

Abstract
Nuclear respiratory factor-1 (NRF-1) is integral to the transcriptional regulation of mitochondrial biogenesis, but its control over various respiratory genes overlaps other regulatory elements including those involved in O2 sensing. Aerobic metabolism generally suppresses hypoxia-sensitive genes, e.g. via hypoxia-inducible factor-1 (HIF-1), but mutations in Complex II-succinate dehydrogenase (SDH), a tumor suppressor, stabilize HIF-1, producing pseudo-hypoxia. In aerobic cardiomyocytes, which rely on oxidative phosphorylation, we tested the hypothesis that NRF-1 regulates Complex II expression and opposes hypoxia-inducible factor-1. NRF-1 gene silencing blocked aerobic succinate oxidation, increasing nuclear HIF-1α protein prior to the loss of Complex I function. We postulated that NRF-1 suppression either specifically decreases the expression of one or more SDH subunits and increases succinate availability to regulate HIF-1 prolyl hydroxylases, or stimulates mitochondrial reactive oxygen production, which interferes with HIF-1α degradation. Using promoter analysis, gene silencing, and chromatin immunoprecipitation, NRF-1 was found to bind to the gene promoters of two of four nuclear-encoded Complex II subunits: SDHa and SDHd, but the enzyme activity was dynamically regulated through the catalytic SDHa flavoprotein. Complex II was inactivated by SDHa silencing, which led to aerobic HIF-1α stabilization, nuclear translocation, and enhanced expression of glucose transporters and heme oxygenase-1. This was unrelated to mitochondrial ROS production, reversible by high α-ketoglutarate concentrations, and coherent with regulation of HIF-1 by succinate reported in tumor cells. These findings disclose a novel role for NRF-1 in the transcriptional control of Complex II and prevention of pseudo-hypoxic gene expression in aerobic cardiac cells.
Transcriptional regulation of mitochondrial electron transport chain (ETC)2 proteins requires the expression of both nuclear- and mitochondrial-encoded genes that are specifically coordinated by a small number of nuclear factors (e.g. NRF-1, NRF-2, and ERRα) and co-activators of the PGC-1 family (e.g. PGC-1α and PRC) (1-3). However, these nuclear factors vary in their degree of control of various mitochondrial oxidative pathways (4, 5). The NRF-1 transcription factor binds as a homodimer to promoter sites in nuclear-encoded genes involved in the regulation of mitochondrial biogenesis as well as genes encoding selected subunits of the five respiratory complexes (6). NRF-1 also binds regulatory elements in genes involved in cell cycle control and proliferation, overlapping most notably with E2F target genes involved in DNA replication, mitosis, and cytokinesis (7). NRF-1 occupancy has been reported in 5% of gene promoters in living human cells (7), and it is strongly up-regulated by inflammation and mtDNA damage (8). Although NRF-1 has been implicated in nuclear activation of genes encoding subunits of all five respiratory complexes (1), its role in the integrative expression of different mitochondrial electron transport chain proteins is neither clearly elucidated nor is it known whether it has a role in oxygen (O2) sensing.
Mammalian cells sense O2 deprivation by activating the transcription of adaptive hypoxia-inducible genes: glucose transporters, vascular endothelial growth factor, and erythropoietin (9-11). This response facilitates glycolytic ATP production until O2 delivery improves via enhanced erythropoiesis and angiogenesis. O2 sensing is regulated in part by hypoxia-inducible factor-1 (HIF-1), a heterodimer of HIF-1α and HIF-1β of the ARNT family of transcription factors (10). Both subunits are required for DNA binding and gene activation, but only HIF-1α is regulated by the cellular O2 tension. In the presence of O2, the HIF-1α subunit is targeted for proteosomal degradation by prolyl hydroxylases (PHD 1-3) that utilize α-ketoglutarate in the presence of ferrous iron and ascorbate to hydroxylate specific proline residues catalytically and liberate CO2 and succinate (11).
The stabilization of HIF-1α under aerobic conditions, called pseudo-hypoxia, is a feature of some tumors related to inactivation of mitochondrial Complex II, succinate dehydrogenase (SDH; succinate:ubiquinone oxidoreductase) (12-14), highlighting its role as a mitochondrial tumor suppressor. HIF-1 activation by SDH inactivation has been proposed to involve one of two mitochondrial mechanisms, one entails mitochondrial reactive oxygen species (ROS) generation, while the other requires egress of mitochondrial succinate; either would inhibit cytoplasmic HIF PHDs and stabilize the α-subunit (12, 15). In hypoxia, the connections between the mitochondrial ETC, ROS production, and HIF-1α expression are physiologically important, and the mechanisms have generated some controversy (15-18).
Understanding ETC involvement in hypoxic gene activation is also necessary to comprehend respiratory chain anomalies involved in disease pathogenesis, including human cardiomyopathy (19, 20). Complex II inhibition or the generation of ROS at Complex III leading to HIF-1 activation have implicated ETC disturbances to hypoxic gene expression; moreover, Complex I or IV defects that allow the molecular O2 concentration in the cell to rise may destabilize HIF-1α, e.g. as shown by nitric oxide binding to the oxidase (21). Under such conditions, down-regulation of NRF-1-regulated gene expression could increase HIF-1α stability by multiple mechanisms. Here we tested the hypothesis that NRF-1 interference promotes HIF-1 activity in aerobic cardiomyocytes by either: (i) decreasing specific SDH subunit expression and increasing succinate availability, thereby regulating prolyl hydroxylase activity, or (ii) enhancing mitochondrial reactive oxygen species (ROS) production at Complex II or III that prevents the degradation of the HIF-1 α-subunit.
EXPERIMENTAL PROCEDURES
Cell Culture—Two cell lines were used: H9c2 cells derived from embryonic rat ventricle (American Type Culture Collection) and primary ventricular cardiomyocytes isolated from rat heart. H9c2 cells were seeded at 5 × 105 cells/100-mm plate and cultured at 37 °C in a 5% CO2 humidified atmosphere in Dulbecco's modified Eagle's medium (DMEM) supplemented with 0.2 mm glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (high serum, growth-promoting medium) renewed every second day. Cells were switched to DMEM supplemented with 1% fetal bovine serum (low serum, differentiation-promoting medium, DM) for differentiation after reaching subconfluence (70-80%, 3-4 days). H9c2 cell morphology was followed daily by phase contrast microscopy to confirm differentiation, including myotube formation and expression of myogenic transcription factors, calcium channel proteins, and LIM protein FHL2 (22, 23). Rat ventricular cardiomyocytes were isolated using a kit (Worthington, Lakewood, NJ) from 3-day-old Sprague-Dawley rats under an approved animal protocol and the cardiomyocytes cultured at 37 °C (density of 6 × 104 cells/cm2) in DMEM/F12 supplemented with antibiotics, 10% horse serum (Sigma-Aldrich), 5% heat-inactivated fetal bovine serum (HyClone, Logan, UT), and 10 μg/ml fibronectin. These cardiomyocytes beat spontaneously, and their purity was >97% by immunochemical staining with anti-cardiac sarcomeric α-actinin and fluorescence microscopy.
Cells for transfection were grown in standard culture dishes, and those for immunofluorescence and fluorescent oxidative stress assays were grown on Nunc Lab-Tech Permanox glass chamber two-well slides (Fisher Scientific). All experiments were performed in separate triplicate cultures, and ten groups were analyzed: control cells grown in air + 5% CO2 (aerobic conditions); cells grown in aerobic conditions plus dichloromethane (DCM, 50-100 μm for 24 h); aerobic cells exposed to DMOG (1 mm) for 24 h; aerobic cells fed α-ketoglutarate (2 mm); aerobic cells transfected with small interfering (si) SDHa, siNRF-1, or SDHa, or siNRF-1 plus SDHa or with scrambled siRNA; or aerobic cells incubated with 5 μm MG132 for 24 h. Final doses were chosen from preliminary studies.
