

Abstract
There are three distinct members of the myosin V family in vertebrates, and each isoform is involved in different membrane trafficking pathways. Both myosin Va and Vb have demonstrated that they are high duty ratio motors that are consistent with the processive nature of these motors. Here we report that the ATPase cycle mechanism of the single-headed construct of myosin Vc is quite different from those of other vertebrate myosin V isoforms. KATPase of the actin-activated ATPase was 62 μm, which is much higher than that of myosin Va (∼1 μm). The rate of ADP release from actomyosin Vc was 12.7 s-1, which was 2 times greater than the entire ATPase cycle rate, 6.5 s-1. Pi burst size was 0.31, indicating that the equilibrium of the ATP hydrolysis step is shifted to the prehydrolysis form. Our kinetic model, based on all kinetic data we determined in this study, suggests that myosin Vc spends the majority of the ATPase cycle time in the weak actin binding state in contrast to myosin Va and Vb. Consistently, the two-headed myosin Vc construct did not show processive movement in total internal reflection fluorescence microscope analysis, demonstrating that myosin Vc is a nonprocessive motor. Our findings suggest that myosin Vc fulfills its function as a cargo transporter by different mechanisms from other myosin V isoforms.
Class V myosins function as actin-based motors for various cargo transportations. Class V myosins have been widely found in species from lower organisms, such as yeast, Caenorhabditis elegans, and Drosophila, to vertebrates. In vertebrates, there are three distinct subclasses of myosin V. Among them, most of the research has focused on myosin Va, which plays a critical role in membrane trafficking, including melanosome transport (1, 2) and endoplasmic reticulum transport (3–6). In contrast to myosin Va expressed mainly in brain and melanocytes, myosin Vb and Vc are widely expressed in a variety of tissues, although the relative expression level is distinct from each other. For instance, myosin Vb is most abundant in kidney, whereas myosin Vc is significantly expressed in epithelial and glandular tissues, including pancreas, colon, and stomach (7).
Evidence has shown that myosin V is a cargo transporter and serves as a membrane transporter in various biological processes. Myosin Va is involved in melanosome transportation in melanocytes (1, 2) and synaptic vesicle movement in neuronal cells (3–6). On the other hand, myosin Vb is involved in the plasma membrane recycling systems of transferrin receptor (8), chemokine receptor CXCR2 (9), and M4 muscarinic acetylcholine receptor (10). Although the tissue distribution of the expression of myosin Vb and Vc is overlapped, it is thought that they have a distinct physiological relevance. Myosin Vb directly interacts with the small GTP-binding protein, Rab11a (8), whereas myosin Vc colocalizes with Rab8, but not Rab11a (7), suggesting that each myosin V isoform has specific target cargo molecules and is involved in different membrane trafficking pathways.
The most intriguing finding was that myosin Va moves processively on actin filaments for a long distance without dissociating from actin (11, 12). Since myosin Va has a long neck domain consisting of six IQ motifs and a coiled-coil domain to form a two-headed structure, it is thought that myosin Va moves in a hand-over-hand fashion in which each head alternatively steps on an actin monomer at a half-helical pitch ahead on the filament. The enzyme kinetic studies have shown that myosin Va is a high duty ratio motor that spends the majority of the ATPase cycle in the strong actin binding state (13), which is thought to be required for processive myosins to prevent the head dissociating away from the track during the movement on actin.
Although the domain structure of myosin Va is common in all class V myosins from lower eukaryotes to vertebrates, whether or not the processive nature of myosin Va is common among all class V myosins has been controversial. It has been reported by multimolecule in vitro motility assays that two yeast class V myosins, Myo2p and Myo4p, are nonprocessive motors (14), although these myosins participate in membrane trafficking (15–17) and mRNA transport (18, 19), which suggests that they serve as cargo transporters. On the other hand, it was shown recently by using single molecule assays that Myo4p is a processive motor (20). Moreover, a recent kinetic study showed that Drosophila myosin V is a low duty ratio motor (21), suggesting that it is not a processive motor. Since there is no clear evidence that Drosophila myosin V is implicated in vesicle transport, myosin V may not be involved in the vesicle transporting process in Drosophila. The question is whether all three myosin V isoforms expressed in vertebrates are processive motors and support vesicular transportation in cells. Quite recently, we reported that human myosin Vb is a high duty ratio and processive motor like myosin Va (22). In the present study, we analyzed the detailed ATP hydrolysis mechanism of human myosin Vc for the first time. Our kinetic study demonstrated that myosin Vc is a low duty ratio motor. Consistently, the single molecule assays of total internal reflection fluorescence (TIRF)3 microscopy revealed that myosin Vc does not move processively on actin filaments. It is thought that myosin Vc serves as a membrane transporter in cells, but our results suggests that it would be achieved by a quite different mechanism from myosin Va and Vb.
EXPERIMENTAL PROCEDURES
Materials—Restriction enzymes and modifying enzymes were purchased from New England Biolabs (Beverly, MA). Purine nucleoside phosphorylase, 7-methylguanosine, phosphoenolpyruvate, pyruvate kinase, apyrase, ATPγS, and AMPPNP were purchased from Sigma. Actin was prepared from rabbit skeletal muscle according to Spudich and Watt (23). Pyrene-actin was prepared as described (24). 7-Diethylamino-3-((((2-maleimidyl)-ethyl)amino)carbonyl)coumarin-labeled phosphate-binding protein (MDCC-PBP) was prepared as described (25, 26). Recombinant calmodulin was expressed in Escherichia coli and purified as described previously (27). GFP-tagged myosin Va HMM was expressed in insect cells and purified as described previously (28).
Cloning, Expression, and Purification of Human Myosin Vc Protein—Human myosin Vc cDNA was obtained from human kidney total RNA by reverse transcription-PCR. The nucleotide sequence was determined by direct DNA sequencing to confirm the authenticity of the DNA sequence of the clone. The cDNA fragments encoding Met1–Gln787 (subfragment 1 construct) and Met1–Glu1129 (HMM construct) were subcloned into modified pFastBac1 baculovirus transfer vector (Invitrogen) containing a FLAG tag sequence at the 5′ end of the polylinker region and GFP sequence for the HMM construct. The subfragment 1 construct (M5CIQ1) encodes the motor domain and the first IQ motif, and the HMM construct (GFP-M5CHMM) encodes the motor domain, all six IQ motifs, and the first long coiled-coil region. To express the recombinant myosin Vc proteins, Sf9 cells were co-infected with two viruses expressing myosin Vc heavy chain and calmodulin. The expressed proteins were purified through an anti-FLAG M2 affinity column (Sigma) as previously described (29). The purified proteins were dialyzed against buffer A (25 mm KCl, 20 mm MOPS-KOH (pH 7.5), 2 mm MgCl2, 1 mm EGTA, and 1 mm DTT). The proteins were stored on ice and used within 2 days. Protein concentration was determined by the densitometry analysis of SDS-PAGE using smooth muscle myosin heavy chain as a standard. In addition, the active site concentration of M5CIQ1 was determined by using the [3H]ADP·vanadate trap technique as described previously (30).
ATPase Assay—The steady-state ATPase activity was measured in the presence of the ATP regeneration system (20 units/ml pyruvate kinase and 2 mm phosphoenolpyruvate) at 25 °C. The reaction was carried out in buffer A with 0.2 mg/ml calmodulin. The liberated pyruvate was determined as described (31).
Stopped-flow Measurements—Kinetic measurements were performed in buffer A with 0.2 mg/ml calmodulin at 25 °C using a KinTek SF-2001 apparatus (KinTek Co., Clarence, PA). Mant-nucleotides (λex = 280 nm) and pyrene-actin (λex = 365 nm) were monitored through a 400 nm cut-off filter. Intrinsic tryptophan residues in M5CIQ1 were excited at 295 nm, and the fluorescence was monitored through a 340 nm cut-off filter. Light-scattering was monitored at 420 nm. MDCC-PBP (λex = 465 nm) was monitored through a 450 nm cut-off filter. To achieve nucleotide-free conditions, M5CIQ1 was incubated with 0.05 units/ml apyrase. In the measurement of phosphate release, all solutions and syringes were preincubated with 3 μm MDCC-PBP, 0.02 units/ml purine nucleoside phosphorylase, and 0.2 mm 7-methylguanosine. The volume ratio of the syringe was 1:1 in all single mixing experiments and 1:1:1 in double mixing experiments. Kinetic simulations were performed using STELLA software (Isee Systems, Lebanon, NH).
Quenched-flow Measurement—Quenched-flow measurement was performed in buffer A with 0.2 mg/ml calmodulin at 25 °C using a KinTek RQF-3 apparatus (KinTek Co., Clarence, PA) as described (32).
