

Abstract
We have recently demonstrated that specific oxidized phospholipids (oxPCCD36) accumulate at sites of oxidative stress in vivo such as within atherosclerotic lesions, hyperlipidemic plasma, and plasma with low high-density lipoprotein levels. oxPCCD36 serve as high affinity ligands for the scavenger receptor CD36, mediate uptake of oxidized low density lipoprotein by macrophages, and promote a pro-thrombotic state via platelet scavenger receptor CD36. We now report that oxPCCD36 represent ligands for another member of the scavenger receptor class B, type I (SR-BI). oxPCCD36 prevent binding to SR-BI of its physiological ligand, high density lipoprotein, because of the close proximity of the binding sites for these two ligands on SR-BI. Furthermore, oxPCCD36 interfere with SR-BI-mediated selective uptake of cholesteryl esters in hepatocytes. Thus, oxidative stress and accumulation of specific oxidized phospholipids in plasma may have an inhibitory effect on reverse cholesterol transport.
Atherosclerosis is a chronic inflammatory disease in which macrophage accumulation of cholesterol and subsequent foam cell formation are critical events. Accumulation of cholesterol in macrophages is a result of failure to adequately adjust cellular cholesterol efflux in conditions of dramatically increased cholesterol acquisition. Efflux of excess of cholesterol from macrophages to HDL2 and to free apolipoproteins and subsequent delivery to the liver for excretion, a process called reverse cholesterol transport, are critical for the maintenance of cholesterol balance. One of the key steps in reverse cholesterol transport is binding of HDL in the liver to hepatocyte SR-BI followed by selective uptake of cholesteryl esters (CE) from HDL. Multiple studies employing various murine models of atherosclerosis demonstrated that hepatic SR-BI is atheroprotective (1–4). Processes that interfere with SR-BI-mediated selective uptake of CE are of particular interest because they are potentially proatherogenic and may represent novel mechanisms contributing to the development of atherosclerosis.
SR-BI belongs to the evolutionarily conserved CD36 family of proteins, sharing 30% sequence identity with CD36 (5). SR-BI is an 82-kDa membrane glycoprotein containing a large extracellular domain and two transmembrane domains with short cytoplasmic amino- and carboxyl-terminal tails (6). Similar to CD36 it is a multifunctional protein; however, its major function is selective uptake of CE from HDL in steroidogenic tissues. SR-BI-mediated selective uptake of HDL CE is a two-step process. The first step involves lipoprotein binding to the extracellular domain of SR-BI, and the second step is the selective transfer of lipid from HDL to the plasma membrane. SR-BI shares with CD36 an affinity for a wide array of ligands, including native and modified lipoproteins, advanced glycation end products, and anionic phospholipids (7, 8).
Atherosclerosis is associated with oxidative stress and the generation of biologically active oxidized lipids. It has been previously demonstrated that these biologically active oxidized phospholipids initiate and modulate many of the cellular events attributed to the pathogenesis of atherosclerosis (9). We have previously described a novel family of oxidized choline glycerophospholipids (oxPCCD36) that are formed during the oxidation of LDL by multiple distinct pathways and are present in vivo in human and animal atherosclerotic lesions and also accumulate in hyperlipidemic plasma and in plasma of subjects with low HDL levels (10–12). oxPCCD36 serve as ligands for the scavenger receptor CD36, mediate highly specific cellular recognition and internalization of targets containing complex mixtures of biologically active oxidized lipids, thus directly induce foam cell formation via CD36 (13, 14). Based on the close protein similarity between CD36 and SR-BI, we hypothesized that oxPCCD36 species would also serve as ligands for SR-BI and therefore could interfere with SR-BI-dependent processes.
In this study we demonstrate that oxPCCD36 bind specifically to SR-BI and that binding of oxPCCD36 prevents HDL association because of the close proximity of the binding sites for these two ligands on SR-BI. We then demonstrate that oxPCCD36 is a potent inhibitor of SR-BI-mediated selective uptake of cholesteryl esters in hepatocytes. Thus, our results suggest that specific oxidized phospholipids accumulated in vivo in oxidative stress may inhibit reverse cholesterol transport and contribute to the development of hypercholesterolemia and atherosclerosis.
EXPERIMENTAL PROCEDURES
Materials
Tissue culture media and additives were purchased from Invitrogen. Na125I was supplied by ICN Biomedicals, Inc. (Costa Mesa, CA). [3H]Cholesteryl oleate ether (COE) and 1,2-[3H]dihexadecanoyl-sn-glycero-3-phosphocholine (DPPC) were from American Radiolabel Chemicals, Inc. (St. Louis, MO). 1-Hexadecanoyl-2-eicosatetra-5′,8′,11′,14′-enoyl-sn-glycero-3-phosphocholine (PAPC), 1-hexadecanoyl-2-octadec-9′-enoyl-sn-glycero-3-phosphocholine (POPC), 1-hexadecanoyl-2-octadec-9′,12′-dienoyl-sn-glycero-3-phosphocholine (PLPC), and DPPC were purchased from Avanti Polar Lipids (Alabaster, AL). The 9-keto-10-dodecendioic acid and 5-keto-6-octendioic acid esters of 2-lyso-PC (KDdiA-PC and KOdiA-PC, respectively) were purchased from Cayman, Inc. (Ann Arbor, MI). Anti-SR-BI blocking antibody was purchased from Novus Biologicals (Littleton, CO). All other reagents were obtained from Sigma unless otherwise specified.
Methods
General Procedures—All buffers were passed over a column of Chelex-100 resin and supplemented with diethylenetriaminepentaacetic acid to remove any potential transition metal ions, which might catalyze phospholipid oxidation during incubations. Protein content was determined by the Markwell-modified Lowry protein assay with bovine serum albumin as standard (15). LDL and HDL were isolated from fresh plasma by sequential ultracentrifugation, and iodination with Na125I was performed as described previously (16). Total synthesis of γ-keto-α,β-unsaturated aldehydic phospholipid, the 5-keto-8-oxo-6-octenoic acid esters of 2-lyso-PC (KOOA-PC), was performed as described earlier (14).