Antibodies and Other Reagents—A specific polyclonal anti-NRF-1 was developed and characterized using a unique peptide of amino acid sequence 262-279 of human NRF-1, highly homologous to the mouse and rat (24, 25). The peptide was conjugated to keyhole limpet hemocyanin and injected into rabbits. Antiserum was screened for specificity against recombinant NRF-1 produced by in vitro translation in reticulocyte lysate (Promega, Madison, WI). The antibody was adsorbed to the peptide and tested by Western blot analysis to confirm NRF-1 specificity.
Anti-tubulin and anti-β-actin were obtained from Sigma, and anti-myogenin, Glut1, Glut2, and Glut4 antibodies from Santa Cruz Biotechnology. Anti-HIF1α was from either Transduction Laboratories (Lexington, KY) or Novus Biologicals (Littleton, CO). Anti-Hyp-P564 antibody was obtained from Genaxis. All other chemicals and reagents, including RPMI and DMEM with 25 mm Hepes and 4.5 g/liter glucose, were from Sigma.
Gene Silencing—These experiments were performed using small interfering NRF-1 (siNRF-1) or siSDHa duplexes to target sequences in the open-reading frames of NRF-1 or SDHa mRNA. Multiple nucleotide sense and antisense siRNAs were synthesized and obtained in annealed form from Ambion. SiRNA target sequences were submitted to BLAST searches against other rat genome sequences to ensure specificity. After preliminary studies, one pair of siRNA sequences was selected and transfected at concentrations of 40 or 80 μm into cells using FuGENE 6 (Roche Applied Sciences) sense NRF-1, 5′-GGAGGUUAAUUCAGAGCUG-3′, and antisense NRF-1, 5′-CAGCUCUGAAUUAACCUCC-3′ and sense SDHa, 5′-GCAAGAAGGCAUCCGCUAAtt-3′, and antisense SDHa, 5′-UUAGCGGAUGCCUUCUUGCtc-3′. A scrambled negative control siRNA was also used. The experiments were conducted at transfection efficiencies of 60-80% determined by co-transfection with fluorescent double-stranded oligonucleotides. Inhibition of NRF-1 by siNRF-1 was established by Western blot analysis and inhibition of SDHa by siSDHa by RT-PCR using specific primers for rat SDHa and by Western blot with mouse monoclonal anti-human SDHa and SDHb (Invitrogen). Cardiomyocytes were transfected with SDHa cloned in pCMV6-XL5 expression vector (OriGene, Rockville, MD) 48 h after plating using FuGENE 6 and transfection efficiency was checked by co-transfection with a green fluorescent protein construct. Cells were exposed to the transfection mix for 8 h, then grown in PC-1/F-12 minimal essential medium for 24, 48, or 72 h.
Nuclear and Mitochondrial Proteins and Respiration—The nuclear and mitochondrial fractions were obtained from disrupted cells by differential centrifugation. Nuclear extracts were prepared in 0.3 m NaCl, 1 m urea, 1% Nonidet P-40, 25 mm HEPES (pH 7.9), and 1 mm dithiothreitol (26). Respiration was measured by polarography at 35 °C in digitonin (0.02%)-permeabilized cells in a standard respiration buffer with or without additions of 0.5 mm ADP, uncoupler 100 μm carbonylcyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP), Complex III inhibitor 50 μm antimycin A, or substrates: 1 mm ascorbate + 100 μm TMPD, 5-10 mm succinate, or 2.5 mm glutamate + 2.5 mm malate (25, 27). Protein concentrations were determined by the Bradford method (Bio-Rad).
Western Blot Analysis—Cells were transfected with siNRF-1 or siSDHa or scrambled RNA (scRNA) using FuGENE 6 and grown for 24, 48, or 72 h post-transfection. For Western blots, cells were lysed in 50 mm Tris-HCl at pH 8.0, 150 mm NaCl, 1% Triton, 0.1 μg/ml phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, and 150 mm mercaptoethanol. The samples were sonicated, boiled in Laemmli buffer (10-20 μg of protein), followed by electrophoresis on 8 or 12% sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinyldifluoride membranes. Nuclear extracts were also prepared from cultured cells to check for HIF-1α nuclear protein (24). For electrophoresis, 15-20 μg of cellular, nuclear, or mitochondrial protein per lane were used, membranes blocked with 5% dry de-fatted milk, incubated with monoclonal anti-HIF-1α antibody or anti-NRF-1 or anti-SDH, followed by secondary antibody and detection using enhanced chemiluminescence (Amersham Biosciences).
Immunoprecipitation—Cell lysates were immunoprecipitated using anti-HIF-1 antibody conjugated to protein A-Sepharose. The precipitates were washed in 80 mm Tris-HCl, pH 7.4, 200 mm NaCl, 20 mm EDTA, 50 mm NaF, 2 mm Na3VO4, 1% (v/v) Nonidet P-40, 50 mg/ml phenylmethylsulfonyl fluoride, 1 mg/ml leupeptin, and 10 mg/ml aprotinin and further washed with 10 mm Tris-HCl, pH 7.4, 100 nm NaCl, and 100 mm Na3VO4. The samples were boiled in Laemmli buffer and separated on 8% SDS-PAGE, transferred to nitrocellulose, and blotted with Anti-Hyp-P564. The blots were developed, and spots quantified on digitized images from the mid-dynamic range. Analysis of band intensities was performed by Image-Quant (Bio-Rad).
RNA Extraction and Quantitative Real-time RT-PCR—Total RNA was extracted from cells followed by reverse transcription with a first-strand RT-PCR kit (Promega) using the manufacturer's instructions. Preliminary assays were done by conventional RT-PCR using the primer sequences in Table 1. Quantitative real-time PCR was performed with TaqMan gene expression and premix assays for SDHa, -b, -c, and -d (Applied Biosystems) on an ABI Prism 7000 (Applied Biosystems) Real-time PCR system. To detect SDH subunit induction, relative expression for each assay was normalized against GAPDH (TaqMan® Endogenous Controls) using the recommended comparative threshold cycle CT method. Because of the exponential PCR reaction, a difference of n in Ct values represents a 2-fold difference in transcript levels. PCR was performed in triplicate, and experiments were repeated in triplicate.
TABLE 1.
Gene primers used for RT-PCR and PCR
| Gene | Oligonucleotide (5′-3′) | Annealing temperature |
|---|---|---|
| °C | ||
| SDHa (mRNA) | AACACTGGAGGAAGCACACC | 60 |
| GCAACTCGAGTCCCTCACAT | ||
| SDHb (mRNA) | GGAGGGCAAGCAACAGTATC | 60 |
| TGCAATTGCTTTTCCTGGAT | ||
| SDHc (mRNA) | TTGGTTCTTGCAGTGCTGTC | 60 |
| CAAGAAGCAGCACAAAGCTG | ||
| SDHd (mRNA) | CACATCCACCTGTCACCAAG | 60 |
| AAGTAGCAAAGCCCAGCAAA | ||
| SDHa (DNA) | CACTTACCCCTGGAGCAAAG | 68 |
| CTGGTGTGGCGCTTGGAAG | ||
| SDHb (DNA) | TTCCCTCTTTCCCGCTGCTC | 68 |
| TTTTGGCTCCTGACGTCAGCC | ||
| SDHc (DNA) | ATGCACTAACTAATCACTC | 68 |
| GGACGCTGTCATGTGACACC | ||
| SDHd (DNA) | GGCCATGGTCAGCGTCAAT | 68 |
| CCCAGCTTTAAGAGAACCGCC |
In-gel Enzyme Activity Staining—In-gel staining of SDH activity relies on formazan precipitation as purple crystals derived from the reduction of nitro-blue tetrazolium (NBT). The colorless dye accepts hydrogen during oxidation by NADH and FADH2 dehydrogenases. NBT accepts hydrogen directly from NADH systems, e.g. Complex I, whereas dye reduction by SDH is enhanced by an artificial electron carrier, phenazine methosulfate (PMS). In control H9c2 cells or those transfected with siSDHa for 24 h, the plasma membrane was disrupted, the nuclei removed, and the mitochondrial complexes separated by non-denaturing PAGE. The gels were rinsed in distilled H2O and incubated in PMS and NBT buffer for varying times in the dark at room temperature (28). The reactions were stopped by fixation in 45% (v/v) methanol and 10% (v/v) acetic acid and the gels de-stained overnight in the same solution.
