

Abstract
The acquisition of new traits through horizontal gene transfer depends on the ability of the recipient organism to express the incorporated genes. However, foreign DNA appears to be silenced by the histone-like nucleoid-structuring protein (H-NS) in several enteric pathogens, raising the question of how this silencing is overcome and the acquired genes are expressed at the right time and place. To address this question, we investigated transcription of the horizontally acquired ugtL and pagC genes from Salmonella enterica, which is dependent on the regulatory DNA-binding proteins PhoP and SlyA. We reconstituted transcription of the ugtL and pagC genes in vitro and determined occupancy of their respective promoters by PhoP, H-NS, and RNA polymerase in vivo. The SlyA protein counteracted H-NS-promoted repression in vitro but could not promote gene transcription by itself. PhoP-promoted transcription required SlyA when H-NS was present but not in its absence. In vivo, H-NS remained bound to the ugtL and pagC promoters under inducing conditions that promoted RNA polymerase recruitment and transcription of the ugtL and pagC genes. Our results indicate that relief of H-NS repression and recruitment of RNA polymerase are controlled by different regulatory proteins that act in concert to express horizontally acquired genes.
Horizontal gene transfer contributes significantly to the genetic diversity of bacteria. The importance of this process in bacterial evolution is underscored by the fact that it allows microorganisms to rapidly acquire new traits, such as those involved in virulence, resistance to antibiotics, or the ability to live in new niches (1). However, the inappropriate expression of newly acquired genes can be detrimental and even place a microorganism at a competitive disadvantage (2). Enteric bacteria have solved this problem, in part, by using the DNA-binding histone-like nucleoid structuring protein (H-NS)2 to silence the expression of foreign genes in a process referred to as “xenogeneic silencing” (3). In vivo, H-NS preferentially binds to sequences that are AT-rich, which results in increased binding to horizontally acquired DNA sequences (4–6) and silencing of their transcription. Although silencing foreign DNA sequences may avoid potential negative effects, the acquired genes must be expressed if they are to contribute to an organism's lifestyle. This implies that bacteria must have the means to counteract the H-NS silencing effects and to transcribe the acquired genes when their products are needed.
Expression of a large number of horizontally acquired genes is controlled by the Mg2+-responding PhoP/PhoQ two-component regulatory system in the Gram-negative pathogen Salmonella enterica serovar Typhimurium (7) (Fig. 1). The DNA-binding protein PhoP regulates gene expression directly by binding to its target promoters and indirectly by governing the production and/or activity of other regulatory proteins (7). One of the PhoP-regulated targets is the DNA-binding protein SlyA, which is required for expression of a subset of PhoP-regulated genes (8) that exhibit a restricted phylogenetic distribution (often with no BLAST matches outside Salmonella species), suggesting that they have been acquired recently by the Salmonella lineage through horizontal gene transfer from unidentified sources.
FIGURE 1.

Model depicting transcriptional control of the horizontally acquired pagC and ugtL genes by the regulatory proteins H-NS, PhoP, and SlyA.
We analyzed the regions of the Salmonella genome that have been reported to be bound by the H-NS protein in vivo (4, 5) and found that a subset of these regions overlaps with the set of genes known to be co-regulated by the PhoP and SlyA proteins. This suggested that these two regulatory proteins may provide a means to overcome the H-NS-mediated silencing of horizontally acquired genes. Thus, to understand this process, we investigated the expression of the PhoP- and SlyA-dependent ugtL and pagC genes, which are normally bound by the H-NS protein (4, 5). BLAST searches with the UgtL and PagC protein sequences retrieved no homologs for the former and only low similarity sequences (<48% identity) for the latter, indicating that the respective genes have no orthologs outside of Salmonella spp. The ugtL gene encodes an inner membrane protein that promotes the formation of monophosphorylated lipid A in the lipopolysaccharide and is required for resistance to the antimicrobial peptides magainin 2 and polymyxin B (9). The pagC gene encodes an outer membrane protein implicated in serum resistance (10).
Using a combination of in vivo expression and promoter occupancy assays with in vitro transcription and DNA binding experiments, we now report the roles that the regulatory proteins PhoP and SlyA play in promoting transcription of the ugtL and pagC genes. Our findings indicate that H-NS repression relief and RNA polymerase recruitment are events controlled by different regulatory proteins that act in distinct ways to allow regulated gene expression. This may be applicable to other horizontally acquired genes that require the PhoP and SlyA proteins for transcription.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Growth Conditions—Bacterial strains and plasmids used in this study are listed in Table 1. All S. enterica serovar Typhimurium strains are derived from wild-type strain 14028s and were constructed by phage P22-mediated transductions as described (11). Bacteria were grown at 37 °C in N-minimal medium (12) buffered in 50 mm BisTris, pH 7.7, supplemented with 0.1% casamino acids, 38 mm glycerol, and 10 μm or 10 mm MgCl2. Escherichia coli strain DH5α was used as the host for the preparation of plasmid DNA. Ampicillin and kanamycin were used at 50 μg/ml, chloramphenicol at 20 μg/ml, and tetracycline at 10 μg/ml.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| S. enterica | ||
| 14028s | Wild type | ATCC |
| EG14078 | ΔslyA::CmR | Ref. 9 |
| EG15598 | ΔphoP/phoQ::CmR | Ref. 14 |
| EG13918 | phoP-HA | Ref. 14 |
| EG18825 | phoP-HA ΔslyA::CmR | This work |
| EG18603 | phoP-HA PhoP box mut in pagC pr | This work |
| EG18817 | phoP-HA SlyA/H-NS box mut in pagC pr | This work |
| EG17828 | ΔrpoS::CmR Δhns::KmR | This work |
| EG18482 | ΔrpoS phoP7953::Tn10 Δhns::KmR | This work |
| EG18483 | ΔrpoS ΔslyA::CmR Δhns::KmR | This work |
| E. coli | ||
| DH5α | F–supE44 ΔlacU169 (φ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Ref. 46 |
| ER2566 | fhuA2 [lon] ompT lacZ::T7 gene1 gal sulA11 Δ(mcrC-mrr)114::IS10 R(mcr-73::miniTn10-TetS)2 R(zgb-210::Tn10-TetS) endA1 [dcm] | New England Biolabs |
| EG17025 | ER2566 ΔphoPQ::KmR | This work |
| EG17246 | ER2566 ΔslyA::KmR | This work |
| Plasmids | ||
| pT7-7 | repPMB1 ApR pT7 | Ref. 47 |
| pT7.7-His6-H-NS | repPMB1 ApR pT7 His6-hns | This work |
| pT7.7-His6-SlyA | repPMB1 ApR pT7 His6-slyA | This work |
| pT7.7-PhoP-His6 | repPMB1 ApR pT7phoP-His6 | Ref. 48 |
| pKD3 | repR6Kγ ApR FRT CmR FRT | Ref. 13 |
| pKD4 | repR6Kγ ApR FRT KmR FRT | Ref. 13 |
| pKD46 | reppSC101 ts ApR paraBAD γ β exo | Ref. 13 |
| pCP20 | reppSC101 ts ApR CmRcl857 λPRflp | Ref. 49 |
Construction of Strains with Nucleotide Substitutions in Regulatory Sites and/or Deletions in the Chromosome—Point mutations in the PhoP binding site in the pagC promoter were introduced in the Salmonella chromosome as follows. First, we introduced a KmR cassette immediately downstream of the stop codon of the pagD gene, which is divergently transcribed from pagC, using a PCR product generated with primers 7885 and 7886 (primer sequences are described in Table S1) and plasmid pKD4 as template (13). Chromosomal DNA from the resulting strain was used as template to generate a PCR product with primers 7886 and 7977. A second PCR product was generated with primers 2896 and 7978 and 14028s chromosomal DNA as template. These two DNA fragments were combined in a third PCR reaction with primers 2896 and 7886. The resulting product was introduced into wild-type Salmonella cells carrying plasmid pKD46 as previously described (13). The structure of the generated mutant was verified by colony PCR and DNA sequencing; the KmR cassette was removed using plasmid pCP20 as described (13). The presence of the “scar” sequence downstream of the stop codon of the pagD gene had no effect on the levels of pagC or pagD transcripts as determined by real time PCR experiments (data not shown).