Lipoprotein Modification—LDL was modified utilizing the
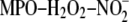 system by incubating LDL (0.2 mg of protein/ml) at 37 °C in 50
mm sodium phosphate (pH 7.4), 100 μm
diethylenetriaminepentaacetic acid, 30 nm MPO, 100 μg/ml
glucose, 20 ng/ml glucose oxidase, and 0.5 mm NaNO2 for
8 h (16). Oxidation reactions
were terminated by addition of 40 μm butylated hydroxytoluene
and 300 nm catalase to the reaction mixture. LDL acetylation and
oxidation of LDL by copper ions were performed as described previously
(17).
system by incubating LDL (0.2 mg of protein/ml) at 37 °C in 50
mm sodium phosphate (pH 7.4), 100 μm
diethylenetriaminepentaacetic acid, 30 nm MPO, 100 μg/ml
glucose, 20 ng/ml glucose oxidase, and 0.5 mm NaNO2 for
8 h (16). Oxidation reactions
were terminated by addition of 40 μm butylated hydroxytoluene
and 300 nm catalase to the reaction mixture. LDL acetylation and
oxidation of LDL by copper ions were performed as described previously
(17).
Phospholipid Vesicle Preparation and Modification—Stock solutions (2 mg/ml) of small unilamellar vesicles consisting of PAPC, PLPC, or POPC were prepared in argon-sparged sodium phosphate buffer by extrusion (11 times) through a 0.1-μm polycarbonate filter using an Avanti Mini-Extruder Set (Avanti Polar Lipids) at 37 °C (14). For direct binding experiments, [3H]DPPC (25 μCi/mg of phospholipids) was added to phospholipids (PAPC or PLPC) with equimolar amounts of specific synthetic oxidized phospholipids (oxPCCD36). Phospholipid vesicles oxidized by reactive nitrogen species were prepared from PAPC or PLPC vesicles (0.2 mg lipid/ml) by exposure to the MPO-H2O2-NO–2 system as described (16).
Cells—Hep G2 cells were from American Type Culture Collection (Manassas, VA). CHO cells lacking LDL receptor activity (ldlA7 cells) and a subcloned cell line expressing scavenger receptor class B type I (ldlA7-SR-BI) (a gift from Dr. M. Krieger, MIT, Boston) were cultured as described elsewhere (18). For generating the SR-BI overexpressing HEK-293T cell line, full-length human SR-BI was PCR-amplified from liver Marathon-Ready cDNA (BD Biosciences) and cloned into the pEF6V5-His vector (Invitrogen). HEK-293T cells were stably transfected with Lipofectamine 2000 (Invitrogen) using blasticidine as the selection marker. Clones overexpressing SR-BI were selected based on Western blot for SR-BI. Experiments were performed on confluent cell monolayer in Dulbecco's modified Eagle's medium/F-12 medium containing 10% fetal bovine serum, butylated hydroxytoluene (20 μm), diethylenetriaminepentaacetic acid (100 μm), and catalase (300 nm).
Selective Uptake of Cholesteryl Ester in Hepatocytes—Phospholipid liposomes containing [3H]cholesteryl oleate ether (COE) were prepared as described elsewhere (19) and incubated with HDL in the presence of excess cholesteryl ester transfer protein and 0.5% BSA at 37 °C for 18 h. HDL was then re-isolated by ultracentrifugation and dialyzed. Specific activity of resulting [3H]COE HDL was 4 cpm/ng HDL protein. Hep G2 cells were cultured for 18 h in the presence of 0.1 μm adrenocorticotropic hormone in Dulbecco's modified Eagle's/F-12 media containing 5% lipoprotein-deficient serum. Selective uptake of cholesteryl esters was determined as described before (19). Briefly, parallel experiments to assess HDL uptake were performed using [3H]COE HDL and 125I-HDL. After 5 h of incubation at 37 °C with indicated lipoproteins, the medium was removed, and the cells washed with warm PBS. Following a 30-min chase with 100 μg/ml unlabeled HDL, cells were lysed and cell-associated radioactivity was quantified. Degradation of 125I-HDL was determined as described before (16), and total uptake of HDL protein was calculated as the sum of the 125I associated with cells plus the noniodide-, nontrichloroacetic acid-precipitable 125I in the medium. Selective uptake was calculated as the difference between total lipid uptake (3H radioactivity) and the amount of lipid incorporation that could be accounted for by whole lipoprotein uptake as determined by the accumulated cellular 125I.
Cloning and Purification of GST-SR-BI Ligand Binding Domain Fusion Protein—The sequence encoding full-length human SR-BI was amplified by PCR from liver Marathon-Ready cDNA and subcloned into the PCR-blunt vector (Invitrogen). A fragment containing a putative human HDL ligand-binding site, an extracellular amino-terminal domain of SR-BI (amino acids 144–205), was amplified by PCR with appropriate primers and cloned between BamHI and XbaI sites of the pGST-Parallel1 vector. The junction with GST was sequenced to confirm insertion of SR-BI-(144–205) in-frame. GST fusion protein expression in transformed Escherichia coli BL21(DE3) was induced by isopropyl 1-thio-β-d-galactopyranoside; cells were lysed, and fusion protein was isolated by affinity chromatography using glutathione-Sepharose 4B beads (GE Healthcare). The size, amount, and purity of the fusion protein were examined by SDS-PAGE. The molecular weight was found to be close to the predicted value, and purity was typically >95%.