Chromatin Immunoprecipitation Assays—H9c2 cells (∼4.0 × 106) cultured in 15-cm plates were treated 100 μm DCM or transfected with siNRF-1. After 24 h, cells were treated with 1% formaldehyde (cross-linker) for 7.5 min, harvested, sonicated (into ∼500 bp fragments) in media to achieve a final concentration of 1%, and incubated at 37 °C for 30 min. Formaldehyde was quenched with 0.125 m glycine. Cells were washed with PBS, harvested, and processed with the ChIP IT Assay Kit (Active Motif) and NRF-1 antibody. After ethanol precipitation, DNA was resuspended in 200 μl/107 cells, and 2-5 μl used for the PCR template. Input samples representing 1% of total DNA were diluted at 1:5 and IP fractions 1:2. PCR was carried out on 1-μl samples using validated primer sets for SDHa, SDHb, SDHc, and SDHd 5′-untranslated regions (Table 1). Total input DNA was also tracked through the assay by PCR using primers that amplify 223 bp from the β-actin promoter. The primer mix came from Active Motif (Carlsbad, CA) and was used as recommended. Preimmune rabbit serum (IgG) was used for IP and purified DNA for PCR of the SDH subunits (Table 1). PCR products were analyzed on 2% ethidium bromide-stained agarose gels and normalized to total DNA in each sample.
Immunofluorescence Microscopy—H9c2 cell were grown in two-well chamber slides to 60-70% confluence to avoid hypoxia. Cells were transfected with siSDHa for 24 h or transfected with siSDHa and incubated with 2 mm α-ketoglutarate for 6 h. Cells were treated with MitoTracker Green (1 μm) for 10 min at 37 °C, fixed in 4% paraformaldehyde, and then 1% Triton X-100 in PBS for 15 min at room temperature. Cells were incubated for 30 min at room temperature with 10% normal horse serum (Dako Corp.) in PBS and for 2 h with anti-HIF-1α at 1:100 in PBS containing 5% horse serum. After three 15-min washes in PBS with gentle agitation, cells were labeled for 1 h with goat anti-mouse FluoroLink Cy3-labeled (Molecular Probes) at a 1:100 dilution in PBS containing 5% horse serum, washed three times in PBS, cover-slipped using Slow Fade (Invitrogen) mounting medium, and observed by confocal microscopy (Zeiss 410 LSCM). For all samples, images were collected at ×620 magnification in the fluorescence mode using identical settings, and images merged electronically.
ROS Production—To localize ROS production, cells were transfected or exposed to DCM/CO and incubated 24-h later with MitoSox (1 μm) and MitoTracker Green (Molecular Probes) for 10 min followed by fluorescence confocal microscopy at 568 nm (MitoSox) and 488 nm (MitoTracker) on a Model 410 LSCM (Carl Zeiss MicroImaging, Inc). Control and treated samples were scanned at ×620 using the same parameters, and the images collected in fluorescence mode and merged electronically.
Statistics—Grouped data were expressed as means ± S.E. Statistical analyses were performed by two-way analysis of variance using commercial software. A value of p < 0.05 was accepted as significant.
RESULTS
Respiration and Mitochondrial Complex II Activity—Oxidative phosphorylation substrate utilization in H9c2 cardiomyocytes was examined before and after NRF-1 silencing. As a positive control, NRF-1 expression, which in these cells involves mitochondrial oxidant production, was stimulated with DCM/CO (25). At the baseline, the cells expressed immunoreactive nuclear NRF-1 protein, which was increased 2-fold by DCM/CO and blocked (>90%) 24 h after transfection with siRNA (Fig. 1A). scRNA had no effect on NRF-1 protein expression.
FIGURE 1.

NRF-1 knockdown using siRNA targeting of the NRF-1 open-reading frame in H9c2 cells. A, histogram and Western blot for nuclear NRF-1 protein normalized to tubulin. First column and lane 1 show NRF-1 in H9c2 cells after DCM treatment for 24 h to induce NRF-1 (positive control). Second column and lane 2 show nuclear NRF-1 expression in control cells. Third column and lane 3 show the effect of siRNA (80 μm) on nuclear NRF-1 protein at 24 h. (Mean + S.E. for n = 3; symbols indicate p < 0.05 compared with control). B, respiration studies demonstrating the early loss of FAD-linked before NADH-linked substrate oxidation at 24 h after siNRF-1 with 40 or 80 μm oligonucleotide. Substrates were 5 mm succinate or 2.5 mm malate + 2.5 mm glutamate.
Within 24 h after NRF-1 silencing, cell respiration became compromised; however, loss of FADH-linked respiration appeared early and was more sensitive to siNRF-1 than NADH-linked (malate + glutamate) respiration (Fig. 1B). scRNA had no effect on respiration, and the oxidation of ascorbate-TMPD (+antimycin A) was intact 24 h after NRF-1 silencing (data not shown). NRF-1-dependent gene expression is thus necessary for carbon entry into the ETC in rat cardiomyocytes, and Complex II is exquisitely sensitive to loss of NRF-1 gene expression.
SDH Subunit Gene Expression—The effects of NRF-1 silencing on succinate oxidation and the association of Complex II activity with HIF-1α stabilization implicated NRF-1 in the prevention of pseudo-hypoxia through transcriptional regulation SDH, for which all four subunits are nuclear-encoded. On the basis of a bioinformatics promoter analysis, we focused on NRF-1 (and did not investigate NRF-2); moreover, NRF-1 is not up-regulated by mild hypoxia, and the NRF-1 promoter does not contain a canonical HIF-1 consensus motif. Using RT-PCR, we evaluated the extent to which NRF-1 regulates steady-state mRNA levels for SDHa, -b, -c, and -d in H9c2 cardiomyocytes and whether suppressing it deregulates one or more subunits. As a positive control, DCM/CO (50-100 μm) was used to stimulate NRF-1; this increased SDHa mRNA content 3-fold in cardiomyocytes, but SDHb, -c, and -d mRNA less than 2-fold (Fig. 2A). siNRF-1 transfection suppressed mRNA expression for two of four SDH subunits: a and d, for 48 h (Fig. 2B; 18 S rRNA is the densitometry reference for subunit mRNA) but the effect was far greater for SDHa mRNA (∼17-fold). This implies a large dynamic range for NRF-1 in regulating the catalytic flavoprotein that converts succinate to fumarate in Complex II.
FIGURE 2.