The deletion of 12 nucleotides corresponding to the SlyA/H-NS binding site ∼100 nt upstream the pagC transcription start site was carried out as follows. First, we replaced the region upstream of the pagC promoter, including the pagD gene, with a TetR cassette generated with primers 7984 and 7888 and genomic DNA of Salmonella strain (EG16459) harboring a Tn10 insertion as template. Second, a PCR product containing a KmR cassette was generated with primers 7886 and 8293 and template DNA from a Salmonella strain harboring a KmR cassette immediately downstream of the stop codon of the pagD gene (described above). This product was introduced into the TetR strain carrying plasmid pKD46, and KmR transformants were selected and then screened for being TetS. The structure of the generated mutant was verified by DNA sequencing; the KmR cassette was removed using pCP20 as described (13).
Strain EG17828, which has a deletion of both the rpoS and hns genes was constructed by the one-step gene inactivation method (13) as follows. A KmR cassette was amplified using primers 7068 and 7069 and pKD4 as template and recombined into the hns region in a strain with deletion of the rpoS gene (EG14749). The structure of the generated mutant was verified by colony PCR as described (13).
Plasmid Constructs—Plasmid pT7.7-His6-H-NS was constructed by cloning between the NdeI and HindIII sites of plasmid pT7.7 a DNA fragment generated by PCR with primers 7239 and 7252 and genomic DNA from wild-type Salmonella as template.
Plasmid pT7.7-His6-SlyA was constructed by cloning between the NdeI and HindIII sites of pT7.7, a DNA fragment generated by PCR with primers 7070 and 7065, and genomic DNA from wild-type Salmonella as template.
PCRs were carried out with AccuPrime™ TaqDNA Polymerase High Fidelity (Invitrogen), and the correct sequence of the constructs was verified by DNA sequencing.
RNA Isolation and Real Time PCR to Determine Transcript Levels—Cells were grown in N-minimal medium containing 10 mm MgCl2 to A600 ∼ 0.7. 3 ml of cell culture were washed with Mg2+-free medium and inoculated into 10 ml of fresh medium containing either 10 μm or 10 mm MgCl2. Cells were grown with vigorous shaking at 37 °C for 30 min. 1-ml samples were collected and used to prepare total RNA using the SV Total RNA Isolation System (Promega). cDNA was synthesized using TaqMan (Applied Biosystems) and random hexamers following the manufacturer's instructions. Quantification of transcripts was performed by real time PCR using SYBR Green PCR Master Mix (Applied Biosystems) in an ABI 7000 sequence detection system (Applied Biosystems). The pagC and ugtL transcripts were each detected with two sets of primers (which gave similar results): primers 6684 and 6685 and primers 6492 and 6493 were used to quantify the pagC transcript. Primers 7108 and 7114 and primers 6494 and 6495 were used to quantify the ugtL transcript. The mgtA transcript was detected with primers 4443 and 4446. Results were normalized to the levels of 16 S ribosomal RNA, which were estimated using primers 6970 and 6971. The amount of each PCR product was calculated from standard curves obtained from PCRs with the same primers and serially diluted DNA.
Chromatin Immunoprecipitation Assay—Cells were grown in N-minimal medium containing 10 mm MgCl2 to A600 ∼0.7. 6 ml of cell culture were washed with Mg2+-free medium and inoculated into 20 ml of fresh medium containing either 10 mm or 10 μm MgCl2. Cells were then grown with vigorous shaking at 37 °C for 30 min. Chromatin immunoprecipitation assays were carried out as described (14) with the following modifications. PhoP-HA-, H-NS-, and RpoB-cross-linked DNA was immunoprecipitated with monoclonal anti-HA sc-7392X (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-H-NS H113 (15), and anti-RpoB WP023 (Neoclone) antibodies, respectively. The anti-HA antibodies were captured with Protein A-agarose beads (Pierce), whereas Protein G-Sepharose (GE Healthcare) was used to capture anti-H-NS and anti-RpoB antibodies. After reversal of cross-linking, the immunoprecipitated and input DNA were purified using QIAquick columns (Qiagen) following the manufacturer's instructions.
Quantification of the immunoprecipitated and input DNA was performed by real time PCR using SYBR Green PCR Master Mix (Applied Biosystems) in an ABI 7000 sequence detection system (Applied Biosystems). For amplification of the rpoD, mgtA, pagC, and ugtL promoter regions, primers 4149 and 4150, primers 5852 and 5853, primers 7857 and 7858, and primers 7855 and 7856 were used, respectively. The amount of each PCR product was calculated from standard curves obtained from PCRs with the same primers and serially diluted DNA. The level of protein binding to a particular promoter under each condition was calculated as follows,
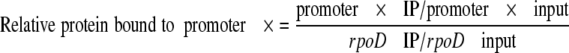 |
where IP represents immunoprecipitate. The rpoD promoter neither binds to nor is regulated by the PhoP protein.
Immunoblotting Analysis—Cells were grown in 15 ml of N-minimal medium to A600 ∼0.4, washed with Tris-buffered saline twice, resuspended in 400 μl of Tris-buffered saline, and opened by sonication. Whole-cell lysates were electrophoresed on 10% NuPAGE BisTris gels (Invitrogen) with MES running buffer, transferred to nitrocellulose membranes, and analyzed by immunoblotting with anti-HA or anti-RpoB (Neoclone) monoclonal antibodies or an anti-PagC polyclonal antibody (10). Blots were developed by using anti-mouse or anti-rabbit IgG horseradish peroxidase-linked antibodies (Amersham Biosciences) and the ECL detection system (Amersham Biosciences).