Lipoprotein and Phospholipid Vesicle Binding Experiments—Cells were washed with serum-free medium, and the indicated amounts of 125I-labeled lipoproteins were added in 250 μl of the appropriate media containing 10% fetal bovine serum. Lipoprotein binding to SR-BI expressing CHO cells (ldlA7-SR-BI) and control vector cells (ldlA7) was determined following 3 h of incubation at 37 °C with the indicated concentrations of native or modified forms of 125I-labeled lipoproteins (17). In competition experiments, 125I-labeled lipoproteins (5 μg/ml) were incubated with cells in the presence of the indicated concentrations of unlabeled competitor.
For phospholipid vesicle binding experiments, cells (0.25–0.3 mg of cell protein/well) were incubated with the corresponding radiolabeled phospholipid vesicles (10 μg/ml, 250 μl of Dulbecco's modified Eagle's medium) for 3 h at 37 °C in the presence of 40-fold excess of unlabeled POPC vesicles to block nonspecific binding. Unbound vesicles were then washed from cells with PBS/BSA; cells were solubilized with 0.1 m NaOH, and bound radioactivity was quantified.
GST-SR-BI binding experiments were performed by incubating GST or GST-SR-BI-(144–205) protein beads (1 μg of protein/tube) with the corresponding phospholipid vesicles containing the radioactive tracer (14) at 10 μg lipid/ml in PBS for 3 h at 25 °C with gentle shaking. Unbound vesicles were removed from the beads by repeated washing with PBS, and then bound radioactivity was quantified. HDL binding and competition assays were performed by incubating 125I-HDL (5 μg/ml) in PBS with GST or GST-SR-BI-(144–205) beads (1 μg of protein/tube) for 3 h at 25 °C. In competition experiments, unlabeled competitors were added at 20-fold excess. Where indicated, the beads were preincubated for 15 min with anti-SR-BI blocking antibody. HDL binding parameters for HEK-293T cells overexpressing SR-BI and GST-SR-BI-(144–205) fusion protein were obtained from nonlinear regression analysis in Prism 4 software (GraphPad Software Inc., San Diego).
Statistics—Each experimental point represents the mean ± S.D. for triplicate determinations of a representative experiment performed at least three times. Statistical analyses were made using a paired Student's t test. For all hypotheses, the significance level was 0.05. When multiple comparisons were made, a Bonferoni correction to the significance criterion for each test was made.
RESULTS
oxPCCD36 Represent Ligands for SR-BI—SR-BI is
closely related to scavenger receptor CD36 and shares with CD36 several
ligands, including oxLDL (13,
20,
21). We hypothesized that
oxLDL could bind to SR-BI via specific oxidized phospholipids
(oxPCCD36) that were previously shown to be ligands for CD36. LDL
oxidized by reactive nitrogen species generated by the
MPO-H2O2-NO2-system (NO2-LDL) is a
carrier of oxPCCD36 and represents a biologically relevant and well
characterized model of oxidized LDL that selectively binds to CD36
(13,
14). First, we tested whether
NO2-LDL was recognized by SR-BI and whether oxPCCD36
present in NO2-LDL serve as ligands for SR-BI.
125I-Labeled NO2-LDL binding to SR-BI-overexpressing CHO
cells was significantly higher than control cells
(Fig. 1a). Binding of
125I-acLDL (a specific ligand for the scavenger receptor class A)
was low (data not shown). Native LDL and LDL oxidized by alternative methods
(e.g. Cu2+) also bound to SR-BI-overexpressing cells in
agreement with previous reports
(22,
23). The specificity of
SR-BI-mediated recognition of 125I-labeled NO2-LDL was
verified by competition experiments. Excess amounts of unlabeled
NO2-LDL effectively competed with 125I-labeled
NO2-LDL for binding to SR-BI-overexpressing cells
(Fig. 1b). In
contrast, a 40-fold molar excess of native LDL had no significant effect
(Fig. 1b). An
anti-SR-BI blocking antibody significantly inhibited binding of
125I-labeled NO2-LDL to SR-BI-overexpressing cells,
whereas an isotype-matched nonimmune antibody had no effect
(Fig. 1b). These
findings confirmed that LDL modified by the
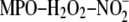 system was recognized specifically by SR-BI. We then tested whether SR-BI
recognized NO2-LDL via its oxidized phospholipid moiety. Small
unilamellar vesicles comprised of synthetic homogeneous phosphatidylcholine
molecular species were oxidized by the
system was recognized specifically by SR-BI. We then tested whether SR-BI
recognized NO2-LDL via its oxidized phospholipid moiety. Small
unilamellar vesicles comprised of synthetic homogeneous phosphatidylcholine
molecular species were oxidized by the
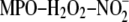 system (NO2-PAPC) and then tested for their ability to compete for
125I-labeled NO2-LDL binding to SR-BI-overexpressing
cells. NO2-PAPC significantly inhibited binding of
125I-labeled NO2-LDL
(Fig. 1b) indicating
that free (not bound covalently to protein) oxidized phospholipid, in
NO2-LDL, served as at least one of the ligands for SR-BI. A similar
pattern of recognition was demonstrated by our group previously for CD36
(11,
14). Because CD36 and SR-BI
belong to the same family of proteins and share evolutionally conserved
domains, these results suggest that CD36 and SR-BI recognize
NO2-LDL via similar mechanisms. We next examined individual
synthetic phospholipids previously identified by our group as specific
endogenous ligands for CD36 (oxPCCD36)
(14), such as KOOA-PC (the
5,8-dioxooct-6-enoic acid) and KODA-PC (9,12-dioxododec-10-enoic acid ester of
2-lyso-PC), for their ability to compete for the binding to SR-BI. As shown in
Fig. 1b,
oxPCCD36 (data for KOOA-PC are shown), but not their unoxidized
phospholipid precursors (data for PAPC are shown), inhibited binding of
125I-labeled NO2-LDL to SR-BI-expressing cells.