Mitochondrial SDH/Complex II gene expression in H9c2 cells. A, gel star-stained 2% agarose gels of Complex II gene products measured by conventional RT-PCR using rat primers for SDHa, SDHb, SDHc, and SDHd in Table 1. The 18 S rRNA product was used to control for RNA loading. Cells were controls or incubated with 100 μm DCM/CO for 24 h or transfected with 80 μm NRF-1 siRNA. B, quantitative real time RT-PCR of mRNA expression for Complex II subunits. Values are mean ± S.D. (n = 4) Asterisks indicate p < 0.05 compared with control. Note 17-fold range for SDHa. C, nuclear HIF-α in control cells or cells transfected with NRF-1 siRNA, scRNA, or exposed to DCM/CO (100 μm) for 24 h. Tubulin is a loading control. D, knockdown experiments with siRNA targeting the open-reading frames of NRF-1 and SDHa in rat cardiomyocytes. Western blots for nuclear HIF-1α and NRF-1 protein are compared with tubulin and SDHa with β-actin. Lane 1, protein expression in control cells. Lane 2, protein expression 24 h after DCM/CO used as a positive control to induce NRF-1. Lane 3, SDHa siRNA (80 μm) increases HIF-1α in cardiomyocytes, but has no effect on NRF-1 and decreases SDHa protein expression. Lane 4, NRF-1 siRNA (80 μm) increases HIF-1α and decreases NRF-1 and SDHa protein expression. Lane 5, scrambled siRNA has no effect on HIF-1α, NRF-1, or SDHa protein expression.
NRF-1 Silencing Increases Nuclear HIF-1α Accumulation and Arrests Cardiomyocyte Differentiation—Studies were done under aerobic conditions to measure the influence of NRF-1 on HIF-1 without the O2 limitation of hypoxia. Aerobic H9c2 cells transfected with siNRF-1 had significant increases in nuclear HIF-1α protein (Fig. 2C). By comparison, DCM/CO, which inhibits cytochrome c oxidase and stimulates mitochondrial ROS in these cells, had the converse effect on nuclear HIF-1α.
After serum removal, embryonic H9c2 rat heart cells proliferate in high serum medium and differentiate over 96 h. Here the use of siNRF-1 inhibited myotubule formation (not shown), thus making it possible that HIF-1α behaves differently in differentiated myocytes. In primary rat cardiomyocytes, nuclear HIF-1α nuclear protein content was stimulated 10-fold by siNRF-1 or siSDHa (Fig. 2D), connecting NRF-1, via mitochondrial ETC function or cell cycle interactions, or both, with HIF-1 regulation throughout cardiac differentiation.
Identification of NRF-1 Recognition Sites in Rat SDH Subunit Promoter Regions—To identify NRF-1-binding motifs in the four SDH subunit genes, the rat SDHa, -b, -c, and d promoters were cloned and sequenced (Fig. 3A). A search of the proximal SDHa promoter for transcription factor-binding sites demonstrated multiple and tandem consensus NRF-1 recognition sites. The rat SDHb promoter revealed two NRF-2 recognition sites and a partial but not a consensus NRF-1 site, and the SDHc promoter revealed no NRF-1, one NRF-2, and multiple tandem Sp1 recognition sites as well as a USF-1 consensus domain (not shown). The proximal SDHd promoter region revealed one NRF-1 and three tandem NRF-2 recognition sites. The NRF-1-binding sites in the SDHa and -d promoters are located within 150 bp of the transcription start site, while the SDHc promoter has three upstream Sp1 sites flanked by NRF-2 sites. A similar configuration of Sp1 and NRF sites has been observed in other genes involved in respiratory protein expression. The five NRF-1 sites are nearly complete matches for canonical NRF-1 binding and contain the invariant nucleotide consensus motif for functional NRF-1 recognition (24).
FIGURE 3.

Nucleotide sequences of the upstream regions of SDHa, SDHb, SDHc, and SDHd in H9c2 cells. A, consensus sequences for transcription factor binding located with DNASIS (Hitachi Software; Alameda, CA) and MatInspector (Genomatix Software; München, Germany). Putative NRF-1 and NRF-2 canonical binding sites of 92-100% homology are underlined (EntrezGene data base ID numbers SDHa 157074, SDHb 298596, SDHc 289217, SDHd 363061). The arrows indicate major transcription start sites. Uppercase letters indicate promoter and intron sequences, and lowercase letters are exon sequences. a, SDHa 5′-region within the 180-nt upstream sequence and the NRF-1 sites underlined; b, SDHb 5′-region within the 180-nt upstream sequence and NRF-2 sites underlined; the underlined sites at -90 to -77 is a partial NRF-1 consensus sequence; c, SDHc 5′-region within the 180-nt upstream sequence, the NRF-2 site is underlined; no NRF-1 site is found; d, SDHd 5′-region within the 180-nt upstream sequence, the NRF-2 sites and single NRF-1 site are underlined. B, ChIP assay for NRF-1 binding to the SDHa, SDHb, SDHc, and SDHd promoters. H9c2 cells were transfected with scRNA or siRNA directed at NRF-1 or exposed to 100 μm DCM/CO or medium alone and subjected to ChIP assays using anti-NRF-1. Input lanes show chromatin PCR product before immunoprecipitation. Precipitated DNA was analyzed by PCR with specific primer sets. Data are from one of three experiments.
In Vivo SDHa, -b, -c, and -d Promoter Binding by NRF-1—The ChIP assay was performed to evaluate NRF-1 binding to SDH gene promoters in the H9c2 cardiomyocyte in vivo (Fig. 3B). Fig. 3B demonstrates that NRF-1 binds specifically to the promoter regions of both SDH a and d. When the ChIP is performed after siNRF-1, these promoter fragments are no longer detected by PCR, and no NRF-1-dependent products are obtained with primers for SDHc promoter segments that lack NRF-1 recognition site (Fig. 3B), or when performing the assay with primers specific for the SDHa and -d 5′-flanking regions upstream of the NRF-1 recognition sites (not shown). NRF-1 binding to the SDHa and SDHd promoter regions is enhanced in cells that receive DCM/CO.
SDHa Subunit Silencing, SDH Enzyme Activity and HIF-1α Nuclear Translocation—The role of SDHa in Complex II function and HIF-1α nuclear localization was checked in H9c2 cells using siRNA, and cells undergoing SDHa silencing exhibited a ∼90% decline in SDHa mRNA (at 48 h) compared with controls (Fig. 4A). Gene silencing markedly attenuated SDHa but not SDHb protein by Western blot (Fig. 4B); thus, one of the two subunits required for electron transfer was targeted specifically. Similarly, siNRF-1 also decreased SDHa but not SDHb protein expression (not shown), but importantly SDHa silencing blocked in-gel Complex II activity (Fig. 4C), confirming the SDHa requirement in succinate oxidation and Complex II activity. SDHa silencing also led to pronounced nuclear HIF-1α accumulation and a trend for less NRF-1 nuclear translocation (Fig. 2D; p = 0.07, but no trend in primary cardiomyocytes, data not shown).
FIGURE 4.