Overproduction and Purification of Proteins—Histidine-tagged H-NS, SlyA, and PhoP proteins were overproduced in E. coli strain EG17246 harboring pT7-7-His6-H-NS or pT7.7-His6-SlyA and in E. coli strain EG17025 harboring pT7-7-PhoP-His6, respectively. Protein purification was performed as described (16) with the following modifications. After purification, the buffer of the eluate was exchanged with 10 mm Hepes (pH 8.0), 10% (v/v) glycerol (for the H-NS protein, the buffer contained 200 mm NaCl), and the proteins were concentrated using an Amicon Ultra-15 column (Mr 10,000; Millipore). Proteins were stored at -80 °C. Protein concentration was determined with a BCA protein assay (Pierce) using bovine serum albumin as a standard. Protein preparations were >99% pure as determined by SDS-PAGE followed by Coomassie Blue staining (Fig. S1).
In Vitro Single Round Transcription Assays—Linear DNA templates for in vitro transcription assays were generated by PCR using primers 7756 and 7758 for pagC and 7193 and 7194 for ugtL and genomic DNA of wild-type Salmonella as template. The DNA fragments were then gel-purified with QIA-quick columns (Qiagen). The in vitro transcription reactions were carried out under standard conditions as described (17, 18). Briefly, a mixture of template DNA (9 nm), purified His-tagged proteins, and RNA polymerase holoenzyme (Epicentre) were incubated in 15 μl of transcription buffer (50 mm Tris-HCl (pH 8.0), 50 mm NaCl, 3 mm MgCl2, 0.1 mm EDTA, 0.1 mm dithiothreitol, and 25 μg/ml bovine serum albumin) for 30 min at 37 °C to form open complexes. A 5-μl mixture of substrate and heparin was then added to make a final concentration of 160 μm each of ATP, GTP, and CTP; 50 μm UTP; 2 μCi of [α-32P]UTP; and 200 μg/ml heparin. After 10 min of incubation at 37 °C, reactions were stopped by adding TBE-urea loading buffer (Invitrogen) and resolved in 10% TBE-urea gels (Invitrogen).
DNase I Footprinting and Gel Mobility Shift Assays—DNase I footprinting with the PhoP protein was performed as reported (19). The pagC promoter region was amplified with primers 7756 and 7758 and genomic DNA of wild-type Salmonella as template. Primer 7756 was labeled for the coding strand, and primer 7758 was labeled for the noncoding strand. Footprinting with the H-NS and SlyA proteins was carried out as described (20) with several modifications as follows. Labeled primer 7756 and unlabeled primer 7758 were used to generate the DNA fragment containing the pagC promoter. After purification with QIAquick columns (Qiagen), the labeled fragment (9 nm) was incubated with the indicated amount of H-NS and/or SlyA proteins for 20 min at room temperature in 20 μl of 40 mm Hepes (pH 8.0), 8 mm MgCl2, 60 mm potassium glutamate, 5 mm dithiothreitol, 0.05% Nonidet P-40, and 0.1 mg/ml bovine serum albumin (Ambion). DNase I (Epicentre) (0.02 units), 10 mm CaCl2, and 10 mm MgCl2 were added and incubated for 3 min or 2.5 min (in the absence of protein). The reaction was stopped by the addition of 100 μl of phenol (pH 8.0), and the aqueous phase was precipitated. Samples were analyzed by electrophoresis on a 6% polyacrylamide, 7.5 m urea gel and compared with a Maxam-Gilbert A + G DNA ladder generated from the same DNA probe.
The pagC DNA fragments for gel mobility shift assays were generated by PCR using primers 7756 and 7758 and genomic DNA of wild-type or mutant (EG18603) Salmonella as template. The DNA fragments were then gel-purified with QIAquick columns (Qiagen) and 100 ng of DNA labeled with T4 polynucleotide kinase and [γ-32P]ATP. Unincorporated [γ-32P]ATP was removed using G-50 microcolumns (Amersham Biosciences). 1 × 104 cpm of labeled probe (∼6 fmol), 200 ng of poly(dI-dC)·poly(dI-dC) (Amersham Biosciences), and purified His-tagged PhoP were mixed with binding buffer (20 mm Hepes (pH 8.0), 10 mm KCl, 2 mm MgCl2, 0.1 mm EDTA, 0.1 mm dithiothreitol, 50 μg/ml bovine serum albumin, and 10% glycerol) in a total volume of 20 μl and incubated for 20 min at room temperature. Samples were then electrophoresed on 4–20% TBE gels (Invitrogen), and the gels were dried and autoradiographed.
RESULTS
Transcription of the Horizontally Acquired pagC and ugtL Genes Is PhoP- and SlyA-dependent—We examined the mRNA levels of the PhoP-activated pagC, ugtL, mgtA, pagP, rstA, and slyB genes following bacterial growth under inducing (10 μm) and repressing (10 mm) Mg2+ concentrations in isogenic wild-type, phoP, and slyA Salmonella strains. Transcription of all six genes was detected in the wild-type strain following growth in low Mg2+ but not in high Mg2+, and it was absent from the phoP mutant regardless of the Mg2+ concentration (Figs. 2 and S2), which is in agreement with previous results (21, 22). A functional slyA gene was necessary for expression of the horizontally acquired pagC (Fig. 2A) and ugtL (Fig. 2B) genes but not of the ancestral mgtA, pagP, rstA, and slyB genes (Figs. 2C and S2).
FIGURE 2.

The slyA gene is necessary to promote transcription of the horizontally acquired PhoP-regulated pagC and ugtL genes but not the ancestral PhoP-regulated mgtA gene. A–C, transcript levels corresponding to the pagC (A), ugtL (B), and mgtA (C) genes as determined by quantitative real time PCR in wild-type (14028s), phoPQ (EG15598) and slyA (EG14078) cells grown in N-minimal medium containing 10 mm (H) or 10 μm (L) MgCl2. Shown are the mean and S.D. values of at least three independent experiments.
The PhoP Protein Promotes pagC Transcription through Direct Interaction with the pagC Promoter Region—A requirement for both the PhoP and SlyA proteins to activate pagC and ugtL transcription could be due to these two regulatory proteins directly interacting with the promoter regions of the pagC and ugtL genes. Alternatively, PhoP and SlyA could be part of a regulatory cascade whereby one of these proteins controls the expression and/or activity of the other. It appears that both of these mechanisms may be operating because the PhoP and SlyA proteins have been shown to footprint the ugtL promoter in vitro (23) and because expression of the slyA gene is PhoP-dependent under certain conditions (23, 24).