Actin Cosedimentation Assay—Prior to the assay, M5CIQ1 was centrifuged at 300,000 × g for 10 min to remove any potential aggregates, and the supernatant was used in the actin cosedimentation assay. Various concentrations (2–60 μm) of actin were mixed with 1 μm M5CIQ1 in buffer A with 0.2 mg/ml calmodulin and incubated for 10 min at room temperature. Immediately after adding 2.5 mm Mg-ATPγS or 5 mm Mg-AMPPNP, the reaction mixtures were centrifuged at 300,000 × g for 10 min. The supernatants and dissolved pellets were subjected to the densitometry analysis of SDS-PAGE.
Multimolecule in Vitro Motility Assay—The actin gliding velocity was measured by an in vitro actin gliding assay as described previously (22, 30). The experiment was done with a buffer containing 25 mm KCl, 5 mm MgCl2, 1 mm EGTA, 25 mm MOPS-KOH (pH 7.5), 0.2 mg/ml calmodulin, 1 mm ATP, 10 mm DTT, and oxygen scavenger system (216 μg/ml glucose oxidase, 36 μg/ml catalase, 4.5 mg/ml glucose).
Single Molecule Assays—TIRF microscopy was set up as previously described (33). The fluorescently labeled actin filaments containing 0.5% biotinylated G-actin were adsorbed onto an avidin-coated quartz surface. GFP-tagged myosin Vc HMM and myosin Va HMM in buffer A containing 10 μm or 1 mm MgATP, 0.2 mg/ml calmodulin, and oxygen scavenger system were added to the actin filament-coated surface. The frame rate was 20 ms, which was almost equal to the exposure time of the CCD camera. The movements of the individual fluorescent spots were analyzed using special software (G-Track, G-Angstrom, Japan), which could find and track the position of the fluorescent spots automatically.
RESULTS
Expression and Purification of Myosin Vc Construct—In the kinetic study, we used a human myosin Vc single-headed construct having the entire motor domain and the first IQ motif (M5CIQ1). M5CIQ1 and calmodulin were co-expressed in Sf9 cells and purified with anti-FLAG M2 affinity chromatography. The isolated M5CIQ1 was co-purified with calmodulin (Fig. 1A). The mobility change of the low molecular mass peptide on SDS-PAGE increased with Ca2+, suggesting that it is calmodulin (data not shown). It is known that the essential light chain was co-purified with chicken brain myosin Va (34, 35) and is thought to be a part of the light chains of myosin Va. Therefore, we examined whether the essential light chain binds to M5CIQ1. M5CIQ1 was co-expressed with nonmuscle essential light chain (LC17b) that is more widely distributed among various tissues than LC1sa and LC17a, and the purified M5CIQ1 was subjected to SDS-PAGE analysis. As shown in Fig. 1B, the low molecular mass peptide co-purified with M5CIQ1 showed a Ca2+-dependent change in the migration, a characteristic of calmodulin. The results suggest that endogenous calmodulin, but not LC17b, was co-purified with M5CIQ1. The results indicate that calmodulin has a much higher affinity for the first IQ motif of myosin Vc than LC17b. It should be noted that the light chains of mammalian myosin Va and Vb are calmodulin but not LC17 (22, 36). In the present study, we used M5CIQ1 and GFP-M5CHMM with calmodulin for all experiments.
FIGURE 1.

Purification of human myosin Vc construct. A, SDS-PAGE of the purified human myosin Vc (M5CIQ1). M5CIQ1 expressed in Sf9 cells was extracted and purified through an anti-FLAG affinity column, and the purified proteins were analyzed by SDS-PAGE. HC and CaM, heavy chain of M5CIQ1 and calmodulin, respectively. A, M5CIQ1 co-expressed with calmodulin; B, M5CIQ1 co-expressed with LC17b. Lane 1, EGTA condition; lane 2, calcium condition. Molecular mass markers are shown on the left.
Steady-state ATPase Activity of M5CIQ1—The steady-state ATPase activity of M5CIQ1 was markedly activated by actin with a hyperbolic saturation curve (Fig. 2). The Vmax of the steady-state ATPase activity was 6.5 ± 0.4 s-1. Surprisingly, the KATPase was 62 ± 9 μm at 25 mm KCl, which was much higher than that of myosin Va (1 μm at 50 mm KCl) (13). The KATPase value of myosin Vc is rather similar to that of conventional myosin IIs, such as cardiac muscle, smooth muscle, and nonmuscle myosins (37–41).
FIGURE 2.

Steady-state actin-activated ATPase activity of M5CIQ1. The actin-activated ATPase activity of M5CIQ1 (20 nm) was measured in buffer A with 2 mm phosphoenol pyruvate, 20 units/ml pyruvate kinase, 0.2 mg/ml calmodulin, and 2 mm MgATP. The assay was done at 25 °C. The solid line is the hyperbola fit with Vmax of 6.5 ± 0.4 s-1 and KATPase of 62 ± 9 μm. The broken line is simulated based on our kinetic model in this study (see “Discussion”), giving a Vmax value of 6.3 s-1 and KATPase of 50 μm.
ATP Binding to M5CIQ1 and Acto-M5CIQ1—The fluorescent
nucleotide, dmant-ATP, was used to measure the rate of ATP binding to M5CIQ1
and acto-M5CIQ1 (Fig. 3). The
time courses of the fluorescence enhancement followed single exponential
kinetics for both M5CIQ1 and acto-M5CIQ1
(Fig. 3, insets). The
observed rate constants (kobs) showed a linear dependence
on the dmant-ATP concentration. The second order rate constants, obtained from
the slopes of the lines, were 2.4 ± 0.1 μm-1
s-1 for M5CIQ1 (K1k+2) and
1.6 ± 0.1 μm-1 s-1 for acto-M5CIQ1
(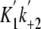 ).
).
FIGURE 3.

Kinetics of dmant-ATP binding to M5CIQ1 and acto-M5CIQ1. The
experiment was done in the same conditions as described in the legend to
Fig. 2 except for the absence
of the ATP regeneration system. A, in the absence of actin. 0.3
μm M5CIQ1 was mixed with various concentrations of dmant-ATP.
The second order rate constant for dmant-ATP binding
(K1k+2) was 2.4 ± 0.1
μm-1·s-1. Inset, a typical
recording of the binding of 5 μm dmant-ATP to M5CIQ1. The
solid line is the best fit to single exponential kinetics with
kobs of 12.6 s-1. B, in the presence
of actin. 0.3 μm M5CIQ1 in the presence of 0.4 μm
actin was mixed with various concentrations of dmant-ATP. The second order
rate constant
(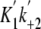 ) of
1.6 ± 0.1 μm-1·s-1 was
obtained. Inset, a typical recording of the binding of 5
μm dmant-ATP to acto-M5CIQ1. The solid line is the best
fit to single exponential kinetics with kobs of 8.1
s-1. The error bars represent the S.E. from 3–5
independent experiments.
) of
1.6 ± 0.1 μm-1·s-1 was
obtained. Inset, a typical recording of the binding of 5
μm dmant-ATP to acto-M5CIQ1. The solid line is the best
fit to single exponential kinetics with kobs of 8.1
s-1. The error bars represent the S.E. from 3–5
independent experiments.
ATP-induced Acto-M5CIQ1 Dissociation—The kinetics of the
ATP-induced transition to the weak actin binding state and the dissociation of
M5CIQ1 from actin was monitored by measuring the changes in the pyrene
fluorescence and light scattering, respectively
(Fig. 4). The increase in the
pyrene fluorescence upon the formation of the weak actin-binding form of
M5CIQ1 and the decrease in light scattering upon dissociation of
pyrene-acto-M5CIQ1 followed single exponential kinetics
(Fig. 4, inset). The
kobs of both signals showed almost identical hyperbolic
saturation curves on the ATP concentration
(Fig. 4). These results suggest
that the process of ATP binding to acto-M5CIQ1 is composed of two steps
(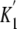 and
and
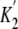 in
Fig. 13A), and the
dissociation of M5CIQ1 from actin occurs immediately after the transition from
a strong actin binding state to a weak actin binding state. The initial slopes
of the curves represent a second order rate constant of ATP binding to
acto-M5CIQ1
(
in
Fig. 13A), and the
dissociation of M5CIQ1 from actin occurs immediately after the transition from
a strong actin binding state to a weak actin binding state. The initial slopes
of the curves represent a second order rate constant of ATP binding to
acto-M5CIQ1
( ). The
). The
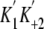 values,
obtained from the change in pyrene fluorescence and light scattering, were 1.8
± 0.1 and 1.6 ± 0.1 μm-1
s-1, respectively. These values are similar to that obtained from
dmant-ATP binding (Fig. 3),
suggesting that the mant moiety does not significantly influence the ATP
binding rate to M5CIQ1. The maximal rates of the hyperbolic curves gave
values,
obtained from the change in pyrene fluorescence and light scattering, were 1.8
± 0.1 and 1.6 ± 0.1 μm-1
s-1, respectively. These values are similar to that obtained from
dmant-ATP binding (Fig. 3),
suggesting that the mant moiety does not significantly influence the ATP
binding rate to M5CIQ1. The maximal rates of the hyperbolic curves gave
 of >300 s-1.
of >300 s-1.