Knockdown and transfection experiments in H9c2 cells using siRNA to sequences in the open-reading frame of SDHa mRNA. A, RT-PCR for SDHa normalized to 18 S rRNA. Lanes 1 and 2 shows SDHa mRNA expression in control cells. Lanes 3, 4, lanes 5, 6, and lanes 7, 8 show effects of SDHa siRNA (80 μm) on mRNA expression at 24, 48, and 72 h, respectively. The histogram represents SDHa induction by real time PT-PCR (n = 3; *, p < 0.05 compared with control). B, Western blotting for mitochondrial succinate-ubiquinol oxidoreductase (SDH) subunit A and B proteins, the two functionally active subunits of Complex II. The blot shows controls (lanes 1 and 2) and cells transfected with siRNA to the SDHa subunit (lanes 3-8). Porin is used as a loading comparison. Lanes 3-6 show SDHa siRNA transiently inhibits SDHa but not SDHb protein expression. C, in-gel quantification of Complex II enzyme activity in control and SDHa-silenced H9c2 cells at two concentrations of SDHa siRNA (40 and 80 μm). Native gels were incubated in reaction buffer containing 10 mm succinate and developed with nitroblue tetrazolium. D, nuclear HIF-1α and NRF-1 in SDHa-silenced cells. SDHa silencing increases HIF-1α but not NRF-1 nuclear protein accumulation. Cells were transfected with either scRNA (controls) or siRNA to SDHa subunit. After transfection, nuclear HIF1α and NRF-1 were analyzed by Western blot. Tubulin is shown for a loading comparison. E, PHD activity in SDHa-silenced cells. PHD activity was assessed by detection of hydroxylated proline 564 of HIF-1α (Hyp-564) by immunoprecipitation followed by Western blot analysis. Proteasomal degradation was inhibited with 5 μm MG132 (2-h incubation). Aerobic H9c2 cells were incubated with 1 mm DMOG for 2 h to inhibit PHD. Cells were transfected with either scRNA or siRNA for the SDHa subunit. The last two lanes show the effect of siNRF-1. Total HIF-1α is shown for comparison. F, SDHa transfection and NRF-1 knockdown in primary rat cardiomyocytes. Left shows Western blot analysis of nuclear HIF-1α and NRF-1 relative to tubulin. SDHa protein is shown relative to β-actin. Lane 1, control cells. Lane 2, SDHa overexpression and SDHa, NRF-1, and HIF-1α protein expression at 48 h. Lane 3, SDHa, NRF-1, and HIF-1α expression 48 h after siNRF-1. Lane 4, co-transfection of SDHa-pCMV6-XL5 plasmid with siNRF-1 (low NRF-1 and high SDHa expression) prevents nuclear HIF-1α protein accumulation. The histogram (right) shows HIF-1α nuclear protein relative to tubulin by densitometry. Data are means of n = 4 (*, p < 0.05 compared with control).
PHD activity was assessed by Western blot analysis using anti-Hyp-564 of -HIF-1α in aerobic cells. In some runs, proteasomal HIF-1α degradation was inhibited with MG132, and PHD activity inhibition with DMOG (positive control) (29) blocked Hyp-564 accumulation (Fig. 4E). SDHa silencing decreased Hyp-564 compared with controls and with cells treated with scRNA (Fig. 4E). Thus, loss of SDHa activates HIF-1α and promotes aerobic nuclear HIF-1 accumulation. Fig. 4E also shows that siNRF-1 (which disrupts SDHa expression) inactivates PHD.
To check for SDHa-independent effects of loss of NRF-1 on nuclear HIF-1α accumulation, we overexpressed SDHa alone or followed by siNRF-1. Fig. 4F shows that SDHa overexpression minimizes nuclear HIF-1 expression, but NRF-1 silencing while SDHa is overexpressed allows HIF-1α to still accumulate in the nucleus as in control cells. SDHa overexpression, therefore, overcomes the dominant siNRF-1 effect on nuclear HIF-1α accumulation.
SDHa Silencing and HIF-1 Target Gene Expression—That either NRF-1 or SDHa silencing causes nuclear HIF-1α accumulation was corroborated functionally using HIF-1α target gene activation. HIF-1 regulates glucose transporter-1 protein (Glut1) expression, and Glut1 and Glut4 are markers of HIF-1 activity. Glut1 and -4 levels were used to monitor HIF-1α function before and after SDHa silencing in H9c2 cells and Glut1 protein increased 2.25-fold (Fig. 5A). Glut4 increased 2.8-fold after siSDHa with no change in Glut2 (Fig. 5A). Other important HIF-1 target genes were also up-regulated by siSDHa in H9c2 cells, including heme oxygenase-1 (Fig. 5B; HO-1, 2.9-fold, p < 0.05) and hexokinase (2-fold; data not shown). Finally, siNRF-1 increased Glut4 and HO-1 protein levels within 24 h by an amount comparable to siSDH (Fig. 5C).
FIGURE 5.

SDHa silencing increases HIF-1 target gene expression in H9c2 cells. A, Western blot analysis of glucose transporter proteins Glut1, Glut2, and Glut4 in control lysate (lanes 1 and 2) or lysate from cells transfected with siRNA to SDHa. Lanes 3, 4, lanes 5, 6, and lanes 7, 8 show timed effects of SDHa siRNA on transporter levels. Tubulin is the loading control. The histogram shows Glut 1, 2, and 4 protein levels by densitometry (*, p < 0.05 compared with control; † indicates p < 0.05 compared with control and Glut1). B, Western blot analysis for HO-1 in H9c2 cell lysates described in A shows protein induction after SDHa silencing. (Tubulin is for comparison.) The histogram shows relative HO-1 induction by densitometry (n = 3; *, p < 0.05 compared with control). C, siNRF-1 causes the expected increases in Glut4 and HO-1 protein by Western blot analysis by 24 h. In duplicate experiments, densitometry indicated that both proteins increased after siNRF-1 by an amount comparable to siSDHa (∼2-fold and 3-fold respectively; p < 0.05 for both).
α-Ketoglutarate Inhibition of HIF-1 Nuclear Translocation after SDHa Silencing—PHD1-3 catalyze O2-dependent HIF-1α hydroxylation, providing the signal for von Hippel-Lindau (pVHL)-dependent proteasomal degradation, thereby inactivating PHD in hypoxia, or aerobically by iron chelation (30) or with oxoglutarate analogs (29). To assess the effect of succinate on HIF-1α stabilization after SDHa silencing, α-ketoglutarate (α-KG), the substrate for PHD1-3, was fed to H9c2 cells. Supplementation of α-KG (2.0 mm) blocked both HIF-1α translocation after siSDHa by confocal microscopy (Fig. 6A) and the nuclear accumulation of HIF-1α by Western blot analysis (Fig. 6B). In control studies, α-KG did not alter total superoxide dismutase (SOD) activity in SDHa-silenced cells (not shown). Therefore, PHD substrate/product equilibrium is seen as a key regulator of HIF-1α nuclear translocation after SDHa suppression.
FIGURE 6.

Prevention of HIF-1α nuclear translocation by α-ketoglutarate in SDHa-silenced H9c2 cells. A, confocal images of control H9c2 cells stained with MitoTracker Green (panel a) and Cy3 immunostaining for HIF-1α (panel b, red) and overlay (panel c); or SDHa-silenced cells with α-KG and MitoTracker Green (panel d) and HIF-1α (panel e, red) and overlay (panel f); or SDHa-silencing without α-KG and MitoTracker Green (panel g) and HIF-1α (panel h, red) and overlay (panel j). B, Western blot of nuclear extracts (20 μg) using antibodies to HIF-1α and tubulin is shown.
Mitochondrial Superoxide and Nuclear HIF-1α in Aerobic
Cells—HIF-1α nuclear translocation may occur after PHD
inhibition by ROS, e.g. superoxide
( ) and/or
H2O2. Therefore, the mitochondrial ROS leak rate was
evaluated in control and SDHa-silenced aerobic H9c2 cells media using
fluorogenic mitoSOX dye to track mitochondrial
) and/or
H2O2. Therefore, the mitochondrial ROS leak rate was
evaluated in control and SDHa-silenced aerobic H9c2 cells media using
fluorogenic mitoSOX dye to track mitochondrial
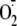 generation. Using
mitoSOX, control H9c2 cells showed minimal mitochondrial
generation. Using
mitoSOX, control H9c2 cells showed minimal mitochondrial
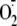 production, but
this was enhanced by DCM/CO, the positive control
(Fig. 7). In contrast, cells
treated with either siSDHa or siNRF-1 did not exhibit measurable mitochondrial
production, but
this was enhanced by DCM/CO, the positive control
(Fig. 7). In contrast, cells
treated with either siSDHa or siNRF-1 did not exhibit measurable mitochondrial
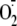 production after
24 h. Thus, silencing functional Complex II in these cardiac cells did not
enhance mitochondrial
production after
24 h. Thus, silencing functional Complex II in these cardiac cells did not
enhance mitochondrial
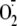 release, and
mitochondrial ROS production did not initiate HIF-1α nuclear
translocation.
release, and
mitochondrial ROS production did not initiate HIF-1α nuclear
translocation.