It has been reported that the SlyA protein, but not the PhoP protein, binds to the pagC promoter (8). However, using a recently developed method to analyze bacterial promoter sequences (25), we could identify a putative PhoP binding site ∼60 nt upstream of the transcription start site of the pagC gene (Fig. 3A), at a position and orientation relative to the -10 region that is shared with other PhoP-regulated promoters (26). We determined that the predicted PhoP box in the pagC promoter is a bona fide PhoP-binding site, because the PhoP protein footprinted this region (Fig. 3B).
FIGURE 3.

The PhoP protein promotes pagC transcription through direct interaction with the pagC promoter region. A, DNA sequence of the pagC promoter region. The transcription start site is indicated by the bent arrow; the PhoP binding site is boxed, and the putative -10 sequence is underlined. Nucleotide substitutions introduced in the PhoP binding site are indicated above the boxes. B, DNase I footprinting analysis of the pagC promoter region performed with probes for the coding and noncoding strands and increasing amounts of PhoP protein (0, 0.1, 0.2, 0.4, and 0.8 μm). The bars indicate protected regions at lower PhoP concentrations. C, electrophoretic mobility shift assay analysis of the pagC promoter region (-178 to +122 nt with respect to the transcription start site) using fragments harboring the wild-type sequence (wild type) or containing point mutations in the PhoP box (mutant). The amounts of PhoP protein used were 0, 1, 2, and 3 μm. D, in vivo PhoP binding to the pagC promoter in a strain with a wild-type pagC promoter (EG13918) or an isogenic strain containing point mutations in the PhoP box (EG18603). Cells were grown in N-minimal medium containing 10 mm (H) or 10 μm (L) MgCl2. PhoP binding was determined by chromatin immunoprecipitation. Shown are the mean and S.D. values of at least three independent experiments. E, pagC transcription in strains EG13918 and EG18603 grown as described in D. Transcript levels were determined by quantitative real time PCR. Shown are the mean and S.D. values of at least three independent experiments. F, Western blot analysis of crude extracts prepared from strains EG13918 and EG18603 grown as described under “Experimental Procedures” to detect the PhoP-HA, PagC, and RpoB proteins.
To test whether binding of the PhoP protein to the PhoP box in the pagC promoter was required for pagC transcription, we constructed a strain harboring nucleotide substitutions in the PhoP box of the pagC promoter in the Salmonella chromosome (Fig. 3A). First, we established that the nucleotide substitutions impaired the ability of the PhoP protein to gel-shift a DNA fragment corresponding to the pagC promoter in vitro (Fig. 3C). Accordingly, PhoP could not bind to the pagC promoter in the mutant strain in vivo when assayed by chromatin immunoprecipitation (Fig. 3D). Moreover, the pagC promoter mutant strain failed to express the pagC gene, since neither the pagC transcript (Fig. 3E) nor the PagC protein (Fig. 3F) was detected under conditions promoting their production in the wild-type strain. In vitro, the nucleotide substitutions also completely abolished the ability of the PhoP protein to promote pagC transcription (Fig. S3). Taken together with previous findings (23, 24), these results demonstrate that transcriptional activation of the pagC and ugtL genes entails binding of both the PhoP and SlyA proteins to the promoters of these two genes.
H-NS Remains Associated with the pagC and ugtL Promoters under Inducing Conditions—The PhoP and SlyA proteins could activate transcription of the pagC and ugtL genes by either displacing H-NS from their respective promoters or by counteracting its repressing effects in a manner not involving removal of H-NS from the promoters. To distinguish these two possibilities, we probed the association of the H-NS protein with the pagC and ugtL promoters in vivo using chromatin immunoprecipitation (which produces DNA fragments mostly 200–500 bp in size). H-NS displayed significantly higher (>10-fold) association with the pagC and ugtL promoter regions compared with regions known not to associate with H-NS, such as the rpoD promoter (which was used to normalized the data) or the ancestral mgtA promoter (Figs. 4, A and B). Interestingly, H-NS displayed similar levels of promoter occupancy under repressing and inducing conditions for the PhoP/PhoQ system (i.e. in cells grown in either high or low Mg2+, respectively). These results indicate that the H-NS protein remains associated with the promoter regions of the pagC and ugtL genes even under conditions in which these genes are transcribed.
FIGURE 4.

Occupancy of the pagC and ugtL promoter regions by H-NS, PhoP, and RNA polymerase in vivo. A–F, occupancy of the pagC (A, C, and E) and ugtL (B, D, and F) promoters by H-NS (A and B), PhoP (C and D), and RNA polymerase (E and F) was determined by chromatin immunoprecipitation in wild type (EG13918) or an isogenic strain lacking the slyA gene (EG18825). Cells were grown in N-minimal medium containing 10 mm (H) or 10 μm (L) MgCl2 as described under “Experimental Procedures.” H-NS occupancy of the mgtA promoter (A and B) represents background levels of nonspecifically immunoprecipitated DNA. Shown are the mean and S.D. values of at least three independent experiments.
RNA Polymerase Recruitment to the pagC and ugtL Promoters—The H-NS protein has been shown to impede transcriptional activation by at least two mechanisms: 1) H-NS binding to a promoter may hinder recruitment of RNA polymerase, and 2) H-NS may prevent mRNA elongation by trapping RNA polymerase in the promoter (27, 28). To determine whether RNA polymerase is recruited to the pagC and ugtL promoters, we examined the in vivo promoter occupancy by RNA polymerase using chromatin immunoprecipitation with an antibody directed against the β subunit of the enzyme. The DNA fragments corresponding to the pagC and ugtL promoter regions were recovered ∼11- or ∼8-fold less in bacteria grown in high Mg2+, which are nonactivating conditions, compared with those grown in low Mg2+ (Fig. 4, C and D), when these genes are transcribed (Fig. 2). These results argue that H-NS does not trap RNA polymerase in the pagC and ugtL promoter regions under noninducing conditions.
SlyA Is Required for PhoP Binding and RNA Polymerase Recruitment to the pagC and ugtL Promoters in Vivo—The PhoP protein binds to its activated promoters and recruits RNA polymerase during growth in low Mg2+ (29), which is a condition that promotes synthesis (Fig. 3F) and activation (29) of the PhoP protein. Then what prevents PhoP from promoting transcription of the pagC and ugtL genes when Salmonella is missing the slyA gene? We investigated the possibility that PhoP may be unable to associate with H-NS-bound promoters in the absence of SlyA by comparing the in vivo promoter occupancy by the PhoP protein in isogenic wild-type and slyA strains. PhoP binding to the pagC and ugtL promoters was severely diminished in the slyA mutant under conditions that resulted in PhoP binding in the slyA+ strain (Fig. 4, E and F). Consistent with the requirement for PhoP to recruit RNA polymerase to its activated promoters, there was no RNA polymerase binding to the pagC and ugtL promoters in the slyA mutant (Fig. 4, C and D). Thus, the SlyA protein is necessary for PhoP binding and RNA polymerase recruitment to the promoters of these horizontally acquired genes.