FIGURE 4.

ATP-induced dissociation of acto-M5CIQ1. Pyrene-acto-M5CIQ1 (0.3
μm M5CIQ1 plus 0.5 μm pyrene-actin) was mixed with
various concentrations of MgATP, and the changes in the pyrene fluorescence or
light scattering were monitored. The second order rate constants for ATP
binding to acto-M5CIQ1
(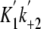 )
estimated from the initial slopes of the linear fits were 1.8 ± 0.1
μm-1·s-1 for pyrene fluorescence
and 1.6 ± 0.1 μm-1·s-1 for
light scattering. Inset, time courses of the pyrene fluorescence and
light-scattering changes after mixing acto-M5CIQ1 with 10 μm
MgATP. The solid lines are the best fits to single exponential
kinetics, with kobs of 14.7 s-1 for pyrene
fluorescence and 13.1 s-1 for light scattering. The experimental
conditions are as described in the legend to
Fig. 3. The error bars
represent the S.E. from 3–5 independent experiments.
)
estimated from the initial slopes of the linear fits were 1.8 ± 0.1
μm-1·s-1 for pyrene fluorescence
and 1.6 ± 0.1 μm-1·s-1 for
light scattering. Inset, time courses of the pyrene fluorescence and
light-scattering changes after mixing acto-M5CIQ1 with 10 μm
MgATP. The solid lines are the best fits to single exponential
kinetics, with kobs of 14.7 s-1 for pyrene
fluorescence and 13.1 s-1 for light scattering. The experimental
conditions are as described in the legend to
Fig. 3. The error bars
represent the S.E. from 3–5 independent experiments.
FIGURE 13.

A, reaction scheme of myosin Vc ATPase cycle. A, actin; M, myosin; T, ATP; D, ADP; P, phosphate. The major kinetic pathway is shown in gray shading. B, actin concentration dependence of the duty ratio of M5CIQ1. The duty ratio was calculated based on our kinetic model using experimentally obtained parameters as described under “Discussion.” The duty ratio was 0.33 at saturating actin concentration.
Enhancement of Intrinsic Tryptophan Fluorescence—Human myosin Vc contains a conserved tryptophan residue (Trp482) located at the rigid relay loop that is responsible for the fluorescence change coupled with ATP-induced conformational change of myosin. The time courses of the intrinsic tryptophan fluorescence change, upon the addition of ATP, followed single exponentials (Fig. 5, inset). The ATP dependence of the kobs showed a hyperbolic saturation curve (Fig. 5), providing the maximum rate of 59 ± 3 s-1. This value is much less than the ATP binding rate at a high ATP concentration, suggesting that the rate constant represents the ATP hydrolysis step in the absence of actin (k+3 + k-3). The rate for myosin Vc is 10-fold slower than that of myosin Va (13). The second order rate constant for ATP binding to M5CIQ1 (K1k+2) determined from the initial slope of the curve was 2.5 ± 0.1 μm-1 s-1, which was consistent with that obtained by direct ATP binding measurement using dmant-ATP (Fig. 3).
FIGURE 5.

ATP-induced intrinsic tryptophan fluorescence change of M5CIQ1. 0.8 μm M5CIQ1 was mixed with various concentrations of MgATP, and the tryptophan fluorescence change was monitored. The observed rates (kobs) were saturated at 59 ± 3 s-1 (k+3 + k-3). The second order rate constant for ATP binding can be estimated from the initial slope of the linear fit, and the obtained value (K1k+2 = 2.5 ± 0.1 μm-1 s-1) was consistent with that obtained in Fig. 3. The inset shows a typical recording of the intrinsic tryptophan fluorescence change after mixing M5CIQ1 with 10 μm MgATP. The solid line is the best fit to single exponential kinetics, with kobs of 23.7 s-1. The experimental conditions are as described in the legend to Fig. 3. The error bars represent the S.E. from 3–5 independent experiments.
ATP Hydrolysis—We measured the Pi burst of M5CIQ1 by using a quenched-flow apparatus. Single turnover experiments were carried out in which the active site concentration exceeded the substrate concentration. Since all given ATP is bound to the ATP binding site of M5CIQ1, the equilibrium of the ATP hydrolysis step can be accurately determined by measuring the fraction of the fast Pi burst phase. The time course of the Pi burst followed double exponential kinetics (Fig. 6). The fast phase corresponds to the initial rapid ATP hydrolysis in which ATP binding is rate-limiting. The slow phase (0.06 ± 0.03 s-1) represents the apparent phosphate release rate (k+4,obs). From the fractional amplitudes of the fast and slow phase, the Pi burst size was estimated to be 0.31 ± 0.02. Therefore, the equilibrium constant of the ATP hydrolysis (K3) was calculated to be 0.45 ± 0.04. The results indicate that the equilibrium of the ATP hydrolysis step is significantly shifted to the prehydrolyzed form (MT). The rate of the ATP hydrolysis was also measured at higher ATP concentration, where multiple turnovers of ATP hydrolysis take place. The Pi burst rate of 55 ± 18 s-1 was obtained with 100 μm ATP (date not shown). Since the ATP binding rate at this ATP concentration does not limit the Pi burst rate, the obtained rate constant represents the rate of ATP hydrolysis (k+3 + k-3). The observed value is consistent with the value obtained by the measurement of intrinsic tryptophan fluorescence change and is 10-fold lower than that of myosin Va, although it is significantly larger than the steady-state ATPase cycle rate, thus not limiting the overall ATPase rate.
FIGURE 6.

Kinetics of ATP hydrolysis of M5CIQ1. A single turnover experiment was carried out by mixing 0.9 μm M5CIQ1 with 0.5 μm [γ-32P]ATP, and the fraction of hydrolyzed ATP was plotted against time. The time course was fitted to double exponential kinetics. The rate constant of the burst phase (kobs = 4.0 s-1) with a fractional amplitude (equal to Afast/(Afast + Aslow)) of 0.31 ± 0.02 was obtained. The rate constant of the slow phase was kobs of 0.064 ± 0.025 s-1. The experimental conditions are as described in the legend to Fig. 3, except no actin was added.
Phosphate Release Rate—The rate of the phosphate release
step was determined by measuring the fluorescence increase of the
fluorescently labeled MDCC-PBP. Double-mixing, single turnover experiments
were carried out in which M5CIQ1 was mixed with substoichometric amounts of
ATP. The mixture was aged for 5 s to advance the ATP binding and hydrolysis
and then mixed with various concentrations of actin. Upon the release of the
bound phosphate, MDCC-PBP rapidly binds to the released phosphate, resulting
in an increase in the fluorescence intensity. The observed fluorescence
increase followed single exponential kinetics in the absence of actin
(Fig. 7A,
inset). The observed rate (k+4,obs) of 0.11 ±
0.01 s-1 was consistent with the rate of the slow phase obtained
from the quenched-flow experiment (Fig.
6). In the presence of actin, the time courses of the fluorescence
increase were best fitted to double exponential kinetics
(Fig. 7A). The fast
phase showed linear actin dependence due to the actin rebinding step
(K9) (Fig.
7B). From the slope of the line,
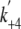 /K9,obs of 0.46
± 0.05 μm-1 s-1 was obtained. The
maximum phosphate release rate from
acto-M5CIQ1·ADP·Pi
(
/K9,obs of 0.46
± 0.05 μm-1 s-1 was obtained. The
maximum phosphate release rate from
acto-M5CIQ1·ADP·Pi
( ) was estimated to be >60
s-1 based upon the lack of curvature on the actin dependence of
kobs. The rates of the slow phase were around 1
s-1 and actin-independent (data not shown). A similar slow phase
was previously observed for myosin X
(30) and myosin VIIa
(29). Homma et al.
(30) found that the slow phase
is due to ATP rebinding to myosin X, because the rates of the slow phase were
linearly increased with myosin concentration. In this study, it was difficult
to estimate the fraction of the slow phase from the obtained kinetic
parameters, because the rate constant for ATP dissociation
(k-2) could not be determined accurately. Since the ATP
hydrolysis step (K3) is largely shifted toward the
prehydrolyzed form (MT) and the rates of the slow phase were close to the ATP
binding rate at the ATP concentration in this experiment (0.75 s-1
at 0.3 μm ATP), it is possible that the slow phase is due to the
ATP rebinding step of M5CIQ1. However, the ATP dissociation step seems to be
slower (Fig. 3), and the origin
of the slow phase may not be the same as myosin X. Another possibility is that
the slow phase is due to the ATP hydrolysis via the actin-associated pathway,
as previously reported (26).
The obtained results indicate that the phosphate release step (>60
s-1) is not the rate-limiting step of the acto-M5CIQ1 ATPase
cycle.
) was estimated to be >60
s-1 based upon the lack of curvature on the actin dependence of
kobs. The rates of the slow phase were around 1
s-1 and actin-independent (data not shown). A similar slow phase
was previously observed for myosin X
(30) and myosin VIIa
(29). Homma et al.