FIGURE 7.

Mitochondrial ROS production in H9c2 cells. Confocal scanning laser microscopy images showing localization patterns of the Mitosox oxidant probe with mitochondrial-selective dye, MitoTracker Green in aerobic cells exposed to 100 μm DCM/CO for 6 h (panels a-c), and controls (panels d-f) or cells transfected with SDHa siRNA (panels g-j) or NRF-1 siRNA 24 h earlier (panels k-m). Histogram (right) quantifies Mitosox red fluorescence in mitochondria relative to MitoTracker Green. SDHa and NRF-1 silencing lack an effect on mitochondrial ROS production (Values are mean ± S.E. of 30 cells from each of three experiments; *, p < 0.01 compared with control).
DISCUSSION
The nuclear transcriptional program for the expression of the proteins of the five respiratory complexes requires NRF-1 (1, 31, 32). NRF-1 is also necessary for mitochondrial biogenesis, has a role in cell growth control (7), and its expression and activity are repressed by cyclin D1 (33). NRF-1 is also necessary for the expression of the mitochondrial transcription factor (Tfam) (2), the TFB1M and M2 isoforms (34) and RNA-processing proteins required for mtDNA transcription and replication (24). Here we find that NRF-1 silencing functionally blocks succinate oxidation in aerobic heart cells prior to NADH-linked substrate, and that loss of NRF-1 expression increases HIF-1α stability and glucose transporter and glycolytic gene expression characteristic of pseudo-hypoxia.
The regulation of Complex II activity by NRF-1 and nuclear HIF-1α accumulation was a point of focus because of the emerging information on the role of SDH in tumorigenesis. Until now, little was known about NRF-1 regulation of the four nuclear genes of Complex II; SDHa, SDHb, SDHc, and SDHd, encode for the hydrophilic flavoprotein (Fp) and iron-sulfur protein, and two hydrophobic membrane anchor subunits, respectively, and SDH activity is integral to both the Krebs cycle and the respiratory chain by coupling matrix succinate to fumarate with the flavin adenine dinucleotide-driven reduction of ubiquinone at the inner membrane (35). The hydrophobic membrane anchor subunits contain heme b and the ubiquinone-binding site, and the centers are arranged to avoid ROS generation by FADH2 in the aerobic state (35, 36).
The evidence that NRF-1 regulates SDHa stems from the multiple NRF-1 but no NRF-2 recognition sites in the proximal nucleotide sequence of the rat gene promoter. NRF-1 silencing abrogates SDHa and SDHd mRNA expression in cardiomyocytes with minimal effect on SDHb or SDHc mRNA levels. ChIP demonstrated strong NRF-1 binding to the SDHa promoter, less binding at SDHd, and no or equivocal binding at SDHc and SDHb. Thus, the proclivity of NRF-1 to regulate HIF-1α stability via SDH promoter binding should be greatest at the flavoprotein, although this does not categorically exclude secondary ETC regulatory sites for the NRF-1 effect.
Although mitochondria are vital for energy provision, they regulate other processes such as apoptosis; moreover, altered mitochondrial function contributes to proliferation in a variety of tumor lines (37, 38) and the loss of SDH activity activates HIF-1 in tumor cells (13). Here in normal heart cells, disruption of a transcriptional pathway in mitochondrial biogenesis causes nuclear HIF-1α accumulation and directly links SDH to HIF-1α stability through PHD inhibition. The aerobic suppression of NRF-1 and loss of SDH activity emulates succinate accumulation in hypoxia, a conserved phylogenic sensor of O2 deficiency in both invertebrate and vertebrate organisms (39).
A diagram of the pathway for NRF-1 and SDHa regulation of HIF-1 gene expression in cardiac cells is provided in Fig. 8. When NRF-1 is down-regulated, SDHa expression is lost, and succinate oxidation is abrogated, thereby blocking FAD-linked electron entry into the respiratory chain. This decreases PHD activity, HIF-1α escapes degradation, and HIF-1α nucleoprotein increases. SDHa overexpression with siNRF-1 reduces HIF-1α stability and demonstrates independent and sufficient rescue. Succinate accumulation reflects electron transport dysfunction, and mitochondrial succinate egress is a hypoxic signal, although NRF-1 down-regulation may exert other unknown effects on HIF-1α stability.
FIGURE 8.

Diagram of the effects of NRF-1 on the regulation of SDHa- and HIF-1-responsive genes. The loss of NRF-1 leads to loss of catalytic SDHa and depresses Complex II activity, which causes mitochondria to release succinate. Succinate inhibits the PHD, and HIF-1α escapes the Von Hippel-Lindau protein (pVHL) to translocate to the nucleus where it engages with HIF-1β in DNA binding and gene activation. Heme oxygenase-1 (Hmox1, HO-1) is shown as an example of an HIF-1-responsive gene.
Human SDH gene mutations produce several clinical phenotypes, e.g. myopathy, encephalopathy, and cancer (13, 14, 40), and SDHa mutations usually produce disorders similar to other Krebs cycle defects while SDHb, SDHc, and SDHd mutations are associated with pheochromocytoma and paraganglioma (13). However, the first cases of disease caused by a nuclear-encoded mitochondrial gene mutation involved SDHa and presented as Leigh syndrome (41), and SDHa activity is defective in some patients with hypertrophic cardiomyopathy (42). SDH expression is low in some gastric and colon carcinomas (40), especially during the transition from Dukes' B to more aggressive stages (43). Of note, H9c2 cell differentiation into myotubules is also prevented by NRF-1 suppression, which has implications for NRF-1 as a tumor suppressor, HIF-1 persistence is arguably tumorigenic, e.g. in neuroendocrine tissues.
Because the energy demand of the heart is met principally by aerobic ATP synthesis, it is especially vulnerable to hypoxia, and cardiac cells respond exquisitely to effectors of O2 sensing. Although HIF-1α is a “master” switch in the mammalian hypoxic response, it is also activated by aerobic stressors, and here in aerobic cardiomyocytes, loss of NRF-1 up-regulates HIF-1-responsive genes including selected glucose transporters, hexokinase, and HO-1 (9, 44, 45), congruent with glycolytic protection of rat cardiomyocytes from hypoxic induction of apoptosis (46). Mitochondrial damage also eliminates non-viable cells by initiating intrinsic apoptosis (47) and resistance to apoptosis is sensitive to changes in anti-apoptotic protein expression (48, 49). In terms of energy provision, ATP depletion alone does not stabilize HIF-1α and singular Complex II inactivation has more limited consequences than hypoxia or defects in Complex I.
Mitochondrial ROS production was checked as a mechanism by which
NRF-1-induced loss of SDH could produce pseudo-hypoxia. In the ETC, ROS
generation occurs notably at Complex III, but in theory could arise from
defective SDH assembly, or via the SDHa, as in certain bacteria, by conversion
to a fumarate reductase (36).