SlyA Counteracts H-NS-promoted Repression in Vitro but Does Not Promote Transcription by Itself—To further investigate the role that the PhoP and SlyA proteins play in expression of the pagC and ugtL genes, we carried out in vitro transcription assays with purified proteins and linear or supercoiled DNA templates. The PhoP protein was required to initiate transcription from the pagC (Fig. 5A) and ugtL (Figs. 5B and S4) promoters even in the absence of H-NS. By contrast, when H-NS was present, PhoP and RNA polymerase were no longer able to promote pagC (Fig. 5C, lanes 4–6) or ugtL (Fig. 5D, lanes 7 and 8, and Fig. S4) transcription, unless SlyA was added to the reaction (Fig. 5, C (lanes 7–15) and D (lanes 9 and 10), and Fig. S4). However, the same amounts of SlyA protein failed to promote pagC or ugtL transcription in the absence of the PhoP protein regardless of whether the H-NS protein was present or absent (Fig. 5, C (lanes 7, 10, and 13) and D (lanes 3–6)).
FIGURE 5.

SlyA counteracts H-NS-promoted repression but does not activate gene transcription by itself. A–D, run-off in vitro transcription assays with linear templates corresponding to the pagC promoter region (-178 to +122 nt with respect to the transcription start site) (A and C) or the ugtL promoter region (-180 to +125 nt with respect to the transcription start site) (B and D), RNA polymerase, and increasing amounts of PhoP protein only (0, 1, 2, and 3 μm in A; 0, 1, and 2 μm in B) or PhoP (0, 1, and 2 μm in C; 0 and 2 μm in D) in combination with H-NS (0 and 1.25 μm) and SlyA (0, 100, 200, and 300 nm in C; 0, 100, and 200 nm in D). The pagC and ugtL transcripts are indicated with arrows. The uppermost band in C corresponds to a ∼150-nt run-off transcript resulting from spurious transcription going in the reverse orientation, whereas the band just on top of the pagC transcript originates ∼5 nt upstream of the transcription start site indicated in Fig. 3. (A transcript originating at this position has not been observed in vivo.)
The slyA Gene Is Dispensable to Promote Transcription of the pagC and ugtL Genes in the Absence of hns in Vivo—Our findings suggest that the role of SlyA in promoting pagC and ugtL transcription is limited to counteracting the silencing effects of H-NS. This predicts that transcription of these genes should become slyA-independent in a strain lacking a functional hns gene. To test this notion, we compared the levels of pagC and ugtL transcripts in three isogenic strains: hns rpoS, hns rpoS phoP, and hns rpoS slyA. (These were carried out in an rpoS mutant background, because the hns gene is essential in Salmonella (5), and the rpoS mutation suppresses the lethality. Moreover, the experiments had to be performed with bacteria grown in LB broth, because the hns rpoS strain is unable to grow in N-minimal medium.) The slyA mutation had either only a small effect or no effect at all on pagC or ugtL transcription, respectively, in the hns rpoS strain (Fig. 6), which is in contrast to what happens in an hns+ background (Fig. 2). On the other hand, the phoP mutation clearly decreased the levels of both the pagC and ugtL transcripts under these conditions (Fig. 6). The lower levels for the pagC and ugtL mRNAs are probably due to the less efficient activation of the PhoP/PhoQ system taking place in LB broth, which contains >200 μm Mg2+, a concentration >20 times higher than in the N-minimal medium with 10 μm Mg2+, which induced higher mRNA levels (Fig. 2). In addition, this could reflect the fact that the strains were defective for rpoS, which can be highly pleiotropic on general cell physiology. Taken together with the results of the in vitro experiments (Fig. 5), these in vivo findings indicate that the PhoP and SlyA proteins are necessary and sufficient to promote pagC and ugtL transcription in the presence of H-NS and that they play different roles in transcriptional activation of these promoters.
FIGURE 6.

The slyA gene is dispensable to promote transcription of the pagC and ugtL genes in the absence of hns. A and B, transcript levels corresponding to the pagC (A) and ugtL (B) genes as determined by quantitative real time PCR in hns rpoS (EG17828), hns rpoS phoP (EG18482), and hns rpoS slyA (EG18483) Salmonella cells grown in LB medium to late log phase at 37 °C. Shown are the mean and S.D. values of three independent experiments.
The SlyA and H-NS Proteins Simultaneously Occupy Sites in the pagC Promoter Region—To explore how the SlyA protein relieves H-NS-promoted gene silencing, we first used DNase I footprinting to identify the regions of the pagC promoter that are bound by the H-NS and SlyA proteins. The H-NS protein protected multiple sites in this region (Fig. 7, A and B), which is a common feature of H-NS-regulated promoters (30). The SlyA protein also protected several sites (Fig. 7, A and C), one of which had been previously reported (8). The presence of multiple binding sites for the SlyA protein has been documented for other promoters as well (31). In addition, we identified several DNase I-hypersensitive sites (Fig. 7C), which could result from SlyA-promoted bending of the DNA or changes in the DNA topology (32).
FIGURE 7.

The H-NS and SlyA proteins bind to several sites in the pagC promoter. A, nucleotide sequence of the promoter region of the pagC gene (-178 to +122 nt with respect to the transcription start site) displaying regions bound by the H-NS (in blue) and SlyA (underlined) proteins. The transcription start site is indicated in boldface type and by the bent arrow, whereas the sequence in green highlights the PhoP box. The red line indicates the 12 nucleotides deleted in strain EG18817 (Fig. 8). B–D, DNase I footprinting analysis of the pagC promoter carried out as described under “Experimental Procedures” with increasing amounts of H-NS (0, 0.2, 0.4, 0.8, 1.2, 1.6, and 3.2 μm) (B) or SlyA (0, 4, 20, 80, 200, 400, and 600 nm) alone (C and D, right side) or H-NS (0.8 μm) in combination with SlyA (40, 80, and 400 nm) (D, left side).