(30) found that the slow phase
is due to ATP rebinding to myosin X, because the rates of the slow phase were
linearly increased with myosin concentration. In this study, it was difficult
to estimate the fraction of the slow phase from the obtained kinetic
parameters, because the rate constant for ATP dissociation
(k-2) could not be determined accurately. Since the ATP
hydrolysis step (K3) is largely shifted toward the
prehydrolyzed form (MT) and the rates of the slow phase were close to the ATP
binding rate at the ATP concentration in this experiment (0.75 s-1
at 0.3 μm ATP), it is possible that the slow phase is due to the
ATP rebinding step of M5CIQ1. However, the ATP dissociation step seems to be
slower (Fig. 3), and the origin
of the slow phase may not be the same as myosin X. Another possibility is that
the slow phase is due to the ATP hydrolysis via the actin-associated pathway,
as previously reported (26).
The obtained results indicate that the phosphate release step (>60
s-1) is not the rate-limiting step of the acto-M5CIQ1 ATPase
cycle.
FIGURE 7.

Kinetics of phosphate release from M5CIQ1. The rate of phosphate release from M5CIQ1 was measured by using MDCC-PBP. 1.2 μm M5CIQ1 was mixed with 0.9 μm MgATP, aged for 5 s, and then mixed with various concentrations of actin. Other experimental conditions are as described in the legend to Fig. 3. A, a typical recording of the MDCC-PBP fluorescence change at 50 μm actin. The time courses were fitted to double exponential kinetics. The fast and slow rates were 23.8 and 1.7 s-1, respectively. The inset shows a typical recording in the absence of actin. The fluorescence change in the absence of actin was best fitted to single exponential kinetics, with kobs of 0.11 s-1. B, actin concentration dependence of the rate of phosphate release. The apparent rate of the fast phase was linearly increased with actin concentration. The apparent second order rate constant was 0.46 ± 0.05 μm-1 s-1. The rates of the slow phase (0.2–1.7 s-1) were actin-independent. The error bars represent the S.E. from 4–6 independent experiments.
Kinetics of ADP Binding to and Dissociation from Acto-M5CIQ1—We employed dmant-ADP as a probe to determine the rate of ADP binding to acto-M5CIQ1. The time courses of an increase in the fluorescence intensity followed single exponential kinetics (Figs. 8A and 9A, insets). In the absence of actin, the kobs showed a hyperbolic saturation curve on the dmant-ADP concentration (Fig. 8A), indicating that the ADP binding is a two-step process (Fig. 13A). The maximum rate and the initial slope of the curve represent k+5 + k-5 = 12.5 ± 0.8 s-1 and k-5/K6 = 2.9 ± 0.1 μm-1 s-1, respectively. From the y intercept of the curve, the rate of dmant-ADP dissociation (k+5) was estimated to be 3.6 ± 0.7 s-1. The rate of dmant-ADP dissociation was also determined by monitoring the fluorescence change after mixing M5CIQ1·dmant-ADP complex with 2 mm MgADP (Fig. 8B). The rate of dmant-ADP dissociation (k+5), obtained by this method, was 3.9 ± 0.1 s-1, which is consistent with that obtained from the dmant-ADP binding experiment (Fig. 8A).
FIGURE 8.

Kinetics of dmant-ADP interaction with M5CIQ1. A, kinetics of dmant-ADP binding to M5CIQ1. 0.3 μm M5CIQ1 was mixed with various concentrations of dmant-ADP. The second order rate constant for dmant-ADP binding (k-5/K6) was 2.9 ± 0.1 μm-1 s-1. From the y intercept, a k+5 value of 3.6 ± 0.7 s-1 was obtained. The inset shows a typical recording of the binding of dmant-ADP (5 μm) to M5CIQ1. The solid line is the best fit to single exponential kinetics, with kobs of 9.2 s-1. B, kinetics of dmant-ADP dissociation from M5CIQ1. 0.5 μm M5CIQ1 in the presence of 5 μm dmant-ADP was mixed with 2 mm MgADP. The solid line is the best fit to single exponential kinetics, with kobs of 3.9 ± 0.1 s-1. Error bars, S.E. from 3–5 independent experiments. Other experimental conditions are as described in the legend to Fig. 3.
FIGURE 9.

Kinetics of dmant-ADP interaction with acto-M5CIQ1. A,
kinetics of dmant-ADP binding to acto-M5CIQ1. 0.3 μm M5CIQ1 in
the presence of 0.4 μm actin was mixed with various
concentrations of dmant-ADP. The second order rate constant
( ) of 6.0
± 0.4 μm-1·s-1 and
) of 6.0
± 0.4 μm-1·s-1 and
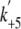 of 17.1 ± 1.3 s-1
from the y intercept were obtained. Inset, a typical
recording of the binding of dmant-ADP (5 μm) to acto-M5CIQ1. The
solid line is the best fit to single exponential kinetics with
kobs of 46.8 s-1. B, kinetics of
dmant-ADP dissociation from acto-M5CIQ1. 0.5 μm M5CIQ1 in the
presence of 5 μm dmant-ADP and 0.6 μm actin was
mixed with 2 mm MgADP. The solid line is the best fit to
single exponential kinetics, with kobs of 17.7 ±
0.6 s-1. C, the rate of ADP dissociation from acto-M5CIQ1
was determined by measuring the time course of change in the light-scattering
intensity of acto-M5CIQ1. Acto-M5CIQ1 (0.3 μm M5CIQ1 and 0.4
μm actin) in the presence of 50 μm MgADP was mixed
with various concentrations of MgATP. The apparent dissociation rates were
saturated at 12.7 ± 0.9 s-1
(
of 17.1 ± 1.3 s-1
from the y intercept were obtained. Inset, a typical
recording of the binding of dmant-ADP (5 μm) to acto-M5CIQ1. The
solid line is the best fit to single exponential kinetics with
kobs of 46.8 s-1. B, kinetics of
dmant-ADP dissociation from acto-M5CIQ1. 0.5 μm M5CIQ1 in the
presence of 5 μm dmant-ADP and 0.6 μm actin was
mixed with 2 mm MgADP. The solid line is the best fit to
single exponential kinetics, with kobs of 17.7 ±
0.6 s-1. C, the rate of ADP dissociation from acto-M5CIQ1
was determined by measuring the time course of change in the light-scattering
intensity of acto-M5CIQ1. Acto-M5CIQ1 (0.3 μm M5CIQ1 and 0.4
μm actin) in the presence of 50 μm MgADP was mixed
with various concentrations of MgATP. The apparent dissociation rates were
saturated at 12.7 ± 0.9 s-1
(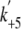 ), which reflected the ADP
dissociation rate from acto-M5CIQ1. The error bars represent the S.E.
from 3–5 independent experiments. Other experimental conditions are as
described in the legend to Fig.
3.
), which reflected the ADP
dissociation rate from acto-M5CIQ1. The error bars represent the S.E.
from 3–5 independent experiments. Other experimental conditions are as
described in the legend to Fig.
3.
In the presence of actin, the kobs were linearly
increased with dmant-ADP concentration
(Fig. 9A). A second
order rate constant
(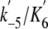 )
for dmant-ADP binding was 6.0 ± 0.4 μm-1
s-1. The y intercept of the line gave the rate constant of
the dmant-ADP dissociation (
)
for dmant-ADP binding was 6.0 ± 0.4 μm-1
s-1. The y intercept of the line gave the rate constant of
the dmant-ADP dissociation (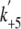 ) of 17.1
± 1.3 s-1. This value was consistent with that determined by
the direct measurement of dmant-ADP dissociation upon the addition of 2
mm MgADP to acto-M5CIQ1·dmant-ADP complex (17.7 ± 0.6
s-1) (Fig.
9B). The rate of ADP dissociation from acto-M5CIQ1 was
also determined by monitoring the light-scattering change of the
acto-M5CIQ1·ADP complex after the addition of ATP. In the presence of
ADP, the rate of ATP-induced dissociation of acto-M5CIQ1 is limited by ADP
release from the acto-M5CIQ1·ADP complex.
Fig. 9C shows the rate
of ATP-induced dissociation of the acto-M5CIQ1·ADP complex as a
function of ATP concentration. In contrast to the results obtained in the
absence of ADP (Fig. 4), the
rate of change in the light-scattering intensity of acto-M5CIQ1 plateaued to
yield the maximum rate of 12.7 ± 0.9 s-1. The observed rate
of dissociation of acto-M5CIQ1 is explained by the ADP dissociation step
(
) of 17.1
± 1.3 s-1. This value was consistent with that determined by
the direct measurement of dmant-ADP dissociation upon the addition of 2
mm MgADP to acto-M5CIQ1·dmant-ADP complex (17.7 ± 0.6
s-1) (Fig.