That conversion, however, requires FAD binding and inhibition of electron flow
from SDHa to iron-sulfur in SDHb and to the inner membrane ubiquinone-binding
site. In aerobic cardiac cells, mitochondrial
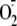 production did not
account for HIF-1α stabilization by either mechanism. Nuclear
HIF-1α is increased by SDHa silencing, but not by an approach
that blocks Complex IV and generates mitochondrial superoxide at Complex III
(Fig. 7)
(25). In fact, as
PO2 rises in the cell this would actually oppose HIF-1α
stabilization. Nuclear translocation of HIF-1α in SDHa-silenced
cells is also opposed by α-KG independently of ROS, thereby excluding
mitochondrial ROS within the assay limits as a factor in HIF-1α
accumulation after aerobic silencing of SDHa.
production did not
account for HIF-1α stabilization by either mechanism. Nuclear
HIF-1α is increased by SDHa silencing, but not by an approach
that blocks Complex IV and generates mitochondrial superoxide at Complex III
(Fig. 7)
(25). In fact, as
PO2 rises in the cell this would actually oppose HIF-1α
stabilization. Nuclear translocation of HIF-1α in SDHa-silenced
cells is also opposed by α-KG independently of ROS, thereby excluding
mitochondrial ROS within the assay limits as a factor in HIF-1α
accumulation after aerobic silencing of SDHa.
That SDHa silencing did not enhance the mitochondrial ROS leak rate in cardiomyocytes concurs with the work of Selak et al. (12, 50) in aerobic tumor cells where SDHd silencing induces HIF-1 stabilization via succinate accumulation with no detectable increase in ROS. There are other possibilities however; Guzy et al. (53) reported that SDHb silencing increased ROS in aerobic tumor cells and stabilized HIF-1 in a ROS-dependent manner. As in our normal heart cells, SDHa suppression did not increase ROS levels, but in contrast, did not increase HIF-1 stabilization. Guzy et al. (53) postulate that SDHb silencing allowed H2O2 to escape the mitochondria and inhibit PHD. The tumor cell differences reported by Selak and Guzy could be due to methodological or to cell-specific effects, but Guzy et al. (53) did not exclude that HIF-1 accumulation associated with loss of SDHa or SDHd operates differently than loss of SDHb. Moreover, the importance of succinate in HIF-1α stabilization is agreed to be sufficient for HIF-1 nuclear translocation in many Complex II activity-deficient tumor cell lines (51).
The product/substrate equilibrium is an important factor in PHD regulation (12, 14) because loss of SDH activity allows succinate accumulation to feedback-inhibit PHD (14), a parsimonious explanation for HIF-1α nuclear translocation. HIF-1α PHD1-3 reside in the cytoplasm, but mitochondrial succinate moves via inner membrane dicarboxylate translocators and outer membrane voltage-dependent anion channels to the cytoplasm (52) where it inhibits the forward HIF-1α hydroxylation reaction because as α-ketoglutarate dioxygenases, the PHDs release succinate upon converting HIF-1α prolyl to the hydroxyprolyl residues. Thus, equilibrium reversal of nuclear HIF-1α accumulation by α-KG connects succinate to activity control of PHD in cardiomyocytes.
In summary, NRF-1 contributes to cytoplasmic HIF-1α instability in aerobic cardiomyocytes by functional occupancy of two of four subunit gene promoters of cardiac mitochondrial Complex II. Specifically, NRF-1 exquisitely regulates the expression levels of the catalytic SDHa flavoprotein required for electron transport into the respiratory chain. NRF-1-dependent loss of SDHa expression allows succinate to accumulate, which promotes nuclear HIF-1α translocation by blocking α-ketoglutarate utilization by the HIF-1 PHD. These findings demonstrate new biological importance for NRF-1 both in the close transcriptional regulation of the SDHa flavoprotein and the avoidance of pseudo-hypoxic gene expression in aerobic cells.
Acknowledgments
We thank John Patterson and Lynn Tatro for technical assistance.
This work was supported in part by NHLBI, National Institutes of Health Grant P01 42-444 and a grant from the Veteran's Administration (to C. A. P.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ETC, electron transport chain; HIF-1, hypoxia-inducible factor-1; NRF, nuclear respiratory factor; PHD, prolyl hydroxylase; ROS, reactive oxygen species; SDH, succinate dehydrogenase; DMEM, Dulbecco's modified Eagle's medium; PBS, phosphate-buffered saline; nt, nucleotide; ChIP, chromatin immunoprecipitation assay; scRNA, scrambled RNA; si, small interfering; DMOG, dimethyloxalylglycine; DCM, dichloromethane; DCM/CO, CO-generating dichloromethane; α-KG, α-ketoglutarate; RT, reverse transcriptase.
References
- 1.Scarpulla, R. C. (2002) Gene (Amst.) 286 81-89 [DOI] [PubMed] [Google Scholar]
- 2.Virbasius, J. V., and Scarpulla, R. C. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 1309-1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vercauteren, K., Pasko, R. A., Gleyzer, N., Marino, V. M., and Scarpulla, R. C. (2006) Mol. Cell. Biol. 26 7409-7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., Troy, A., Cinti, S., Lowell, B., Scarpulla, R. C., and Spiegelman, B. M. (1999) Cell 98 115-124 [DOI] [PubMed] [Google Scholar]
- 5.Barish, G. D., Narkar, V. A., and Evans, R. M. (2006) J. Clin. Investig. 116 590-597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelly, D. P., and Scarpulla, R. C. (2004) Genes Dev. 18 357-368 [DOI] [PubMed] [Google Scholar]
- 7.Cam, H., Balciunaite, E., Blais, A., Spektor, A., Scarpulla, R. C., Young, R., Kluger, Y., and Dynlacht, B. D. (2004) Mol. Cell 16 399-411 [DOI] [PubMed] [Google Scholar]
- 8.Suliman, H. B., Carraway, M. S., Welty-Wolf, K. E., Whorton, A. R., and Piantadosi, C. A. (2003) J. Biol. Chem. 278 41510-41518 [DOI] [PubMed] [Google Scholar]
- 9.Chen, C., Pore, N., Behrooz, A., Ismail-Beigi, F., and Maity, A. (2001) J. Biol. Chem. 276 9519-9525 [DOI] [PubMed] [Google Scholar]
- 10.Wang, G. L., Jiang, B. H., Rue, E. A., and Semenza, G. L. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 5510-5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semenza, G. L. (2003) Nat. Rev. Cancer 3 721-732 [DOI] [PubMed] [Google Scholar]
- 12.Selak, M. A., Duran, R. V., and Gottlieb, E. (2006) Biochim. Biophys. Acta 1757 567-572 [DOI] [PubMed] [Google Scholar]