Some of the regions protected by the H-NS and SlyA proteins overlap either completely or partially (Fig. 7A). This raises the possibility that these two proteins may compete for a common binding site, and suggests that the role of SlyA may be to displace H-NS from its binding site to allow PhoP binding and RNA polymerase recruitment. We tested this possibility by analyzing whether H-NS binding to the pagC promoter was altered in the presence of SlyA. The H-NS footprint pattern in presence of an amount of SlyA protein that was enough to counteract H-NS repression (Fig. 5C) and to footprint the pagC promoter region (Fig. 7, C and D, lanes 6–8) in vitro looked similar to that generated by H-NS alone (Fig. 7D, compare lanes 3, 4, and 5 with lane 2). The SlyA protein footprinted the pagC promoter region between nucleotides +47 and +79 either alone (Fig. 7D, compare lanes 8 and 9) or in the presence of H-NS (Fig. 7D, compare lanes 2 and 5), indicating that both proteins can bind to this promoter simultaneously. H-NS prevented SlyA binding at the site between -110 and -114 (Fig. 7D, compare lanes 5 and 8), but it is unclear whether SlyA binds at the other four sites (between +9 and -6, -77 and -82, -93 and -97, and -102 and -106) in the presence of H-NS (Fig. 7D, compare lanes 5 and 8). Interestingly, the SlyA protein led to the generation of a DNase I-hypersensitive site at position +5 in the presence of H-NS (Fig. 7D, lane 5). In agreement with the footprint data, the SlyA and H-NS proteins could simultaneously occupy the pagC promoter region as determined by gel shift analysis (Fig. S5), suggesting that derepression of the pagC promoter region by SlyA does not involve complete H-NS displacement from the promoter.
A Site Bound by the SlyA and H-NS Proteins in the pagC Promoter Is Dispensable for H-NS-mediated Repression but Necessary for Derepression by the SlyA Protein—If competition were the sole basis for relieving H-NS-promoted repression, elimination of a binding site shared by H-NS and SlyA should result in SlyA-independent pagC transcription. To test this notion, we constructed a strain with a 12-nt chromosomal deletion that removed the overlapping SlyA and H-NS binding site located ∼100 nt upstream from the pagC transcription start site (Fig. 7A). Gel shift analysis of a pagC promoter fragment prepared from this mutant demonstrated that the SlyA protein retained the ability to bind to the mutated promoter, presumably at the other SlyA binding sites (Fig. S6). However, pagC transcription was abolished in the pagC promoter mutant in vivo (Fig. 8A). The pagC promoter mutation does not appear to affect PhoP binding and RNA polymerase recruitment because PhoP was still able to promote transcription in the presence of RNA polymerase in vitro (Fig. 8B). Moreover, it did not affect the ability of H-NS to repress transcription in vitro. By contrast, the SlyA protein was no longer capable of counteracting the H-NS silencing effect (Fig. 8B). These findings, which are consistent with the in vivo results (Fig. 8A), indicate that the investigated site is required for SlyA function but not essential for H-NS-promoted silencing.
FIGURE 8.

An overlapping SlyA and H-NS binding site in the pagC promoter is dispensable for H-NS repression but necessary for derepression by SlyA. A, transcript levels corresponding to the pagC gene as determined by quantitative real time PCR in wild type (EG13918) or the isogenic strain EG18817 with a 12-nt deletion in the SlyA/H-NS binding site located ∼100 nt upstream of the pagC transcription start site (see Fig. 7A). Cells were grown in N-minimal medium containing 10 mm (H) or 10 μm (L) MgCl2. Shown are the mean values and S.D. of at least three independent experiments. B, run-off in vitro transcription assays with linear templates corresponding to the wild-type pagC promoter region (-178 to +122 nt with respect to the transcription start site) (top) or an equivalent DNA fragment with a 12-nt deletion in the SlyA/H-NS binding site (bottom), RNA polymerase, PhoP (0 or 2 μm), H-NS (0 or 1.25 μm), and SlyA (0, 200, and 300 nm). The pagC transcript (lower band) is indicated by an arrow. The uppermost band in both panels corresponds to a ∼150-nt run-off transcript resulting from spurious transcription going in the reverse orientation, whereas the band just on top of the pagC transcript originates ∼5 nt upstream of the transcription start site indicated in Fig. 3. (A transcript originating at this position has not been observed in vivo.)
DISCUSSION
The acquisition of DNA sequences by horizontal gene transfer provides bacteria with the opportunity for “quantum leap evolution” in the development of new traits (33). For this to happen, bacteria must regulate the acquired genes so that they are expressed at the right time and place. It has been estimated that a new sequence is acquired and stably maintained by enteric bacteria on an average of only once every several hundred thousand years (3), suggesting that only rarely is a new sequence favorably selected, and thus its expression has to be regulated. In the case of enteric bacteria, this regulation involves two distinct aspects; on the one hand, it entails counteracting the silencing effects that the H-NS protein imposes on newly acquired DNA; on the other hand, it requires reprogramming the expression of the foreign genes so that it is coordinated with that of the recipient bacterial genome. We have now established that, at least for a subset of horizontally acquired genes in Salmonella, these two aspects are governed by two different proteins, SlyA and PhoP, whose concerted action allows Salmonella to display some of its pathogenic properties, since these proteins govern the expression of a large number of virulence genes residing within pathogenicity islands and islets (7, 8).
The critical role played by the PhoP and SlyA proteins in promoting transcription of horizontally acquired genes, such as pagC and ugtL investigated here, is supported by several lines of evidence. First, transcription of the pagC and ugtL genes was abolished in bacteria lacking the phoP or slyA genes (Fig. 2, A and B); second, both the PhoP and SlyA proteins footprinted and gel-shifted DNA fragments harboring the pagC and ugtL promoters (Fig. 3, B and C) (23); and third, the PhoP and SlyA proteins were necessary and sufficient to promote transcription from the pagC and ugtL promoters in the presence of H-NS in vitro (Fig. 5, C and D). The SlyA protein was required to promote transcription of the horizontally acquired pagC and ugtL genes but not of the ancestral mgtA, pagP, rstA, and slyB genes (Figs. 2C and S2) (8). Since the latter group of genes is also PhoP-dependent, our results suggest that the joint requirement for PhoP and SlyA in gene expression may be specific to foreign DNA sequences in Salmonella.