9B). The rate of ADP dissociation from acto-M5CIQ1 was
also determined by monitoring the light-scattering change of the
acto-M5CIQ1·ADP complex after the addition of ATP. In the presence of
ADP, the rate of ATP-induced dissociation of acto-M5CIQ1 is limited by ADP
release from the acto-M5CIQ1·ADP complex.
Fig. 9C shows the rate
of ATP-induced dissociation of the acto-M5CIQ1·ADP complex as a
function of ATP concentration. In contrast to the results obtained in the
absence of ADP (Fig. 4), the
rate of change in the light-scattering intensity of acto-M5CIQ1 plateaued to
yield the maximum rate of 12.7 ± 0.9 s-1. The observed rate
of dissociation of acto-M5CIQ1 is explained by the ADP dissociation step
( ) from acto-M5CIQ1, since the ATP
binding step does not limit the dissociation rate at these ATP concentrations.
The obtained ADP dissociation rate from acto-M5CIQ1 was significantly larger
than the Vmax of the steady-state ATPase cycle rate of
M5CIQ1, in contrast to myosin Va and Vb, whose rates of ADP dissociation
explain the overall actin-activated ATPase cycling rate
(13,
22). These results suggest
that the ADP dissociation step is not the rate-limiting step in the ATPase
cycle of myosin Vc, which is quite different from myosin Va and Vb.
) from acto-M5CIQ1, since the ATP
binding step does not limit the dissociation rate at these ATP concentrations.
The obtained ADP dissociation rate from acto-M5CIQ1 was significantly larger
than the Vmax of the steady-state ATPase cycle rate of
M5CIQ1, in contrast to myosin Va and Vb, whose rates of ADP dissociation
explain the overall actin-activated ATPase cycling rate
(13,
22). These results suggest
that the ADP dissociation step is not the rate-limiting step in the ATPase
cycle of myosin Vc, which is quite different from myosin Va and Vb.
Actin Binding to M5CIQ1—The rate of actin binding to M5CIQ1 was measured using pyrene-actin. The fluorescence intensity of pyrene-actin was decreased upon the binding of M5CIQ1 to pyrene-actin, as is known for other myosins (13, 42, 43). The time courses of the change in the pyrene fluorescence intensity followed single exponential kinetics (Fig. 10A, inset), and the kobs increased linearly with actin concentration (Fig. 10A) to yield a second order rate constant (k-12) of 1.11 ± 0.03 μm-1 s-1. The dissociation rate of actin from acto-M5CIQ1 was measured by mixing an excess amount of nonlabeled actin with pyrene-acto-M5CIQ1. The time courses of the fluorescence change followed single exponential kinetics, giving the rate constant of k+12 = 0.011 ± 0.001 s-1 (Fig. 10C). The affinity of M5CIQ1 for actin (K12) was calculated to be 9.9 nm, which is 2000-fold weaker than that of myosin Va (13).
FIGURE 10.

Kinetics of pyrene-actin interaction with M5CIQ1. A, kinetics of pyrene-actin binding to M5CIQ1 in the absence of ADP. The rates of pyrene-actin binding to M5CIQ1 in the absence of ADP were measured as a function of pyrene-actin concentration. M5CIQ1 was mixed with various concentrations of pyrene-actin, and time courses of the decrease in the pyrene fluorescence were monitored. The apparent rate constant obtained by single exponential fitting increased with pyrene-actin concentration to give a second order rate constant (k-12) of 1.11 ± 0.03 μm-1 s-1. Inset, a typical recording of the binding of 4 μm pyrene-actin to M5CIQ1. The solid line is the best fit to single exponential kinetics, with kobs of 4.5 s-1. B, kinetics of pyrene-actin binding to M5CIQ1 in the presence of ADP. The rates of pyrene-actin binding to M5CIQ1 in the presence of 50 μm MgADP were measured as a function of pyrene-actin concentration. The apparent rate constant obtained by single exponential fitting increased linearly with pyrene-actin concentration to give a second order rate constant (k-6) of 0.88 ± 0.02 μm-1 s-1. The inset shows a typical recording of the binding of 4 μm pyrene-actin to M5CIQ1·ADP. The solid line is the best fit to single exponential kinetics, with kobs of 3.5 s-1. C, kinetics of pyrene-actin dissociation from acto-M5CIQ1. The rates of pyrene-actin dissociation from acto-M5CIQ1 or acto-M5CIQ1·ADP were measured by mixing acto-M5CIQ1 (0.5 μm M5CIQ1 and 0.7 μm pyrene-actin) in the presence or absence of 50 μm MgADP with an excess amount of nonlabeled actin (25 μm). The apparent rate constants obtained by single exponential fitting were 0.011 ± 0.001 s-1 (k-12) and 0.0099 ± 0.0015 s-1 (k-10), in the absence and presence of ADP, respectively. The error bars represent the S.E. from 3–5 independent experiments. Other experimental conditions are as described in the legend to Fig. 3.
The rate of actin binding to M5CIQ1 was also measured in the presence of ADP. The second order rate constant (k-10) of 0.88 ± 0.02 μm-1 s-1 was obtained (Fig. 10B). The dissociation rate of actin from the pyrene-acto-M5CIQ1·ADP complex was measured as described above to yield the rate constant of k+10 of 0.0099 ± 0.0015 s-1 (Fig. 10C). The calculated affinity of M5CIQ1·ADP for actin (K10), 11.3 nm, is almost identical to that of myosin Va (13).
Actin Affinity of M5CIQ1 in Weak Binding State—Studies on myosin Va using nonhydrolyzed nucleotide analogues have shown that the actin affinity of myosin Va in the weak binding state is much tighter (10–20-fold) than that of nonprocessive conventional myosin II (44). We examined the affinity of myosin Vc for actin in the weak binding state by using nonhydrolyzed nucleotide analogues, ATPγS and AMPPNP. Actin cosedimentation assays in the presence of ATPγS or AMPPNP were performed in 25 mm KCl (Fig. 11). In the presence of ATPγS, the actin-bound fraction of M5CIQ1 was less than 50% at 70 μm actin, suggesting that the apparent affinity of M5CIQ1 for actin in the presence of ATPγS(Kd(ATPγS)) was >70 μm. The apparent affinity of M5CIQ1 for actin in the presence of AMP-PNP (Kd(AMPPNP)) was 6.5 ± 0.6 μm. These values of M5CIQ1 are 6–20-fold weaker than those of myosin Va in 50 mm KCl (44). These results clearly showed that the affinity of myosin Vc for actin in the weak binding state is much weaker than that of myosin Va and rather similar to that of conventional myosin II.
FIGURE 11.

Apparent binding affinities of M5CIQ1 for actin in the presence of ATP analogues. Actin cosedimentation assays were performed in the presence of 2.5 mm Mg-ATPγS or 5 mm Mg-AMPPNP at 25 °C. The experimental conditions are as described in the legend to Fig. 3. Apparent binding affinities of M5CIQ1 for actin were >70 μm in the presence of ATPγS(Kd(ATPγS)) and 6.5 ± 0.6 μm in the presence of AMPPNP (Kd(AMPPNP)).
Multimolecule and Single Molecule in Vitro Motility Assays—To test the motility activity of myosin Vc, we first performed a conventional multimolecule in vitro motility assay using the two-headed myosin Vc (GFP-M5CHMM) construct (Fig. 12A). A majority of actin filaments were moved, and the velocity was 0.16 ± 0.03 μms-1 (Fig. 12A).
FIGURE 12.

Multimolecule and single molecule motility assay of myosin Vc. A, histogram of multimolecule actin gliding velocity of GFP-M5CHMM. GFP-M5CHMM was attached to a coverslip, and the movement of rhodamine-labeled actin filaments was observed with a fluorescent microscope. The solid line shows a fit to a single Gaussian curve. The mean ± S.D. of the actin sliding velocity was 0.16 ± 0.03 μms-1 (n = 51). B, single molecule movement of GFP-myosin Va HMM monitored by TIRF microscope. Almost all fluorescent spots moved in a continuous line and dissociated from actin filament until the photobleaching. Typical time course of the distance from the binding position of individual GFP-myosin Va HMM. Mean velocity was 370 ± 93 nm s-1, and mean travel distance was 710 ± 25 nm at 20 °C. C, single molecule movement of GFP-M5C HMM monitored by TIRF microscope. The fluorescent spots appeared on the actin filament but disappeared without any movements. Typical time course of the distance from the binding position of individual GFP-M5C HMM. Inset, an expanded view of the lowest trace. D, histogram of dwell time of GFP-M5C HMM on the actin filament monitored by TIRF microscope. The dwell time was defined as the period during binding on the actin filament. We did not count the spots on the actin for less than 5 frames (100 ms). The distribution was followed to the single exponential function (solid line), and the time constant was 0.51 s in 10 μm ATP.
