [DOI] [PubMed] [Google Scholar]
- 20.Gu M, Majerus PW. J Biol Chem. 1996;271:27751–27759. doi: 10.1074/jbc.271.44.27751. [DOI] [PubMed] [Google Scholar]
- 21.Kambe A, Yokota M, Saido TC, Satokata I, Fujikawa H, Tabuchi S, Kamitani H, Watanabe T. Brain Res. 2005;1040:36–43. doi: 10.1016/j.brainres.2005.01.080. [DOI] [PubMed] [Google Scholar]
- 22.Jiang SX, Lertvorachon J, Hou ST, Konishi Y, Webster J, Mealing G, Brunette E, Tauskela J, Preston E. J Biol Chem. 2005;280:33811–33818. doi: 10.1074/jbc.M503113200. [DOI] [PubMed] [Google Scholar]
- 23.Wu HY, Yuen EY, Lu YF, Matsushita M, Matsui H, Yan Z, Tomizawa K. J Biol Chem. 2005b;280:21588–21593. doi: 10.1074/jbc.M501603200. [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Fernandez HL. Biochem Biophys Res Commun. 2005;330:714–721. doi: 10.1016/j.bbrc.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 25.Branca D. Biochem Biophys Res Commun. 2004;322:1098–1104. doi: 10.1016/j.bbrc.2004.07.126. [DOI] [PubMed] [Google Scholar]
- 26.Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Proc Natl Acad Sci U S A. 1997;94:6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen TH, Paul S, Xu Y, Gurd JW, Lombroso PJ. J Neurochem. 1999;73:1995–2001. [PubMed] [Google Scholar]
- 28.Aicher B, Lerch MM, Muller T, Schilling J, Ullrich A. J Cell Biol. 1997;138:681–691. doi: 10.1083/jcb.138.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rock MT, Brooks WH, Roszman TL. J Biol Chem. 1997;272:33377–33383. doi: 10.1074/jbc.272.52.33377. [DOI] [PubMed] [Google Scholar]
- 30.Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Biochem Biophys Res Commun. 1996;229:891–895. doi: 10.1006/bbrc.1996.1898. [DOI] [PubMed] [Google Scholar]
- 31.Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S. Proc Natl Acad Sci USA. 1997;94:14060–14065. doi: 10.1073/pnas.94.25.14060. [DOI] [PMC free article] [PubMed] [Google Scholar]