The role of SlyA in transcription of the pagC and ugtL genes appears to be limited to countering the silencing effects of the H-NS protein, because the purified SlyA protein was unable to promote pagC and ugtL transcription in vitro by itself (i.e. with RNA polymerase but no PhoP) (Fig. 5, C and D), even in the absence of H-NS (Fig. 5D, lanes 3 and 4). Indeed, we determined that the PhoP protein is directly involved in promoting pagC transcription, because PhoP bound to the pagC promoter in vitro (Fig. 3, B and C) and in vivo (Fig. 3D) and because point mutations in the PhoP box that prevented PhoP binding (Fig. 3, C and D) abolished pagC transcription (Fig. 3E). Therefore, the role of PhoP in pagC transcription is 2-fold: as a direct transcriptional activator of the pagC promoter and as a regulator of SlyA at transcriptional (23, 24) and/or post-transcriptional (8) levels. This form of regulation is in striking parallel to that controlling expression of the ugtL gene (23), and it may apply to other PhoP- and SlyA-dependent genes, such as mig-14 and virK (which also seem to have been horizontally acquired), because the PhoP protein has been shown to bind to their respective promoters in vitro and in vivo (26).3
How does SlyA antagonize H-NS-promoted transcriptional silencing? One model posits that SlyA displaces H-NS from the promoters where it binds (34), suggesting that these two proteins compete for shared binding sites (35). In agreement with this model, E. coli SlyA and its ortholog in Yersinia, designated RovA, footprinted DNA segments that overlap with regions protected by H-NS in vitro (36, 37), although it is presently unclear whether SlyA or RovA can remove H-NS from a promoter in vivo. On the other hand, SlyA does not seem to function by displacing H-NS from the Salmonella pagC promoter, because both proteins could simultaneously bind to this promoter in vitro (Figs. 7D and S5), and removal of a binding site shared by the SlyA and H-NS proteins abolished the function of SlyA but not silencing by H-NS (Fig. 8). Furthermore, H-NS remained associated with the pagC and ugtL promoters in vivo (as determined by chromatin immunoprecipitation, which has a maximum resolution of ∼500 bp) when these genes were expressed (Fig. 4, A and B).
We propose an alternative model, whereby SlyA remodels the local structure of the H-NS-DNA complex in ways that allow other DNA binding proteins to be recruited to a promoter. This remodeling would entail rearrangement of the H-NS molecules associated with a particular promoter region or partial disruption of H-NS-mediated DNA duplex bridges (38) but not H-NS removal from a promoter. The apparent simultaneous occupancy of the pagC and ugtL promoter regions by the H-NS, SlyA, and PhoP proteins and even RNA polymerase is not unprecedented, since many regions of the E. coli genome bound by H-NS can also associate with RNA polymerase (6, 39), and binding of H-NS and of SlyA to various promoters is not mutually exclusive, at least in vitro (40, 41) (Fig. S5). Recent findings with the E. coli SlyA and H-NS proteins acting on the hlyE promoter (41) also support this notion. The proposed model takes into account the role of H-NS in the formation and maintenance of topological domain barriers in the bacterial chromosome (42), because overcoming H-NS repression would not involve complete H-NS clearance from a given region, which would be detrimental for the organization of the bacterial nucleoid. Consistent with our proposal, the H-NS footprinting pattern of the pagC promoter was hardly modified in the presence of the SlyA protein, except for the appearance of hypersensitive sites (Fig. 7D), which may reflect changes in the local topology of the promoter DNA. Indeed, changes in temperature and osmolarity have been shown to facilitate derepression of certain H-NS-repressed genes in vivo by altering the degree of local DNA supercoiling (30). This mode of action may not be limited to SlyA because the VirB protein of Shigella flexneri seems to use a similar mechanism to antagonize H-NS (43). Finally, it is also possible that SlyA could mediate the reported chemical modification of H-NS (44).
The widespread distribution of PhoP and SlyA in enteric bacteria raises the possibility that these proteins may relieve H-NS-dependent repression of horizontally acquired genes in species other than S. enterica. For example, PhoP has been shown to regulate the expression of E. coli-specific genes (26) and of horizontally acquired genes in Yersinia.3 However, the PhoP and SlyA homologs may behave in a fashion different from those from Salmonella. For example, the Yersinia RovA protein exhibits both anti-repressor and transcriptional activator functions (45), which might reflect a unique ability of this SlyA homolog. Alternatively or in addition, SlyA might also act as a transcriptional activator of yet-to-be-discovered Salmonella promoters.
In summary, the ability of Salmonella to express certain horizontally acquired genes requires the ordered and sequential participation of the ancestral DNA-binding proteins PhoP and SlyA. Upon experiencing inducing conditions for the PhoP/PhoQ system, the PhoP protein promotes expression (23) and/or activation of the SlyA protein (8). This results in SlyA binding to its target promoters, which, by overcoming H-NS repression, allows the PhoP protein to bind to the promoters and then to recruit RNA polymerase (Figs. 1 and 4). The role of SlyA appears to be limited to antagonizing H-NS silencing, because expression of the pagC and ugtL genes in vivo was rendered slyA-independent in a strain lacking hns (Fig. 6), and transcription of the pagC and ugtL promoters in vitro required the purified SlyA protein only when the H-NS protein was present in the reaction (Fig. 5). By contrast, the PhoP protein was required for transcription of these promoters even in the absence of H-NS both in vivo (Fig. 6) and in vitro (Fig. 5). Therefore, the PhoP and SlyA proteins carry out distinct jobs in the activation of promoters for horizontally acquired genes.
Supplementary Material
Acknowledgments
We thank Jay Hinton for providing antibodies directed against the H-NS protein and Karine Brugirard-Ricaud for collaborating with some of the initial chromatin immunoprecipitation experiments.
This work was supported in part by National Institutes of Health Grant AI49561 (to E. A. G.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S6.
Footnotes
The abbreviations used are: H-NS, histone-like nucleoid structuring protein; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; nt, nucleotide(s); HA, hemagglutinin; MES, 4-morpholineethanesulfonic acid.