To address whether or not myosin Vc is a processive motor, we examined the processivity of myosin Vc using TIRF microscopy. As a control, we used GFP-tagged myosin Va HMM, which is a typical processive motor. We observed the processive movement of GFP-tagged myosin Va HMM with a mean travel length of 710 ± 25 nm and velocity of 370 ± 93 nm s-1 at 20 °C in the presence of 1 mm ATP. 30 of 59 total analyzed moving spots showed more than 1 mm run length. We observed that many molecules traveled over 2 μm. The representative movements of GFP-tagged myosin Va HMM on actin are shown in Fig. 12B. The result was consistent with previous studies (20, 45). Next, we examined GFP-M5CHMM in the same conditions as myosin Va. However, any processive movements of GFP-M5CHMM were not observed. In the absence of ATP, the actin filaments decorated with GFP-M5CHMM were observed under the TIRF microscope. Upon the addition of ATP, the GFP-M5CHMM dissociated from the actin filaments and did not travel on the actin filaments, unlike myosin Va HMM (Fig. 12C). If GFP-M5CHMM moves more than 50 nm, we should be able to see such a movement, but as shown in Fig. 12C, such continuous movements of myosin Vc were not observed. Fig. 12D shows dwell time distribution of GFP-M5CHMM in 10 μm ATP. The ATP hydrolysis turnover rate of acto-M5CHMM at this ATP concentration was 2.5 s-1, which provides the calculated single cycle dwell time of 0.4 s. The observed dwell time from the single molecule TIRF assay (0.5 s) agrees well with the calculated value. If GFP-M5CHMM moves on actin with multiple cycles, the dwell time determined by the TIRF assay should be significantly longer than that shown in Fig. 12D. The tracking accuracy of a single GFP molecule in our optical system was about 30 nm/33 ms, which is calculated from the S.D. value of the tracking traces fixed on the glass surface. Since the kcat of myosin Vc was 6 s-1, GFP-M5CHMM would produce one step per 5 frames (165 ms) on average. Therefore, we should be able to detect the stepwise movements if GFP-M5CHMM generated the step with more than 15 nm, which can be calculated by dividing the tracking accuracy (30 nm) by the route of the number of frames minus 1 (30/(5 - 1)½). However the stepwise movement of GFP-M5CHMM was not detected. The result suggests that while GFP-M5CHMM has the ATP-dependent actin interaction, it cannot remain on the actin filament; thus, it cannot processively travel on the filament. From these results, we concluded that myosin Vc is a nonprocessive motor, which is consistent with the kinetic analyses in this study.
DISCUSSION
Although the overall structures of the various class V myosins resemble each other, the physiological function is thought to be different due to the difference in the target proteins that specify the isoform-specific cargos. In addition, recent studies have raised the question of whether class V myosins have similar motor characteristics, such as processivity. There are three isoforms of myosin V in vertebrates. Although myosin Va and myosin Vb are similar to each other in their motor characteristics that are consistent with their high sequence homology of the motor domain, the mechanoenzymatic characteristics of a less conserved myosin Vc have not been studied. In the present study, we analyzed the ATP hydrolysis mechanism of actomyosin Vc for the first time. All of the rate constants and equilibrium constants obtained in this study are summarized in Table 1. We found several unique features for the actomyosin Vc ATP hydrolysis cycle. First, the ADP release rate from actomyosin Vc was significantly larger than the overall ATPase cycle rate; therefore, it does not solely determine the rate of the ATP hydrolysis cycle. This is quite different from myosin Va and myosin Vb, in which the ADP release step explains 80–90% of the entire cycle rate (13, 22). Second, unlike myosin Va, the Pi burst size is only 0.3, suggesting that the ATP hydrolysis step (K3) is largely shifted to the prehydrolyzed form (MT). Third, the apparent affinity of myosin Vc for actin was much lower than other vertebrate myosin V isoforms. The KATPase of the steady-state ATPase of myosin Vc was 62 μm, which is 44-fold higher than that of myosin Va and 7-fold higher than that of myosin Vb (13, 22) (Table 1). The low affinity of the weak binding state of myosin Vc for actin was also supported by the actin cosedimentation assays in the presence of nonhydrolyzed ATP analogues (Table 1). Since the actin affinity of myosin Vc in the rigor state (K12) was also 2000-fold lower than that of myosin Va (Table 1), the actin-myosin interface participating in the rigor interaction may, in part, contribute to the low affinity of myosin Vc for actin during the ATPase cycle. Although the amino acid sequences in the motor domain among the myosin V family show high homology, a significant difference among the vertebrate myosin V isoforms is found in loop 2. Loop 2 is known to affect the actin binding affinity of myosin, and it has been reported that the charged residues in loop 2 influence the KATPase of myosin Va (46). Therefore, it is plausible that the difference in the loop 2 sequences among the myosin V isoforms is in part responsible for the unique actin affinity of each myosin V isoform. In addition to loop 2, we found that there is a significant sequence difference at the C-loop adjacent to the myopathy loop between the processive myosin (myosin Va and Vb) and myosin Vc. Since it has been shown that the C-loop and the myopathy loop are important for the interaction with actin (41), it is plausible that the sequence difference in the C-loop in addition to the difference in the loop 2 causes the decreased affinity of myosin Vc for actin.
TABLE 1.
Kinetic parameters of M5CIQ1 ATPase cycle and comparison with myosin Va and Vb
| Signal | Myosin Vca | Myosin Vab | Myosin Vbc | |
|---|---|---|---|---|
| Steady state | ||||
| v0 (s–1) | 0.10 ± 0.01 | 0.03 | 0.09 | |
| Vmax (s–1) | 6.5 ± 0.4 | 15 | 9.7 | |
| KATPase (μm) | 62 ± 9 | 1.4 | 8.5 | |
| Kd(ATPγS) (μm) | 70 | 13d | ||
| Kd(AMPPNP) (μm) | 6.5 ± 0.6 | 0.3d | ||
| ATP binding | ||||
| K1k+2 (μm–1 s–1) | Mant-ATP | 2.4 ± 0.1 | 1.6 | 0.78 |
| Tryptophan | 2.5 ± 0.1 | 1.5 | ||
 (μm–1 s–1)
(μm–1 s–1)
|
Mant-ATP | 1.6 ± 0.1 | 0.9 | 0.42 |
| Light scattering | 1.6 ± 0.1 | 0.31 | ||
| Pyrene-actin | 1.8 ± 0.1 | 0.9 | ||
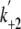 (s–1)
(s–1)
|
Pyrene-actin | >300 | 870 | |
| ATP hydrolysis | ||||
| k+3 + k–3 (s–1) | Tryptophan | 59 ± 3 | 750 | |
| Quenched flow | 55 ± 18 | |||
| K3 | Quenched flow | 0.45 ± 0.04 | 5.3 | |
| Phosphate release | ||||
| k+4,obs (s–1) | MDCC-PBP | 0.11 ± 0.01 | ||
| k+4,obs (s–1) | Quenched flow | 0.064 ± 0.025 | ||
| k+4 (s–1) | k+4,obs(1 + K3)/K3 | 0.34 ± 0.04 | ||
 (s–1)
(s–1)
|
MDCC-PBP | >60 | >250 | |
| ADP binding | ||||
| k+5 (s–1) | Mant-ADP cold chase | 3.9 ± 0.1 | 1.2 | |
| Mant-ADP binding | 3.6 ± 0.7 | |||
| k–5/K6 (μm1 s–1) | Mant-ADP | 2.9 ± 0.1 | 4.6 | |
| K5K6 (μm) | k+5/(k–5/K6) | 1.3 | 0.27 | |
 (s–1)
(s–1)
|
Light scattering (ADP) | 12.7 ± 0.9 | 16e | 12.2 |
| Mant-ADP cold chase | 17.7 ± 0.6 | |||
| Mant-ADP binding | 17.1 ± 1.3 | |||
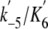 (μm1 s–1)
(μm1 s–1)
|
Mant-ADP | 6.0 ± 0.4 | 12.6 | |
 (μm)
(μm)
|
 |
2.1 | 0.93 | |
| Actin binding | ||||
| k–12 (μm1 s–1) | Pyrene-actin | 1.11 ± 0.03 | 73 | |
| k+12 (s–1) | Pyrene-actin | 0.011 ± 0.001 | 0.00036 | |
| K12 (μm) | k+12/k–12 | 0.0099 | 4.9 × 10–6 | |
| k–10 (μm1 s–1) | Pyrene-actin | 0.88 ± 0.02 | 4.2 | |
| k+10 (s–1) | Pyrene-actin | 0.0099 ± 0.0015 | 0.0032 | |
| K10 (μm) | k+10/k–10 | 0.0113 | 0.0076 |
25 mm KCl, 20 mm MOPS (pH 7.5), 2 mm MgCl2, 1 mm EGTA, 1 mm DTT
Data from Ref. 13; 50 mm KCl, 10 mm imidazole (pH 7.0), 1 mm MgCl2, 1 mm EGTA, 1 mm DTT
Data from Ref. 22; 50 mm KCl, 20 mm MOPS (pH 7.0), 3 mm MgCl2, 1 mm EGTA, 1 mm DTT
Data from Ref. 44; 50 mm KCl, 10 mm imidazole (pH 7.0), 1 mm MgCl2, 1 mm EGTA, 1 mm DTT
Pyrene actin
Based on the obtained rate constants and equilibrium constants of each
elementary kinetic step of the actomyosin Vc ATPase cycle, we carried out the
simulation of the steady-state ATPase activity under a saturating ATP
concentration with an ATP regeneration system as a function of actin
concentration. It should be noted that, since K8,
K9, and 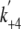 were not
determined directly in this study, we assumed the values of these parameters
to fit the experimentally obtained ATPase activity. The result is shown by the
broken line in Fig. 2.