system (NO2-PAPC) and then tested for their ability to compete for
125I-labeled NO2-LDL binding to SR-BI-overexpressing
cells. NO2-PAPC significantly inhibited binding of
125I-labeled NO2-LDL
(Fig. 1b) indicating
that free (not bound covalently to protein) oxidized phospholipid, in
NO2-LDL, served as at least one of the ligands for SR-BI. A similar
pattern of recognition was demonstrated by our group previously for CD36
(11,
14). Because CD36 and SR-BI
belong to the same family of proteins and share evolutionally conserved
domains, these results suggest that CD36 and SR-BI recognize
NO2-LDL via similar mechanisms. We next examined individual
synthetic phospholipids previously identified by our group as specific
endogenous ligands for CD36 (oxPCCD36)
(14), such as KOOA-PC (the
5,8-dioxooct-6-enoic acid) and KODA-PC (9,12-dioxododec-10-enoic acid ester of
2-lyso-PC), for their ability to compete for the binding to SR-BI. As shown in
Fig. 1b,
oxPCCD36 (data for KOOA-PC are shown), but not their unoxidized
phospholipid precursors (data for PAPC are shown), inhibited binding of
125I-labeled NO2-LDL to SR-BI-expressing cells.
FIGURE 1.

Binding of NO2-LDL and oxPCCD36 to
SR-BI-transfected cells. a, 125I-LDL or various
modified 125I-labeled lipoproteins (5 μg/ml) were incubated with
SR-BI-transfected CHO cells (ldlA7-SR-BI) or control vector-transfected CHO
cells (ldlA7). Cellular binding of lipoproteins was subsequently determined as
described under “Experimental Procedures.”
(Cu2+oxLDL, copper oxidized LDL). *, p
< 0.001 for comparison versus LDL. b,
125I-labeled NO2-LDL was incubated with SR-BI-expressing
cells either in the absence (N.A., no addition) or presence of the
indicated competitors. Anti-SR-BI, anti-SR-BI antibody; N.I.
IgG, nonimmune IgG. *, p < 0.001 for comparison
versus control (no addition). c, small unilamellar vesicles
were generated containing tracer levels of [3H]DPPC (25 μCi/mg
vesicles) and consisting either of the unoxidized parent phospholipid alone
(PAPC), phospholipid oxidized by
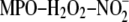 system (NO2-PAPC), or an equimolar mixture of
parent phospholipid and the indicated synthetic oxidized phospholipids
(oxPCCD36). Cells were incubated with phospholipid vesicles, and
cell-associated radioactivity was quantified. *, p < 0.001 for
comparison versus PAPC; #, p < 0.001 for comparison
versus KDdiA-PC and versus KDdiA-PC + nonimmune
(N.I.) IgG.
system (NO2-PAPC), or an equimolar mixture of
parent phospholipid and the indicated synthetic oxidized phospholipids
(oxPCCD36). Cells were incubated with phospholipid vesicles, and
cell-associated radioactivity was quantified. *, p < 0.001 for
comparison versus PAPC; #, p < 0.001 for comparison
versus KDdiA-PC and versus KDdiA-PC + nonimmune
(N.I.) IgG.
To demonstrate that oxPCCD36 directly bind to SR-BI, small unilamellar vesicles containing PLPC or PAPC as a carrier lipid, synthetic oxPCCD36 species, and tracer levels of [3H]DPPC were made and binding assays were performed as described previously (14). Vesicles comprised of PAPC (or PLPC, data not shown) alone failed to demonstrate SR-BI-specific binding (Fig. 1c). However, the NO2-PAPC and NO2-PLPC vesicles bound to SR-BI transfected cells at significantly greater levels than control cells (Fig. 1c and data not shown). Vesicles containing any of the individual synthetic oxPCCD36 (data for KDdiA-PC are shown) readily bound to SR-BI-expressing cells (Fig. 1c). Addition of a blocking anti-SR-BI antibody, but not isotype-matched control antibody, inhibited binding of the oxPCCD36 vesicles further demonstrating specificity. These data demonstrate that SR-BI, similar to CD36, recognizes oxPCCD36.
oxPCCD36 Interfere with HDL Binding to SR-BI Because of Overlap of Binding Sites—We next tested whether NO2-LDL or oxPCCD36 interfered with SR-BI-mediated HDL binding. Binding of HDL to SR-BI-expressing cells was significantly higher than vector-transfected cells as anticipated (Fig. 2a). Excess unlabeled HDL and anti-SR-BI blocking antibody, but not isotype-matched nonimmune antibody, inhibited binding to the SR-BI-transfected cells down to the level of binding to vector-transfected cells, demonstrating specificity. Forty-fold molar excess of unlabeled NO2-LDL competed with 125I-HDL for binding to SR-BI (Fig. 2a). In contrast, native LDL failed to significantly block the binding of HDL to SR-BI-transfected cells. NO2-PAPC significantly inhibited binding of 125I-HDL, in agreement with the results in Fig. 1, b and c. At the same time, native unoxidized PAPC was found to be a weak competitor (Fig. 2a). Finally, small unilamellar vesicles containing individual synthetic oxPCCD36 (data for KDdiA-PC are shown) were potent inhibitors of 125I-HDL binding to SR-BI. Collectively, these results show that LDL and phospholipids vesicles upon oxidation acquire the ability to block HDL binding to SR-BI because of oxPCCD36.
FIGURE 2.