J. C. Perez, T. Latifi, and E. A. Groisman, unpublished results.
References
- 1.Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000) Nature 405 299-304 [DOI] [PubMed] [Google Scholar]
- 2.Dorman, C. J. (2007) Nat. Rev. Microbiol. 5 157-161 [DOI] [PubMed] [Google Scholar]
- 3.Navarre, W. W., McClelland, M., Libby, S. J., and Fang, F. C. (2007) Gene Dev. 21 1456-1471 [DOI] [PubMed] [Google Scholar]
- 4.Lucchini, S., Rowley, G., Goldberg, M. D., Hurd, D., Harrison, M., and Hinton, J. C. D. (2006) PLoS Pathog. 2 746-752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Navarre, W. W., Porwollik, S., Wang, Y. P., McClelland, M., Rosen, H., Libby, S. J., and Fang, F. C. (2006) Science 313 236-238 [DOI] [PubMed] [Google Scholar]
- 6.Oshima, T., Ishikawa, S., Kurokawa, K., Aiba, H., and Ogasawara, N. (2006) DNA Res. 13 141-153 [DOI] [PubMed] [Google Scholar]
- 7.Groisman, E. A. (2001) J. Bacteriol. 183 1835-1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navarre, W. W., Halsey, T. A., Walthers, D., Frye, J., McClelland, M., Potter, J. L., Kenney, L. J., Gunn, J. S., Fang, F. C., and Libby, S. J. (2005) Mol. Microbiol. 56 492-508 [DOI] [PubMed] [Google Scholar]
- 9.Shi, Y. X., Cromie, M. J., Hsu, F. F., Turk, J., and Groisman, E. A. (2004) Mol. Microbiol. 53 229-241 [DOI] [PubMed] [Google Scholar]
- 10.Nishio, M., Okada, N., Miki, T., Haneda, T., and Danbara, H. (2005) Microbiology 151 863-873 [DOI] [PubMed] [Google Scholar]
- 11.Davis, R. W., Bolstein, D., and Roth, J. R. (1980) Advanced Bacterial Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- 12.Snavely, M. D., Gravina, S. A., Cheung, T. T., Miller, C. G., and Maguire, M. E. (1991) J. Biol. Chem. 266 824-829 [PubMed] [Google Scholar]
- 13.Datsenko, K. A., and Wanner, B. L. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 6640-6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shin, D., and Groisman, E. A. (2005) J. Biol. Chem. 280 4089-4094 [DOI] [PubMed] [Google Scholar]
- 15.Sonnenfield, J. M., Burns, C. M., Higgins, C. F., and Hinton, J. C. (2001) Biochimie (Paris) 83 243-249 [DOI] [PubMed] [Google Scholar]
- 16.Wosten, M. M. S. M., and Groisman, E. A. (1999) J. Biol. Chem. 274 27185-27190 [DOI] [PubMed] [Google Scholar]
- 17.Kusano, S., Ding, Q. Q., Fujita, N., and Ishihama, A. (1996) J. Biol. Chem. 271 1998-2004 [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto, K., Ogasawara, H., Fujita, N., Utsumi, R., and Ishihama, A. (2002) Mol. Microbiol. 45 423-438 [DOI] [PubMed] [Google Scholar]
- 19.Winfield, M. D., Latifi, T., and Groisman, E. A. (2005) J. Biol. Chem. 280 14765-14772 [DOI] [PubMed] [Google Scholar]
- 20.Bouffartigues, E., Buckle, M., Badaut, C., Travers, A., and Rimsky, S. (2007) Nat. Struct. Mol. Biol. 14 441-448 [DOI] [PubMed] [Google Scholar]
- 21.Garcia Vescovi, E., Soncini, F. C., and Groisman, E. A. (1996) Cell 84 165-174 [DOI] [PubMed] [Google Scholar]
- 22.Soncini, F. C., Vescovi, E. G., Solomon, F., and Groisman, E. A. (1996) J. Bacteriol. 178 5092-5099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi, Y. X., Latifi, T., Cromie, M. J., and Groisman, E. A. (2004) J. Biol. Chem. 279 38618-38625 [DOI] [PubMed] [Google Scholar]
- 24.Norte, V. A., Stapleton, M. R., and Green, J. (2003) J. Bacteriol. 185 3508-3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zwir, I., Huang, H., and Groisman, E. A. (2005) Bioinformatics 21 4073-4083 [DOI] [PubMed] [Google Scholar]
- 26.Zwir, I., Shin, D., Kato, A., Nishino, K., Latifi, T., Solomon, F., Hare, J. M., Huang, H., and Groisman, E. A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 2862-2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dame, R. T., Wyman, C., Wurm, R., Wagner, R., and Goosen, N. (2002) J. Biol. Chem. 277 2146-2150 [DOI] [PubMed] [Google Scholar]
- 28.Shin, M., Song, M., Rhee, J. H., Hong, Y., Kim, Y. J., Seok, Y. J., Ha, K. S., Jung, S. H., and Choy, H. E. (2005) Gene Dev. 19 2388-2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin, D., Lee, E. J., Huang, H., and Groisman, E. A. (2006) Science 314 1607-1609 [DOI] [PubMed] [Google Scholar]
- 30.Dorman, C. J. (2004) Nat. Rev. Microbiol. 2 391-400 [DOI] [PubMed] [Google Scholar]
- 31.Stapleton, M. R., Norte, V. A., Read, R. C., and Green, J. (2002) J. Biol. Chem. 277 17630-17637 [DOI] [PubMed] [Google Scholar]
- 32.Gourse, R. L., Ross, W., and Gaal, T. (2000) Mol. Microbiol. 37 687-695 [DOI] [PubMed] [Google Scholar]
- 33.Groisman, E. A., and Ochman, H. (1996) Cell 87 791-794 [DOI] [PubMed] [Google Scholar]
- 34.Boyle, E. C., Bishop, J. L., Grassl, G. A., and Finlay, B. B. (2007) J. Bacteriol. 189 1489-1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellison, D. W., and Miller, V. L. (2006) Curr. Opin. Microbiol. 9 153-159 [DOI] [PubMed] [Google Scholar]
- 36.Heroven, A. K., Nagel, G., Tran, H. J., Parr, S., and Dersch, P. (2004) Mol. Microbiol. 53 871-888 [DOI] [PubMed] [Google Scholar]
- 37.Wyborn, N. R., Stapleton, M. R., Norte, V. A., Roberts, R. E., Grafton, J., and Green, J. (2004) J. Bacteriol. 186 1620-1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dame, R. T., Noom, M. C., and Wuite, G. J. (2006) Nature 444 387-390 [DOI] [PubMed] [Google Scholar]
- 39.Grainger, D. C., Hurd, D., Goldberg, M. D., and Busby, S. J. W. (2006) Nucleic Acids Res. 34 4642-4652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corbett, D., Bennett, H. J., Askar, H., Green, J., and Roberts, I. S. (2007) J. Biol. Chem. 282 33326-33335 [DOI] [PubMed] [Google Scholar]
- 41.Lithgow, J. K., Haider, F., Roberts, I. S., and Green, J. (2007) Mol. Microbiol. 66 685-698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noom, M. C., Navarre, W. W., Oshima, T., Wuite, G. J., and Dame, R. T. (2007) Curr. Biol. 17 R913-914 [DOI] [PubMed] [Google Scholar]
- 43.Turner, E. C., and Dorman, C. J. (2007) J. Bacteriol. 189 3403-3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reusch, R. N., Shabalin, O., Crumbaugh, A., Wagner, R., Schroder, O., and Wurm, R. (2002) FEBS Lett. 527 319-322 [DOI] [PubMed] [Google Scholar]
- 45.Tran, H. J., Heroven, A. K., Winkler, L., Spreter, T., Beatrix, B., and Dersch, P. (2005) J. Biol. Chem. 280 42423-42432 [DOI] [PubMed] [Google Scholar]
- 46.Hanahan, D. (1983) J. Mol. Biol. 166 557-580 [DOI] [PubMed] [Google Scholar]
- 47.Tabor, S., and Richardson, C. C. (1985) Proc. Natl. Acad. Sci. U. S. A. 82 1074-1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chamnongpol, S., and Groisman, E. A. (2000) J. Mol. Biol. 300 291-305 [DOI] [PubMed] [Google Scholar]
- 49.Cherepanov, P. P., and Wackernagel, W. (1995) Gene (Amst.) 158 9-14 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.