The contribution of k+4, [M], and [MD] to the overall
ATPase rate was ignored for the simplicity of the simulation. The initial
values, rates, and equilibrium constants employed in the simulation were as
follows. [AM]0 = 20 nm, [AMT]0 =
[MT]0 = [MDP]0 = [AMDP]0 = [AMD]0
= 0 nm,
were not
determined directly in this study, we assumed the values of these parameters
to fit the experimentally obtained ATPase activity. The result is shown by the
broken line in Fig. 2.
The contribution of k+4, [M], and [MD] to the overall
ATPase rate was ignored for the simplicity of the simulation. The initial
values, rates, and equilibrium constants employed in the simulation were as
follows. [AM]0 = 20 nm, [AMT]0 =
[MT]0 = [MDP]0 = [AMDP]0 = [AMD]0
= 0 nm,
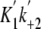 [ATP] =
500 s-1, K8 = 190 μm (rapid
equilibrium), k+3 = 19 s-1,
k-3 = 41 s-1 (K3 = 0.46),
K9 = 340 μm (rapid equilibrium),
[ATP] =
500 s-1, K8 = 190 μm (rapid
equilibrium), k+3 = 19 s-1,
k-3 = 41 s-1 (K3 = 0.46),
K9 = 340 μm (rapid equilibrium),
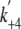 = 155 s-1,
= 155 s-1,
 = 12.7 s-1.
Vmax and KATPase calculated by this
simulation were 6.3 s-1 and 50 μm, respectively.
These values were in agreement with the experimentally obtained values,
supporting the validity of the kinetic model
(Fig. 13A).
= 12.7 s-1.
Vmax and KATPase calculated by this
simulation were 6.3 s-1 and 50 μm, respectively.
These values were in agreement with the experimentally obtained values,
supporting the validity of the kinetic model
(Fig. 13A).
The basal steady-state ATPase activity of myosin Vc is also well explained by the kinetic constants obtained in the present study. The basal steady-state ATPase activity (v0 = 0.10 ± 0.01 s-1) is in agreement with the observed phosphate release rate (k+4,obs) of 0.11 ± 0.01 s-1, which is explained by a combination of the slow phosphate release rate (k+4) and the unfavorable equilibrium of the ATP hydrolysis step (K3).
On the other hand, in the presence of actin, the steady-state ATPase
activity was markedly activated by actin. The ATP hydrolysis rate
(k+3 + k-3), as well as the phosphate
release rate from AMDP complex (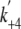 ), is
much faster than the entire ATPase cycle rate, thus not the rate-limiting
steps. Since the ADP release rate is 2-fold faster than the
Vmax, it cannot explain the overall cycle rate, although
it partially limits the ATPase cycle. The overall ATPase cycle rate of
actomyosin Vc can be primarily explained by the unfavorable equilibrium of the
ATP hydrolysis step (K3), the relatively low actin
rebinding rate constant
(
), is
much faster than the entire ATPase cycle rate, thus not the rate-limiting
steps. Since the ADP release rate is 2-fold faster than the
Vmax, it cannot explain the overall cycle rate, although
it partially limits the ATPase cycle. The overall ATPase cycle rate of
actomyosin Vc can be primarily explained by the unfavorable equilibrium of the
ATP hydrolysis step (K3), the relatively low actin
rebinding rate constant
(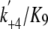 ), and the ADP
release rate from AMD (
), and the ADP
release rate from AMD ( ). The major
kinetic pathway is shown in gray shading in
Fig 13A. These
results suggest that myosin Vc spends a significant time during the ATPase
cycle in the “weak” actin binding forms. We calculated the duty
ratio of myosin Vc based on our kinetic model
(Fig. 13B). In
contrast to myosin Va, the duty ratio was saturated at a high actin
concentration that is consistent with the weak affinity of myosin Vc for
actin. With a saturating actin concentration, the duty ratio of myosin Vc was
estimated to be ∼0.33, which is much lower than those of myosin Va (0.7)
(13) and myosin Vb (∼0.8)
(22). This result supports the
notion described above that myosin Vc spends the majority of the ATP cycle in
the weak actin-binding state, in contrast to myosin Va and Vb, whose major
steady-state intermediates in the ATPase cycle are in the strong actin binding
state. It has been suggested previously that the equilibrium constant of the
hydrolysis step (K3) may be increased at a higher
temperature in cells (35,
47), and this could influence
the duty ratio. However, the Pi burst size of M5CIQ1 at 30 °C
was virtually the same as that at 25 °C (data not shown). Therefore, the
possible increase in the Pi burst size at the physiological
temperature would be minimal, and it is unlikely that myosin Vc becomes a high
duty ratio motor even at body temperature.
). The major
kinetic pathway is shown in gray shading in
Fig 13A. These
results suggest that myosin Vc spends a significant time during the ATPase
cycle in the “weak” actin binding forms. We calculated the duty
ratio of myosin Vc based on our kinetic model
(Fig. 13B). In
contrast to myosin Va, the duty ratio was saturated at a high actin
concentration that is consistent with the weak affinity of myosin Vc for
actin. With a saturating actin concentration, the duty ratio of myosin Vc was
estimated to be ∼0.33, which is much lower than those of myosin Va (0.7)
(13) and myosin Vb (∼0.8)
(22). This result supports the
notion described above that myosin Vc spends the majority of the ATP cycle in
the weak actin-binding state, in contrast to myosin Va and Vb, whose major
steady-state intermediates in the ATPase cycle are in the strong actin binding
state. It has been suggested previously that the equilibrium constant of the
hydrolysis step (K3) may be increased at a higher
temperature in cells (35,
47), and this could influence
the duty ratio. However, the Pi burst size of M5CIQ1 at 30 °C
was virtually the same as that at 25 °C (data not shown). Therefore, the
possible increase in the Pi burst size at the physiological
temperature would be minimal, and it is unlikely that myosin Vc becomes a high
duty ratio motor even at body temperature.
One of the most important issues to evaluate the physiological role of the motor proteins is the processivity, because it is thought that the processive motors are suitable for the cargo transporter in cells, whereas the nonprocessive myosin molecules function better as a force producer by simultaneously interacting with a single actin filament, thus producing a large force. It has been thought that a high duty ratio (>0.5) is required for the processive motor; therefore, our kinetic study that shows myosin Vc to be a low duty ratio motor suggests that this myosin V is a nonprocessive motor. Consistently, the single molecule assays in this study using GFP-M5CHMM indicate that myosin Vc is a nonprocessive motor. Recently, it was reported that Drosophila myosin V is a low duty ratio motor (21), suggesting the presence of a nonprocessive motor in the myosin V family. The present results are consistent with this notion and further suggest the presence of the nonprocessive type of myosin V in vertebrates.
The cell biological studies on myosin Vc showed that myosin Vc colocalizes with Rab8, suggesting the function of myosin Vc in the membrane trafficking (7). How does myosin Vc function as a cargo transporter despite a low duty ratio? One possibility is that myosin Vc forms clusters on the surface of the cargo and transports it processively, although this is not a preferable mechanism for stable cargo transportation, because if the number of molecules in a cluster of myosin Vc is too large, the molecules may interfere with the movement of each other, and if too few, they do not serve continuous movement. Another possibility is that there is an unknown protein that associates with myosin Vc and tethers it to the actin filament, thus acquiring the processive movement. Further biochemical, biophysical, and cell biological studies are required for the function of myosin Vc to provide a clue to this question.
Acknowledgments
We thank Dr. H. D. White (Eastern Virginia Medical School) for providing the PBP cDNA clone.
This work was supported by National Institutes of Health Grants DC006103, AR 048526, AR 048898, and AR 41653. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: TIRF, total internal reflection fluorescence; HMM, heavy meromyosin; AMPPNP, adenosine 5′-(β,γ-imido)triphosphate; ATPγS, adenosine 5′-[γ-thio]triphosphate; MOPS, 4-morpholinepropanesulfonic acid; MDCC, 7-diethylamino-3-((((2-maleimidyl)ethyl)-amino)carbonyl)coumarin; PBP, phosphate-binding protein; dmant-ATP, 2′-deoxy, N-methylanthraniloyl-ATP; GFP, green fluorescent protein; DTT, dithiothreitol; dmant-ADP, 2′-deoxy, N-methylanthraniloyl-ADP.