- 13.Gimenez-Roqueplo, A. P., Favier, J., Rustin, P., Mourad, J. J., Plouin, P. F., Corvol, P., Rotig, A., and Jeunemaitre, X. (2001) Am. J. Hum. Genet. 69 1186-1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollard, P. J., Briere, J. J., Alam, N. A., Barwell, J., Barclay, E., Wortham, N. C., Hunt, T., Mitchell, M., Olpin, S., Moat, S. J., Hargreaves, I. P., Heales, S. J., Chung, Y. L., Griffiths, J. R., Dalgleish, A., McGrath, J. A., Gleeson, M. J., Hodgson, S. V., Poulsom, R., Rustin, P., and Tomlinson, I. P. (2005) Hum. Mol. Genet. 14 2231-2239 [DOI] [PubMed] [Google Scholar]
- 15.Doege, K., Heine, S., Jensen, I., Jelkmann, W., and Metzen, E. (2005) Blood 106 2311-2317 [DOI] [PubMed] [Google Scholar]
- 16.Chandel, N. S., McClintock, D. S., Feliciano, C. E., Wood, T. M., Melendez, J. A., Rodriguez, A. M., and Schumacker, P. T. (2000) J. Biol. Chem. 275 25130-25138 [DOI] [PubMed] [Google Scholar]
- 17.Vaux, E. C., Metzen, E., Yeates, K. M., and Ratcliffe, P. J. (2001) Blood 98 296-302 [DOI] [PubMed] [Google Scholar]
- 18.Srinivas, V., Leshchinsky, I., Sang, N., King, M. P., Minchenko, A., and Caro, J. (2001) J. Biol. Chem. 276 21995-21998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown, M. D., and Wallace, D. C. (1994) J. Bioenerg. Biomembr. 26 273-289 [DOI] [PubMed] [Google Scholar]
- 20.Janssen, R. J., van den Heuvel, L. P., and Smeitink, J. A. (2004) Expert Rev. Mol. Diagn. 4 143-156 [DOI] [PubMed] [Google Scholar]
- 21.Hagen, T., Taylor, C. T., Lam, F., and Moncada, S. (2003) Science 302 1975-1978 [DOI] [PubMed] [Google Scholar]
- 22.Menard, C., Pupier, S., Mornet, D., Kitzmann, M., Nargeot, J., and Lory, P. (1999) J. Biol. Chem. 274 29063-29070 [DOI] [PubMed] [Google Scholar]
- 23.Li, H. Y., Kotaka, M., Kostin, S., Lee, S. M., Kok, L. D., Chan, K. K., Tsui, S. K., Schaper, J., Zimmermann, R., Lee, C. Y., Fung, K. P., and Waye, M. M. (2001) Cell Motil. Cytoskeleton 48 11-23 [DOI] [PubMed] [Google Scholar]
- 24.Piantadosi, C. A., and Suliman, H. B. (2006) J. Biol. Chem. 281 324-333 [DOI] [PubMed] [Google Scholar]
- 25.Suliman, H. B., Carraway, M. S., Tatro, L. G., and Piantadosi, C. A. (2007) J. Cell Sci. 120 299-308 [DOI] [PubMed] [Google Scholar]
- 26.Lavery, D. J., and Schibler, U. (1993) Genes Dev. 7 1871-1884 [DOI] [PubMed] [Google Scholar]
- 27.Suliman, H. B., Welty-Wolf, K. E., Carraway, M., Tatro, L., and Piantadosi, C. A. (2004) Cardiovasc. Res. 64 279-288 [DOI] [PubMed] [Google Scholar]
- 28.Sabar, M., Balk, J., and Leaver, C. J. (2005) Plant J. 44 893-901 [DOI] [PubMed] [Google Scholar]
- 29.Mole, D. R., Schlemminger, I., McNeill, L. A., Hewitson, K. S., Pugh, C. W., Ratcliffe, P. J., and Schofield, C. J. (2003) Bioorg. Med. Chem. Lett. 13 2677-2680 [DOI] [PubMed] [Google Scholar]
- 30.Wang, G. L., and Semenza, G. L. (1993) Blood 82 3610-3615 [PubMed] [Google Scholar]
- 31.Scarpulla, R. C. (2002) Biochim. Biophys. Acta 1576 1-14 [DOI] [PubMed] [Google Scholar]
- 32.Suliman, H. B., Carraway, M. S., and Piantadosi, C. A. (2003) Am. J. Respir. Crit. Care Med. 167 570-579 [DOI] [PubMed] [Google Scholar]
- 33.Wang, C., Li, Z., Lu, Y., Du, R., Katiyar, S., Yang, J., Fu, M., Leader, J. E., Quong, A., Novikoff, P. M., and Pestell, R. G. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 11567-11572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gleyzer, N., Vercauteren, K., and Scarpulla, R. C. (2005) Mol. Cell. Biol. 25 1354-1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cecchini, G., Schroder, I., Gunsalus, R. P., and Maklashina, E. (2002) Biochim. Biophys. Acta 1553 140-157 [DOI] [PubMed] [Google Scholar]
- 36.Yankovskaya, V., Horsefield, R., Tornroth, S., Luna-Chavez, C., Miyoshi, H., Leger, C., Byrne, B., Cecchini, G., and Iwata, S. (2003) Science 299 700-704 [DOI] [PubMed] [Google Scholar]
- 37.Penta, J. S., Johnson, F. M., Wachsman, J. T., and Copeland, W. C. (2001) Mutat. Res. 488 119-133 [DOI] [PubMed] [Google Scholar]
- 38.Mathupala, S. P., Heese, C., and Pedersen, P. L. (1997) J. Biol. Chem. 272 22776-22780 [DOI] [PubMed] [Google Scholar]
- 39.Hohl, C., Oestreich, R., Rosen, P., Wiesner, R., and Grieshaber, M. (1987) Arch Biochem. Biophys. 259 527-535 [DOI] [PubMed] [Google Scholar]
- 40.Habano, W., Sugai, T., Nakamura, S., Uesugi, N., Higuchi, T., Terashima, M., and Horiuchi, S. (2003) Oncol. Rep. 10 1375-1380 [PubMed] [Google Scholar]
- 41.Angelini, C., Melacini, P., Valente, M. L., Reichmann, H., Carrozzo, R., Fanin, M., Vergani, L., Boffa, G. M., Martinuzzi, A., and Fasoli, G. (1993) Jpn. Heart. J. 34 63-77 [DOI] [PubMed] [Google Scholar]
- 42.Bourgeron, T., Rustin, P., Chretien, D., Birch-Machin, M., Bourgeois, M., Viegas-Pequignot, E., Munnich, A., and Rotig, A. (1995) Nat. Genet. 11 144-149 [DOI] [PubMed] [Google Scholar]
- 43.Frederiksen, C. M., Knudsen, S., Laurberg, S., and Orntoft, T. F. (2003) J. Cancer Res. Clin. Oncol. 129 263-271 [DOI] [PubMed] [Google Scholar]
- 44.Lee, P. J., Jiang, B. H., Chin, B. Y., Iyer, N. V., Alam, J., Semenza, G. L., and Choi, A. M. (1997) J. Biol. Chem. 272 5375-5381 [PubMed] [Google Scholar]
- 45.Gao, L., Mejias, R., Echevarria, M., and Lopez-Barneo, J. (2004) FEBS Lett. 569 256-260 [DOI] [PubMed] [Google Scholar]
- 46.Malhotra, R., and Brosius, F. C., 3rd. (1999) J. Biol. Chem. 274 12567-12575 [DOI] [PubMed] [Google Scholar]
- 47.Green, D. R., and Kroemer, G. (2005) J. Clin. Investig. 115 2610-2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hammerman, P. S., Fox, C. J., and Thompson, C. B. (2004) Trends Biochem. Sci 29 586-592 [DOI] [PubMed] [Google Scholar]
- 49.Robey, R. B., and Hay, N. (2005) Cell Cycle 4 654-658 [DOI] [PubMed] [Google Scholar]
- 50.Selak, M. A., Armour, S. M., MacKenzie, E. D., Boulahbel, H., Watson, D. G., Mansfield, K. D., Pan, Y., Simon, M. C., Thompson, C. B., and Gottlieb, E. (2005) Cancer Cell 7 77-85 [DOI] [PubMed] [Google Scholar]
- 51.Briere, J. J., Favier, J., Benit, P., El Ghouzzi, V., Lorenzato, A., Rabier, D., Di Renzo, M. F., Gimenez-Roqueplo, A. P., and Rustin, P. (2005) Hum. Mol. Genet. 14 3263-3269 [DOI] [PubMed] [Google Scholar]
- 52.Mizuarai, S., Miki, S., Araki, H., Takahashi, K., and Kotani, H. (2005) J. Biol. Chem. 280 32434-32441 [DOI] [PubMed] [Google Scholar]
- 53.Guzy, R. D., Sharma, B., Bell, E., Chandel, N. S., and Schumacker, P. T. (2008) Mol. Cell Biol. 28 718-731 [DOI] [PMC free article] [PubMed] [Google Scholar]