NO2-LDL and oxPCCD36 compete for the binding of HDL to SR-BI. a, 125I-HDL (5 μg/ml) was incubated with ldlA7-SR-BI cells or vector control cells, and bound 125I-HDL was quantified. The concentrations of competitors used were 200 μg of protein/ml for lipoproteins, 40 μg of lipid/ml for vesicles, and 20 μg/ml for antibody. Anti-SR-BI, anti-SR-BI antibody; N.I. IgG, nonimmune IgG.*, p < 0.001 for comparison versus control (N.A., no addition). b, 125I-HDL (5 μg/ml) was incubated with GST-SR-BI-(144–205) protein beads, either in the absence (no addition) or presence of 20-fold excess unlabeled indicated competitors in PBS/BSA. Beads were repeatedly washed with PBS to remove unbound lipoproteins, and bound radioactivity was then quantified. *, p < 0.001 for comparison versus control (no addition). c, GST-SR-BI-(144–205) protein beads were incubated with small unilamellar vesicles consisting of PAPC, NO2-PAPC, or the indicated oxPCCD36 with tracer levels of [3H]DPPC (25 μCi/mg vesicles) (10 μg/ml), washed repeatedly with PBS, and assayed for bead-associated radioactivity. *, p < 0.001 for comparison versus PAPC. #, p < 0.001 for comparison versus KDdiA-PC. d, concentration dependence of 125I-HDL binding to SR-BI-overexpressing HEK-293T cell and GST-SR-BI-(144–205) protein. Indicated concentrations of 125I-HDL were incubated with either cells or GST-SR-BI-(144–205) beads at 25 °C for 3 h and then washed, and bound radioactivity was quantified. Specific 125I-HDL binding was determined by subtracting background binding to control vector-transfected cells or GST beads alone, respectively.
SR-BI is a multiligand receptor, one potential mechanism for the observed inhibition is that the binding sites for HDL and oxPCCD36 on SR-BI overlap. To test this hypothesis an extracellular amino-terminal domain of human SR-BI (amino acids 144–205) containing the putative HDL ligand-binding site was expressed as a GST fusion protein, isolated and purified by affinity chromatography on glutathione-Sepharose 4B beads, and used in binding and competition studies. The selection of this SR-BI-(144–205) peptide was based on separate studies of lipoprotein binding domains in CD36, a close relative of SR-BI that can also bind HDL (24, 25). Binding of 125I-HDL to GST-SR-BI-(144–205) was significantly higher than control GST beads (Fig. 2b). Binding was specific because excess unlabeled HDL significantly inhibited binding of 125I-HDL. Importantly, the anti-SR-BI blocking antibody inhibited HDL binding to GST-SR-BI-(144–205) protein to the same extent as in SR-BI-expressing cells (Fig. 2b), suggesting that GST-SR-BI-(144–205) contains the true binding site for HDL. Other properties of the 125I-HDL-binding site on GST-SR-BI-(144–205) mirrored those of the full-length SR-BI expressed on cells. Specifically, NO2-PAPC and vesicles containing individual synthetic oxPCCD36 were potent inhibitors of 125I-HDL binding (results for KDdiA-PC are shown), whereas vesicles consisting of native unoxidized PAPC or PLPC were weak competitors (Fig. 2b, data for PAPC are shown). Direct binding experiments demonstrated that small unilamellar vesicles comprised of PAPC or PLPC alone failed to bind significantly to GST-SR-BI-(144–205) (Fig. 1c and data not shown), whereas NO2-PAPC and vesicles containing any of the individual synthetic oxPCCD36 (data for KDdiA-PC are shown) were found to bind to GST-SR-BI-(144–205) at significantly greater levels than control GST (Fig. 2c). Addition of the anti-SR-BI antibody, but not isotype-matched control antibody, blocked binding of the vesicles containing oxPCCD36. Comparison of SR-BI-overexpressing cells and purified GST-SR-BI-(144–205) fusion protein showed similar saturable HDL binding curves (Fig. 2d). The values for half-maximal concentration for HDL binding were similar, 5.6 ± 0.5 μg/ml for cells overexpressing SR-BI and 4.6 ± 0.1 μg/ml for the GST fusion protein. Together these results suggest that the ligand-binding site of SR-BI lies in the region between amino acids 144 and 205 and that the binding sites for HDL and oxPCCD36 on SR-BI overlap.
oxPCCD36 Bind to Hepatocyte SR-BI and Interfere with SR-BI-mediated Selective Cholesteryl Uptake—Selective uptake of HDL cholesteryl esters in hepatocytes is a major physiological function of SR-BI and is a critical part of the reverse cholesterol transport pathway in the body. Because oxPCCD36 accumulate in plasma under conditions of increased oxidative stress and dyslipoproteinemia, and are associated with atherosclerosis development (10, 11, 14), we tested whether oxPCCD36 can affect SR-BI-mediated selective uptake of cholesteryl esters in hepatocytes. We first used NO2-LDL to test whether hepatocytes recognize this mildly modified form of oxidized LDL via SR-BI and whether oxPCCD36 present in NO2-LDL serve as ligands for hepatocyte SR-BI. Hep G2 cells bound significant amounts of 125I-labeled NO2-LDL (Fig. 3a). SR-BI was the major receptor on Hep G2 cells for 125I-labeled NO2-LDL because an anti-SR-BI blocking antibody but not isotype-matched nonimmune antibody significantly inhibited binding. Excess amounts of unlabeled HDL but not native LDL effectively competed with 125I-labeled NO2-LDL for binding to Hep G2 cells (Fig. 3a). In agreement with the data obtained using SR-BI-overexpressing cells, binding was significantly inhibited by oxPCCD36 but not by native unoxidized phospholipids. These findings demonstrated that LDL modified by the MPO-H2O2-NO–2 system was recognized specifically by SR-BI on hepatocytes via its oxidized phospholipid moiety.
FIGURE 3.