References
- 1.Nascimento, A. A., Amaral, R. G., Bizario, J. C., Larson, R. E., and Espreafico, E. M. (1997) Mol. Biol. Cell 8 1971-1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu, X., Bowers, B., Wei, Q., Kocher, B., and Hammer, J. A., III (1997) J. Cell Sci. 110 847-859 [DOI] [PubMed] [Google Scholar]
- 3.Takagishi, Y., Oda, S., Hayasaka, S., Dekker-Ohno, K., Shikata, T., Inouye, M., and Yamamura, H. (1996) Neurosci. Lett. 215 169-172 [DOI] [PubMed] [Google Scholar]
- 4.Prekeris, R., and Terrian, D. M. (1997) J. Cell Biol. 137 1589-1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bridgman, P. C. (1999) J. Cell Biol. 146 1045-1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tilelli, C. Q., Martins, A. R., Larson, R. E., and Garcia-Cairasco, N. (2003) Neuroscience 121 573-586 [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez, O. C., and Cheney, R. E. (2002) J. Cell Sci. 115 991-1004 [DOI] [PubMed] [Google Scholar]
- 8.Lapierre, L. A., Kumar, R., Hales, C. M., Navarre, J., Bhartur, S. G., Burnette, J. O., Provance, D. W., Jr., Mercer, J. A., Bahler, M., and Goldenring, J. R. (2001) Mol. Biol. Cell 12 1843-1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan, G. H., Lapierre, L. A., Goldenring, J. R., Sai, J., and Richmond, A. (2004) Mol. Biol. Cell 15 2456-2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volpicelli, L. A., Lah, J. J., Fang, G., Goldenring, J. R., and Levey, A. I. (2002) J. Neurosci. 22 9776-9784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehta, A. D., Rock, R. S., Rief, M., Spudich, J. A., Mooseker, M. S., and Cheney, R. E. (1999) Nature 400 590-593 [DOI] [PubMed] [Google Scholar]
- 12.Sakamoto, T., Amitani, I., Yokota, E., and Ando, T. (2000) Biochem. Biophys. Res. Commun. 272 586-590 [DOI] [PubMed] [Google Scholar]
- 13.De La Cruz, E. M., Wells, A. L., Rosenfeld, S. S., Ostap, E. M., and Sweeney, H. L. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 13726-13731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reck-Peterson, S. L., Tyska, M. J., Novick, P. J., and Mooseker, M. S. (2001) J. Cell Biol. 153 1121-1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill, K. L., Catlett, N. L., and Weisman, L. S. (1996) J. Cell Biol. 135 1535-1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schott, D., Ho, J., Pruyne, D., and Bretscher, A. (1999) J. Cell Biol. 147 791-808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossanese, O. W., Reinke, C. A., Bevis, B. J., Hammond, A. T., Sears, I. B., O'Connor, J., and Glick, B. S. (2001) J. Cell Biol. 153 47-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertrand, E., Chartrand, P., Schaefer, M., Shenoy, S. M., Singer, R. H., and Long, R. M. (1998) Mol. Cell 2 437-445 [DOI] [PubMed] [Google Scholar]
- 19.Munchow, S., Sauter, C., and Jansen, R. P. (1999) J. Cell Sci. 112, 1511-1518 [DOI] [PubMed] [Google Scholar]
- 20.Krementsova, E. B., Hodges, A. R., Lu, H., and Trybus, K. M. (2006) J. Biol. Chem. 281 6079-6086 [DOI] [PubMed] [Google Scholar]
- 21.Toth, J., Kovacs, M., Wang, F., Nyitray, L., and Sellers, J. R. (2005) J. Biol. Chem. 280 30594-30603 [DOI] [PubMed] [Google Scholar]
- 22.Watanabe, S., Mabuchi, K., Ikebe, R., and Ikebe, M. (2006) Biochemistry 45 2729-2738 [DOI] [PubMed] [Google Scholar]
- 23.Spudich, J. A., and Watt, S. (1971) J. Biol. Chem. 246 4866-4871 [PubMed] [Google Scholar]
- 24.Kouyama, T., and Mihashi, K. (1981) Eur. J. Biochem. 114 33-38 [PubMed] [Google Scholar]
- 25.Brune, M., Hunter, J. L., Corrie, J. E., and Webb, M. R. (1994) Biochemistry 33 8262-8271 [DOI] [PubMed] [Google Scholar]
- 26.White, H. D., Belknap, B., and Webb, M. R. (1997) Biochemistry 36 11828-11836 [DOI] [PubMed] [Google Scholar]
- 27.Ikebe, M., Kambara, T., Stafford, W. F., Sata, M., Katayama, E., and Ikebe, R. (1998) J. Biol. Chem. 273 17702-17707 [DOI] [PubMed] [Google Scholar]
- 28.Watanabe, T. M., Tanaka, H., Iwane, A. H., Maki-Yonekura, S., Homma, K., Inoue, A., Ikebe, R., Yanagida, T., and Ikebe, M. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 9630-9635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe, S., Ikebe, R., and Ikebe, M. (2006) J. Biol. Chem. 281 7151-7160 [DOI] [PubMed] [Google Scholar]
- 30.Homma, K., and Ikebe, M. (2005) J. Biol. Chem. 280 29381-29391 [DOI] [PubMed] [Google Scholar]
- 31.Reynard, A. M., Hass, L. F., Jacobsen, D. D., and Boyer, P. D. (1961) J. Biol. Chem. 236 2277-2283 [PubMed] [Google Scholar]
- 32.Kambara, T., Rhodes, T. E., Ikebe, R., Yamada, M., White, H. D., and Ikebe, M. (1999) J. Biol. Chem. 274 16400-16406 [DOI] [PubMed] [Google Scholar]
- 33.Nishikawa, S., Homma, K., Komori, Y., Iwaki, M., Wazawa, T., Hikikoshi Iwane, A., Saito, J., Ikebe, R., Katayama, E., Yanagida, T., and Ikebe, M. (2002) Biochem. Biophys. Res. Commun. 290 311-317 [DOI] [PubMed] [Google Scholar]
- 34.Espindola, F. S., Suter, D. M., Partata, L. B., Cao, T., Wolenski, J. S., Cheney, R. E., King, S. M., and Mooseker, M. S. (2000) Cell Motil. Cytoskeleton 47 269-281 [DOI] [PubMed] [Google Scholar]
- 35.De La Cruz, E. M., Wells, A. L., Sweeney, H. L., and Ostap, E. M. (2000) Biochemistry 39 14196-14202 [DOI] [PubMed] [Google Scholar]
- 36.Wang, F., Chen, L., Arcucci, O., Harvey, E. V., Bowers, B., Xu, Y., Hammer, J. A., III, and Sellers, J. R. (2000) J. Biol. Chem. 275 4329-4335 [DOI] [PubMed] [Google Scholar]
- 37.Marston, S. B., and Taylor, E. W. (1980) J. Mol. Biol. 139 573-600 [DOI] [PubMed] [Google Scholar]
- 38.Ikebe, M., Koretz, J., and Hartshorne, D. J. (1988) J. Biol. Chem. 263 6432-6437 [PubMed] [Google Scholar]
- 39.Kovacs, M., Wang, F., Hu, A., Zhang, Y., and Sellers, J. R. (2003) J. Biol. Chem. 278 38132-38140 [DOI] [PubMed] [Google Scholar]
- 40.Wang, F., Kovacs, M., Hu, A., Limouze, J., Harvey, E. V., and Sellers, J. R. (2003) J. Biol. Chem. 278 27439-27448 [DOI] [PubMed] [Google Scholar]
- 41.Ajtai, K., Garamszegi, S. P., Watanabe, S., Ikebe, M., and Burghardt, T. P. (2004) J. Biol. Chem. 279 23415-23421 [DOI] [PubMed] [Google Scholar]
- 42.Taylor, E. W. (1991) J. Biol. Chem. 266 294-302 [PubMed] [Google Scholar]
- 43.De La Cruz, E. M., Ostap, E. M., and Sweeney, H. L. (2001) J. Biol. Chem. 276 32373-32381 [DOI] [PubMed] [Google Scholar]
- 44.Yengo, C. M., De la Cruz, E. M., Safer, D., Ostap, E. M., and Sweeney, H. L. (2002) Biochemistry 41 8508-8517 [DOI] [PubMed] [Google Scholar]
- 45.Sakamoto, T., Wang, F., Schmitz, S., Xu, Y., Xu, Q., Molloy, J. E., Veigel, C., and Sellers, J. R. (2003) J. Biol. Chem. 278 29201-29207 [DOI] [PubMed] [Google Scholar]
- 46.Yengo, C. M., and Sweeney, H. L. (2004) Biochemistry 43 2605-2612 [DOI] [PubMed] [Google Scholar]
- 47.Kodama, T. (1985) Physiol. Rev. 65 467-551 [DOI] [PubMed] [Google Scholar]