Binding of NO2-LDL and oxPCCD36 to Hep G2 cells. a, 125I-NO2-LDL (5 μg/ml) was incubated with Hep G2 cells either in the absence (N.A., no addition) or presence of the indicated competitors at 4 °C for 3 h in the appropriate media. Cellular binding of lipoproteins was subsequently determined as described under “Experimental Procedures.” Anti-SR-BI, anti-SR-BI antibody; N.I. IgG, nonimmune IgG. *, p < 0.001 for comparison versus control (no addition). b, 125I-HDL (5 μg/ml) was incubated with Hep G2 cells either in the absence (no addition) or presence of the indicated competitors. *, p < 0.001 for comparison versus control (no addition). c, small unilamellar vesicles were generated as described in Fig. 1, and Hep G2 cells were incubated with phospholipid vesicles (10 μg/mg of cell protein), following 3 h of incubation at 4 °C and washed, and cell-associated radioactivity was quantified. *, p < 0.001 for comparison versus PAPC. #, p < 0.001 for comparison versus KDdiA-PC and versus KDdiA-PC + nonimmune IgG.
We next investigated the effect of oxPCCD36 on the binding of 125I-HDL to Hep G2 cells. Hep G2 cells bound significant amounts of 125I-HDL, as anticipated. Excess of unlabeled HDL and anti-SR-BI blocking antibody, but not isotype-matched nonimmune antibody, inhibited 125I-HDL binding to Hep G2 cells demonstrating specificity of the binding. Importantly, excess of all competitors containing oxPCCD36, including unlabeled NO2-LDL, NO2-PAPC and vesicles containing individual synthetic oxPCCD36 significantly inhibited binding of 125I-HDL to SR-BI (Fig. 3b), in agreement with the results in Fig. 2. In contrast, native PAPC or PLPC (data for PAPC are shown) had no significant effect on the binding (Fig. 3b).
To demonstrate that oxPCCD36 directly bind to SR-BI on hepatocytes, small unilamellar vesicles containing PLPC or PAPC as a carrier lipid, synthetic oxPCCD36 species, and tracer levels of [3H]DPPC were made, and binding assays were performed as in Fig. 1c. Vesicles comprised of PAPC (or PLPC, data not shown) alone failed to demonstrate significant binding to Hep G2 cells (Fig. 3c). NO2-PAPC vesicles bound to SR-BI-transfected cells at significantly greater levels than PAPC vesicles (Fig. 3c). Vesicles containing individual synthetic oxPCCD36 (data for KOdiA-PC are shown) readily bound to Hep G2 cells (Fig. 3c). Blocking anti-SR-BI antibody, but not isotype-matched control antibody, inhibited binding of the oxPCCD36 vesicles to hepatocytes, demonstrating that SR-BI is the major receptor for oxPCCD36 on hepatocytes.
Finally, we tested the effect of ligands containing oxPCCD36 on SR-BI-mediated [3H]cholesteryl ester association, 125I-HDL uptake, and selective cholesteryl ester uptake using small unilamellar phospholipid vesicles. Native PAPC had a mild effect on cholesteryl ester association, 125I-HDL uptake, and selective uptake of HDL cholesteryl esters in Hep G2 cells (Fig. 4, a–c). At the same time, vesicles containing oxPCCD36 significantly inhibited all tested SR-BI-mediated processes (Fig. 4, a–c). Importantly, oxPCCD36 not only significantly reduced holoparticle uptake of HDL as evidenced by decreased uptake of 125I-HDL (Fig. 4b), it also almost completely inhibited hepatocyte selective uptake of cholesteryl esters (Fig. 4c). This result strongly suggests that oxPCCD36 also interferes with the second step of SR-BI-mediated selective uptake, i.e. lipid transfer from HDL to the cell membrane. An earlier report suggested that reverse cholesterol transport may be proportional to cell surface expression of SR-BI (26). No change in surface expression of SR-BI was observed in Hep G2 cells pretreated with either HDL or with HDL plus oxPCCD36 for the periods of time used in this experiment (data not shown). Taken together, these results support our hypothesis that ligands for SR-BI that contain oxPCCD36 compete for the binding of HDL to SR-BI and disrupt SR-BI-mediated cholesteryl ester selective uptake from HDL.
FIGURE 4.

oxPCCD36 interfere with SR-BI-mediated selective cholesteryl ester uptake. Hep G2 cells were incubated with either [3H]COE HDL (a) or 125I-HDL (b) at a concentration 10 μg/ml for 5 h 37 °C, with indicated additions, and then cells were washed with warm PBS and chased for another 30 min in the presence of 100 μg/ml unlabeled HDL. a, cells were lysed; cell-associated radioactivity was quantified and [3H]COE HDL association determined. b, 125I-HDL uptake was determined as described under “Experimental Procedures.” c, selective uptake was calculated from data in a and b as described under “Experimental Procedures.” *, p < 0.001 for comparison versus control.
DISCUSSION
Pro-atherosclerotic lipid abnormalities are associated with enhanced oxidative stress and the generation of biologically active oxidized lipids. Oxidized phospholipids accumulate at sites of oxidative stress, including plasma and in the vessel wall (10–12). They are involved in all stages of atherosclerosis and can mediate many atherogenic processes from the earliest entry of monocytes into the vessel wall to thrombus formation (9–11, 14, 27). In this study we show that a recently described family of specific oxidized phospholipids represents a ligand for the scavenger receptor BI and interferes with the major physiological function of SR-BI, namely selective uptake of cholesteryl esters from HDL.
We demonstrated the following: (i) oxPCCD36 are recognized by SR-BI when presented in various forms. (ii) oxPCCD36 interfered with HDL binding to SR-BI because of an overlap of binding sites for oxPCCD36 and HDL on SR-BI. (iii) oxPCCD36 prevented SR-BI-mediated selective uptake of cholesteryl esters from HDL in hepatocytes. These observations suggest a novel pro-atherogenic activity of specific oxidized phospholipids: an inhibition of reverse cholesterol transport.
SR-BI belongs to the CD36 family of proteins that is well known for multiligand specificity. SR-BI and CD36 share significant sequence homology and ligands, including HDL, oxidized LDL, and anionic phospholipids (28–30). Oxidized LDL had been shown to interact with SR-BI (20, 21, 23), although the molecular mechanism of this interaction has not been elucidated. We previously reported that a structurally conserved family of oxidized choline glycerophospholipids with an oxidatively truncated sn-2 acyl group incorporating a terminal γ-hydroxy (or oxo)-α,β-unsaturated carbonyl serves as high-affinity ligands for macrophage CD36, mediating recognition of various oxidized forms of LDL (11, 14). oxPCCD36 are present in vivo in human and animal atherosclerotic lesions and accumulate in significant amounts in plasma in dyslipoproteinemia or in normolipidemia associated with low HDL levels (10, 11). oxPCCD36 mediate macrophage foam cell formation and promote platelet hyper-reactivity via scavenger receptor CD36 (10, 11). Based on the close similarity between CD36 and SR-BI, we hypothesized that oxPCCD36 could serve as ligands for SR-BI as well. Using direct binding and competition assays, we demonstrated that SR-BI binds oxidized LDL in the same fashion as CD36, via recognition of oxPCCD36 formed during oxidative modification of LDL.
SR-BI is intimately involved in HDL metabolism in vivo (30, 31). Based on 125I-labeled NO2-LDL and oxPCCD36 binding studies, we hypothesized that oxPCCD36 could interfere with the ability of SR-BI to mediate cholesterol transport to and from HDL. Our competition experiments using 125I-HDL as a ligand and various competitors containing oxPCCD36 demonstrated that oxPCCD36 significantly reduced HDL binding to SR-BI, and thus affect HDL-mediated processes. To elucidate whether the observed inhibition is because of spatial hindrance or more complex cellular events, we performed competition experiments using the extracellular amino-terminal domain of human SR-BI (amino acids 144–205) containing the putative HDL ligand-binding site. Importantly, the Kd value of the binding of HDL to this protein was very similar to that determined for the full-length SR-BI expressed on cells (Fig. 2, a, b, and d). Moreover, an anti-SR-BI blocking antibody inhibited HDL binding to GST-SR-BI-(144–205) peptide, strongly suggesting that it contains the true binding site for HDL. NO2-PAPC and vesicles containing synthetic oxPCCD36 bound specifically to GST-SR-BI-(144–205) with a Kd value similar to that for HDL (Fig. 2c and data not shown). Others have reported that a M158R mutant does not bind either HDL or LDL (32) in agreement with our data. Taken together, these results suggest that the ligand-binding sites for both HDL and oxPCCD36 on SR-BI lie in the region of amino acids 144–205 and that the binding sites for HDL and oxPCCD36 on SR-BI overlap, thus providing the molecular basis for the observed competition between the two ligands on cells.
Our results suggest that under conditions of increased oxidative stress, accumulation of specific oxidized phospholipids may promote atherosclerosis not only by inducing uptake of modified lipoproteins by macrophages via CD36, but can also interfere with reverse cholesterol transport by preventing SR-BI-mediated selective cholesteryl ester uptake in hepatocytes.
Acknowledgments
We thank V. Verbovetskaya for technical assistance.
This work was supported in part by National Institutes of Health Grants HL053315, HL077213 (to E. A. P.), HL53315 (to R. G. S.), and HL70083 (to M. F.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: HDL, high density lipoprotein; SR-BI, scavenger receptor class B, type I; oxPC, oxidized choline glycerophospholipid; PBS, phosphate-buffered saline; GST, glutathione S-transferase; LDL, low density lipoprotein; COE, cholesteryl oleate ether; PAPC, 1-hexadecanoyl-2-eicosatetra-5′,8′,11′,14′-enoyl-sn-glycero-3-phosphocholine; PLPC, -hexadecanoyl-2-octadec-9′,12′-dienoyl-sn-glycero-3-phosphocholine; POPC, 1-hexadecanoyl-2-octadec-9′-enoyl-sn-glycero-3-phosphocholine; DPPC, 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine; BSA, bovine serum albumin; KOOA, 5-keto-8-oxo-6-octenoic acid; CHO, Chinese hamster ovary; KDdiA, keto-6-octendioic acid; CE, cholesteryl ester; MPO, myeloperoxidase.
References
- 1.Arai, T., Wang, N., Bezouevski, M., Welch, C., and Tall, A. R. (1999) J. Biol. Chem. 274 2366–2371 [DOI] [PubMed] [Google Scholar]
- 2.Kozarsky, K. F., Donahee, M. H., Glick, J. M., Krieger, M., and Rader, D. J. (2000) Arterioscler. Thromb. Vasc. Biol. 20 721–727 [DOI] [PubMed] [Google Scholar]
- 3.Ueda, Y., Gong, E., Royer, L., Cooper, P. N., Francone, O. L., and Rubin, E. M. (2000) J. Biol. Chem. 275 20368–20373 [DOI] [PubMed] [Google Scholar]
- 4.Huszar, D., Varban, M. L., Rinninger, F., Feeley, R., Arai, T., Fairchild-Huntress, V., Donovan, M. J., and Tall, A. R. (2000) Arterioscler. Thromb. Vasc. Biol. 20 1068–1073 [DOI] [PubMed] [Google Scholar]
- 5.Acton, S. L., Scherer, P. E., Lodish, H. F., and Krieger, M. (1994) J. Biol. Chem. 269 21003–21009 [PubMed] [Google Scholar]
- 6.Krieger, M. (1999) Annu. Rev. Biochem. 68 523–558 [DOI] [PubMed] [Google Scholar]
- 7.Acton, S., Rigotti, A., Landschulz, K. T., Xu, S., Hobbs, H. H., and Krieger, M. (1996) Science 271 518–520 [DOI] [PubMed] [Google Scholar]
- 8.Ohgami, N., Nagai, R., Miyazaki, A., Ikemoto, M., Arai, H., Horiuchi, S., and Nakayama, H. (2001) J. Biol. Chem. 276 13348–13355 [DOI] [PubMed] [Google Scholar]
- 9.Berliner, J. A., and Watson, A. D. (2005) N. Engl. J. Med. 353 9–11 [DOI] [PubMed] [Google Scholar]
- 10.Podrez, E. A., Byzova, T. V., Febbraio, M., Salomon, R. G., Ma, Y., Valiyaveettil, M., Poliakov, E., Sun, M., Finton, P. J., Curtis, B. R., Chen, J., Zhang, R., Silverstein, R. L., and Hazen, S. L. (2007) Nat. Med. 13 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Podrez, E. A., Poliakov, E., Shen, Z., Zhang, R., Deng, Y., Sun, M., Finton, P. J., Shan, L., Febbraio, M., Hajjar, D. P., Silverstein, R. L., Hoff, H. F., Salomon, R. G., and Hazen, S. L. (2002) J. Biol. Chem. 277 38517–38523 [DOI] [PubMed] [Google Scholar]
- 12.Hoff, H. F., O'Neil, J., Wu, Z., Hoppe, G., and Salomon, R. L. (2003) Arterioscler. Thromb. Vasc. Biol. 23 275–282 [DOI] [PubMed] [Google Scholar]
- 13.Podrez, E. A., Febbraio, M., Sheibani, N., Schmitt, D., Silverstein, R. L., Hajjar, D. P., Cohen, P. A., Frazier, W. A., Hoff, H. F., and Hazen, S. L. (2000) J. Clin. Investig. 105 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Podrez, E. A., Poliakov, E., Shen, Z., Zhang, R., Deng, Y., Sun, M., Finton, P. J., Shan, L., Gugiu, B., Fox, P. L., Hoff, H. F., Salomon, R. G., and Hazen, S. L. (2002) J. Biol. Chem. 277 38503–38516 [DOI] [PubMed] [Google Scholar]
- 15.Markwell, M. A., Haas, S. M., Bieber, L. L., and Tolbert, N. E. (1978) Anal. Biochem. 87 206–210 [DOI] [PubMed] [Google Scholar]
- 16.Podrez, E. A., Schmitt, D., Hoff, H. F., and Hazen, S. L. (1999) J. Clin. Investig. 103 1547–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldstein, J. L., Ho, Y. K., Basu, S. K., and Brown, M. S. (1979) Proc. Natl. Acad. Sci. U. S. A. 76 333–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashkenas, J., Penman, M., Vasile, E., Acton, S., Freeman, M., and Krieger, M. (1993) J. Lipid Res. 34 983–1000 [PubMed] [Google Scholar]
- 19.Greene, D. J., Skeggs, J. W., and Morton, R. E. (2001) J. Biol. Chem. 276 4804–4811 [DOI] [PubMed] [Google Scholar]
- 20.Gillotte-Taylor, K., Boullier, A., Witztum, J. L., Steinberg, D., and Quehenberger, O. (2001) J. Lipid Res. 42 1474–1482 [PubMed] [Google Scholar]
- 21.Han, J., Nicholson, A. C., Zhou, X., Feng, J., Gotto, A. M., Jr., and Hajjar, D. P. (2001) J. Biol. Chem. 276 16567–16572 [DOI] [PubMed] [Google Scholar]
- 22.Rigotti, A., Acton, S. L., and Krieger, M. (1995) J. Biol. Chem. 270 16221–16224 [DOI] [PubMed] [Google Scholar]
- 23.Nishikawa, K., Arai, H., and Inoue, K. (1990) J. Biol. Chem. 265 5226–5231 [PubMed] [Google Scholar]
- 24.Calvo, D., Gomez-Coronado, D., Suarez, Y., Lasuncion, M. A., and Vega, M. A. (1998) J. Lipid Res. 39 777–788 [PubMed] [Google Scholar]
- 25.Gu, X., Trigatti, B., Xu, S., Acton, S., Babitt, J., and Krieger, M. (1998) J. Biol. Chem. 273 26338–26348 [DOI] [PubMed] [Google Scholar]
- 26.Shetty, S., Eckhardt, E. R., Post, S. R., and van der Westhuyzen, D. R. (2006) Arterioscler. Thromb. Vasc. Biol. 26 2125–2131 [DOI] [PubMed] [Google Scholar]
- 27.Chang, M. K., Binder, C. J., Miller, Y. I., Subbanagounder, G., Silverman, G. J., Berliner, J. A., and Witztum, J. L. (2004) J. Exp. Med. 200 1359–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jessup, W., Gelissen, I. C., Gaus, K., and Kritharides, L. (2006) Curr. Opin. Lipidol. 17 247–257 [DOI] [PubMed] [Google Scholar]
- 29.Connelly, M. A., and Williams, D. L. (2004) Curr. Opin. Lipidol. 15 287–295 [DOI] [PubMed] [Google Scholar]
- 30.Rigotti, A., Miettinen, H. E., and Krieger, M. (2003) Endocr. Rev. 24 357–387 [DOI] [PubMed] [Google Scholar]
- 31.Yancey, P. G., Bortnick, A. E., Kellner-Weibel, G., de la Llera-Moya, M., Phillips, M. C., and Rothblat, G. H. (2003) Arterioscler. Thromb. Vasc. Biol. 23 712–719 [DOI] [PubMed] [Google Scholar]
- 32.Gu, X., Kozarsky, K., and Krieger, M. (2000) J. Biol. Chem. 275 29993–30001 [DOI] [PubMed] [Google Scholar]